
CXCR4 and CXCR7 Inhibition Ameliorates the Formation of Platelet–Neutrophil Complexes and Neutrophil Extracellular Traps through Adora2b Signaling

 ,
,  and
and 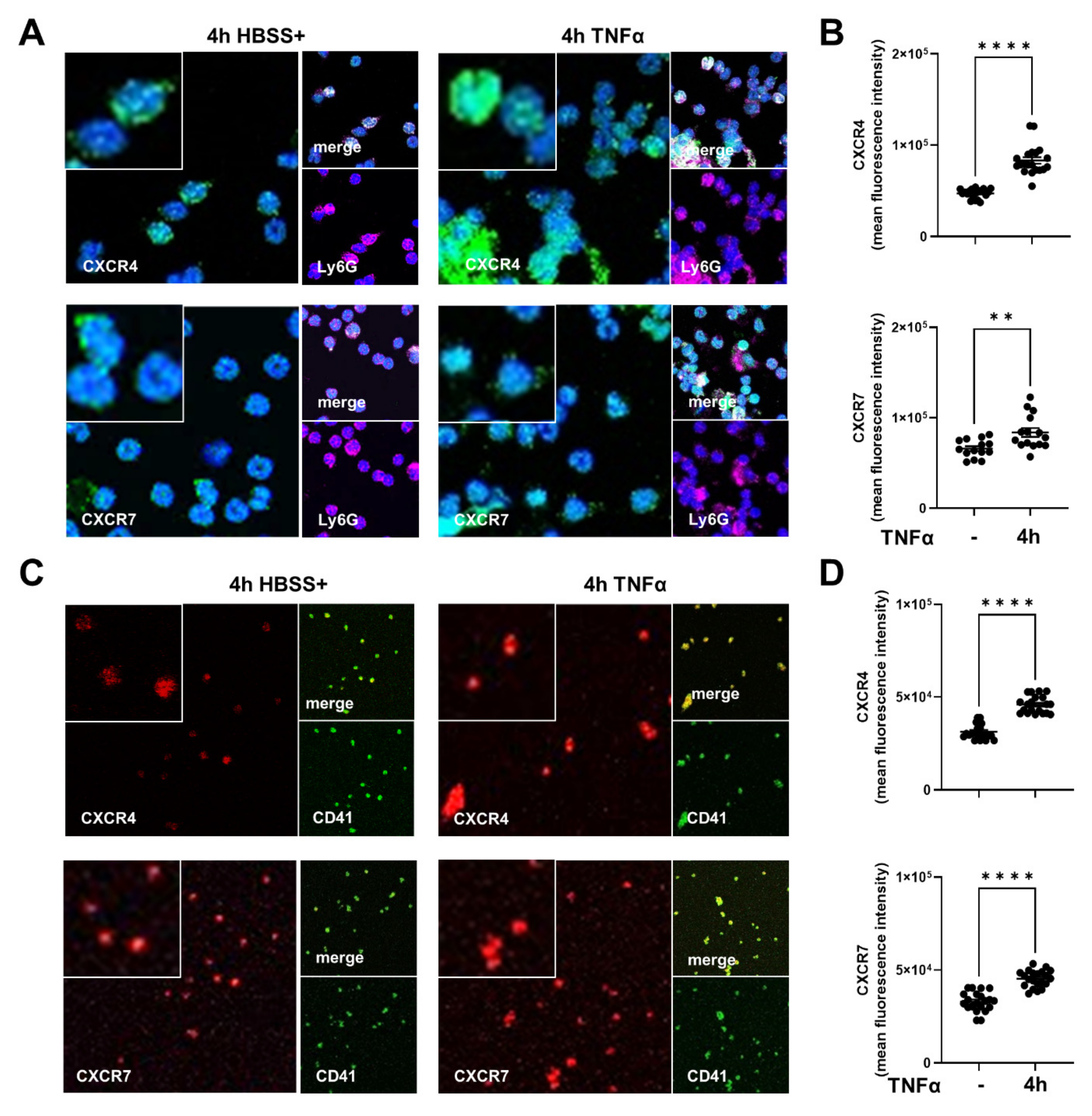{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
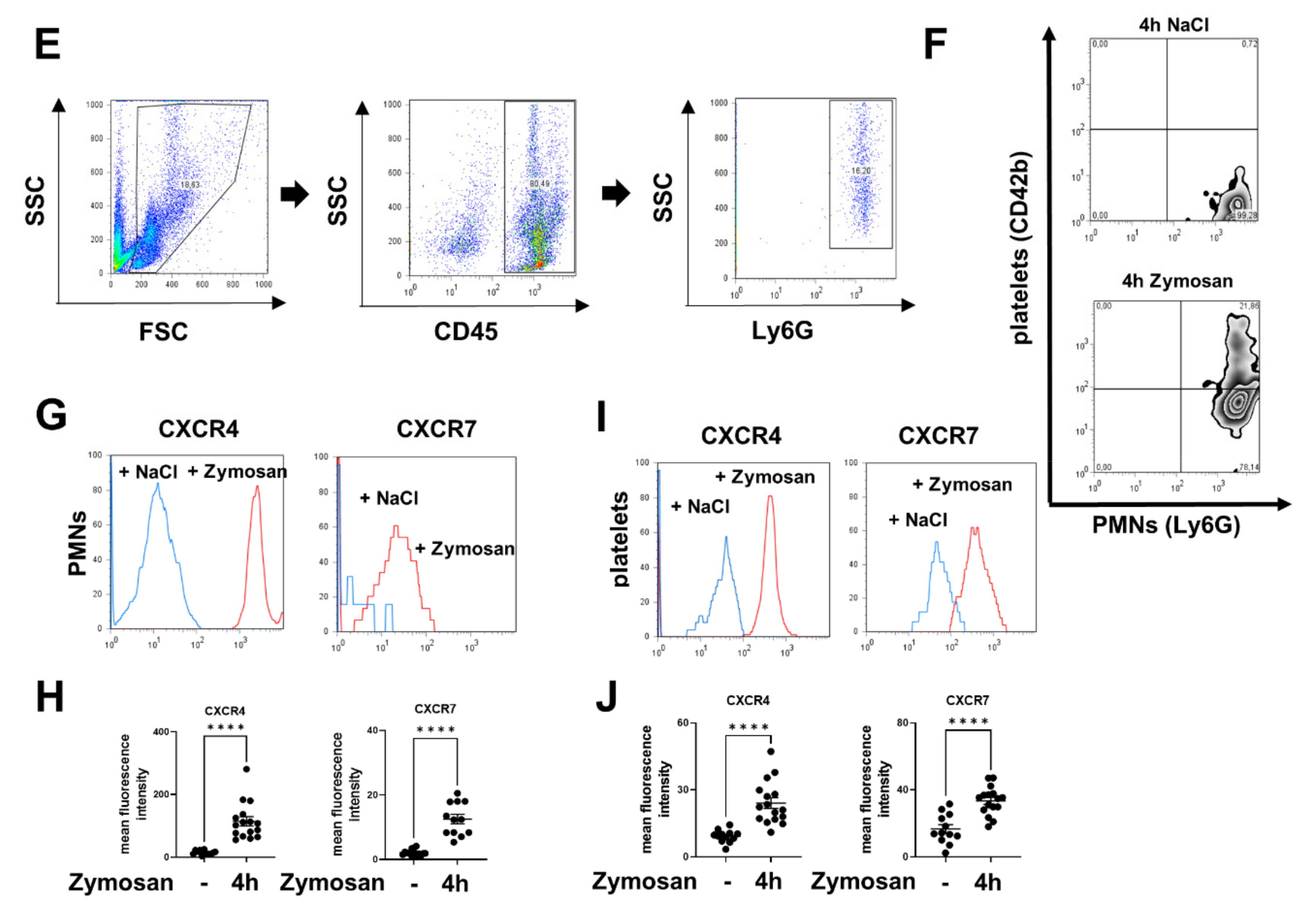
2.1. Expression of CXCR4 and CXCR7 in Platelets, Neutrophils, and Platelet–Neutrophil Complexes In Vivo and In Vitro
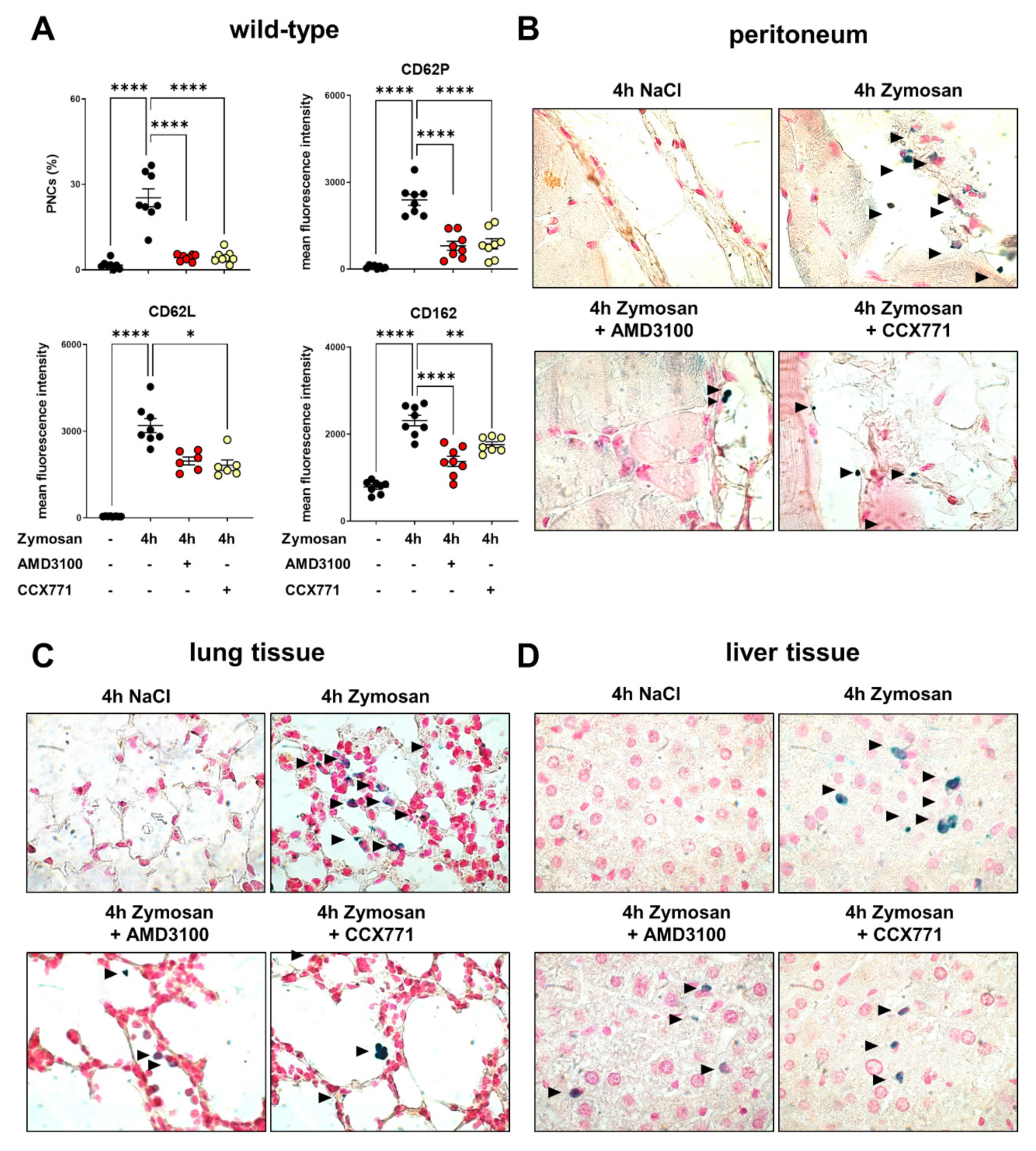
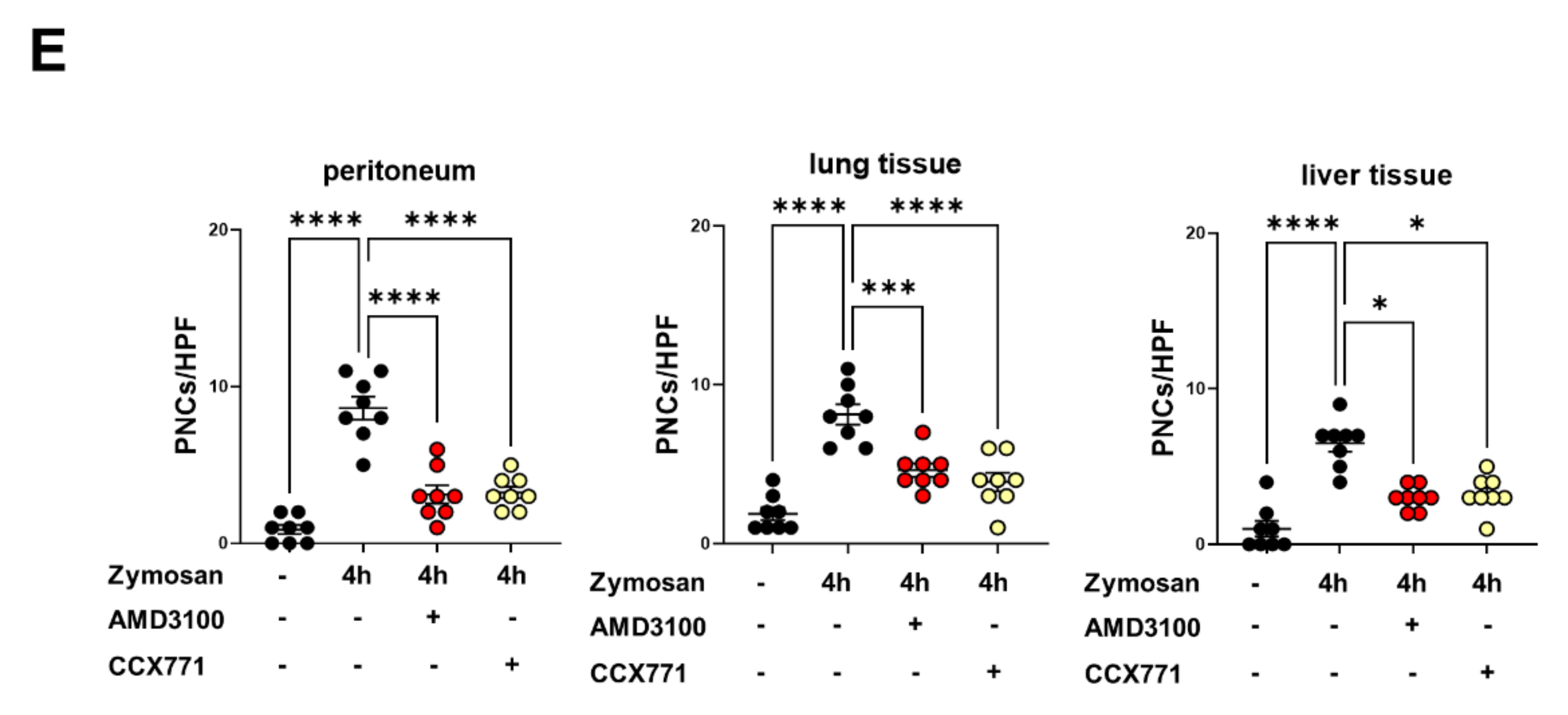
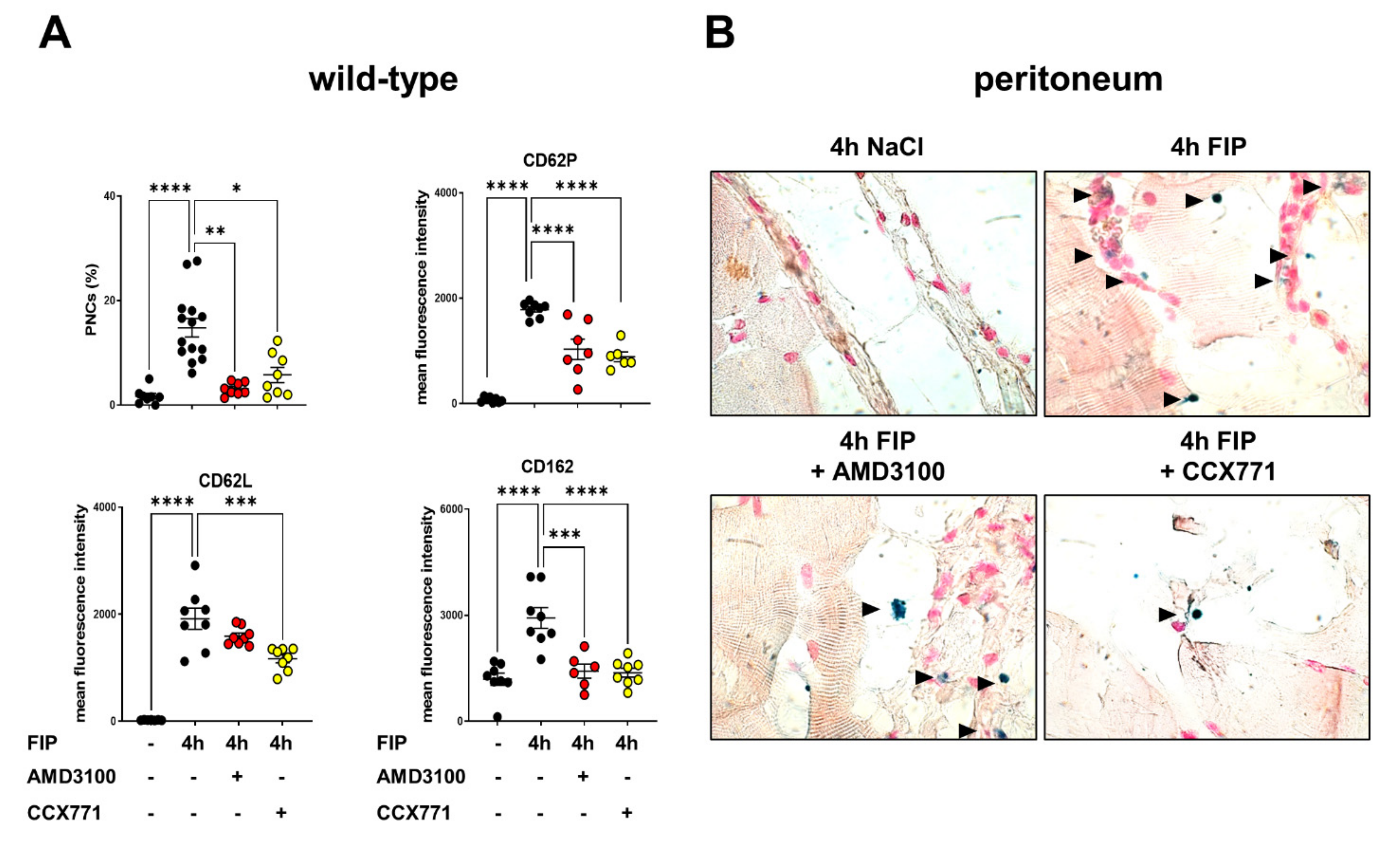
2.2. CXCR4 and CXCR7 Inhibition Ameliorates the Formation of Platelet–Neutrophil Complexes during Acute Inflammation
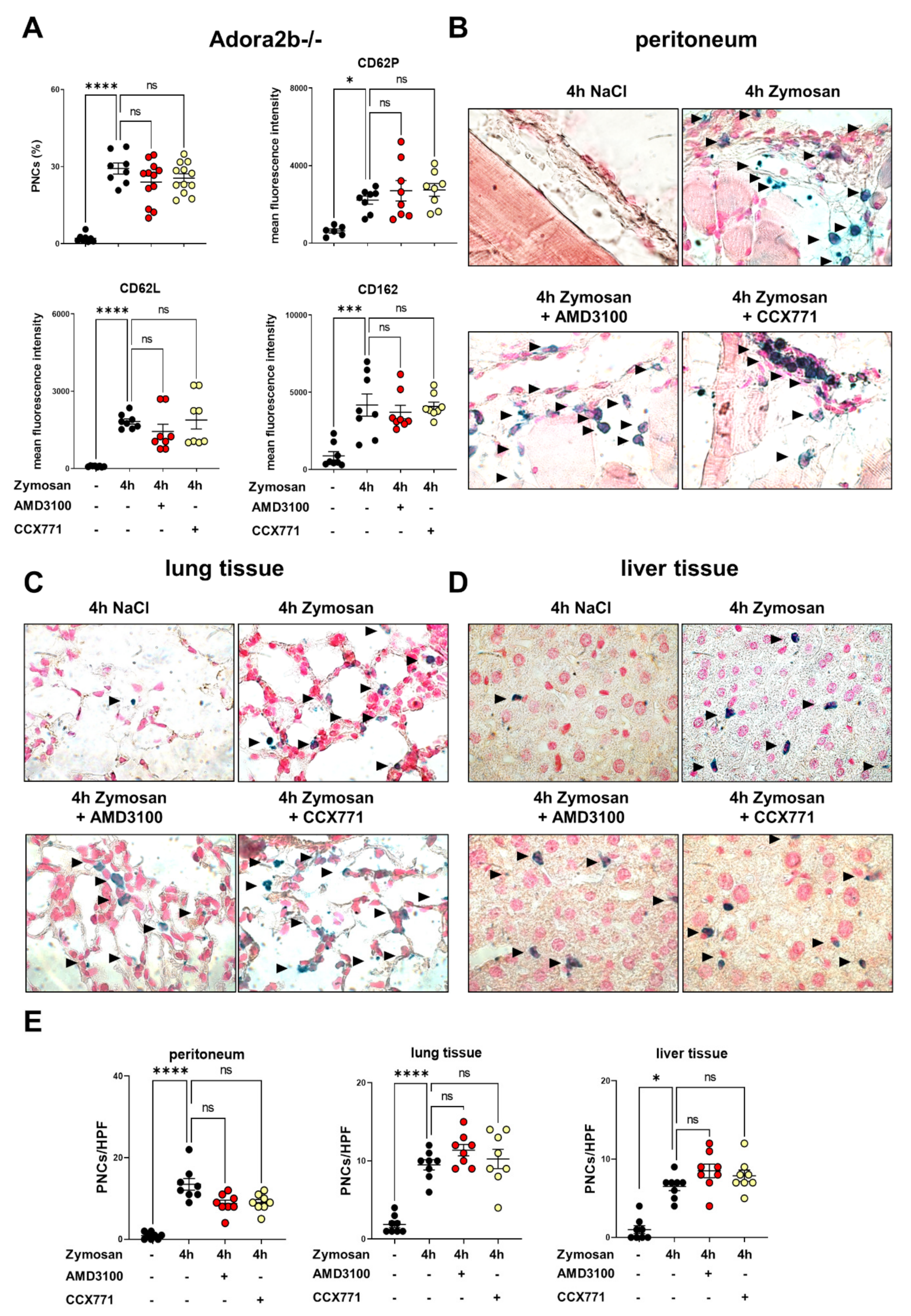
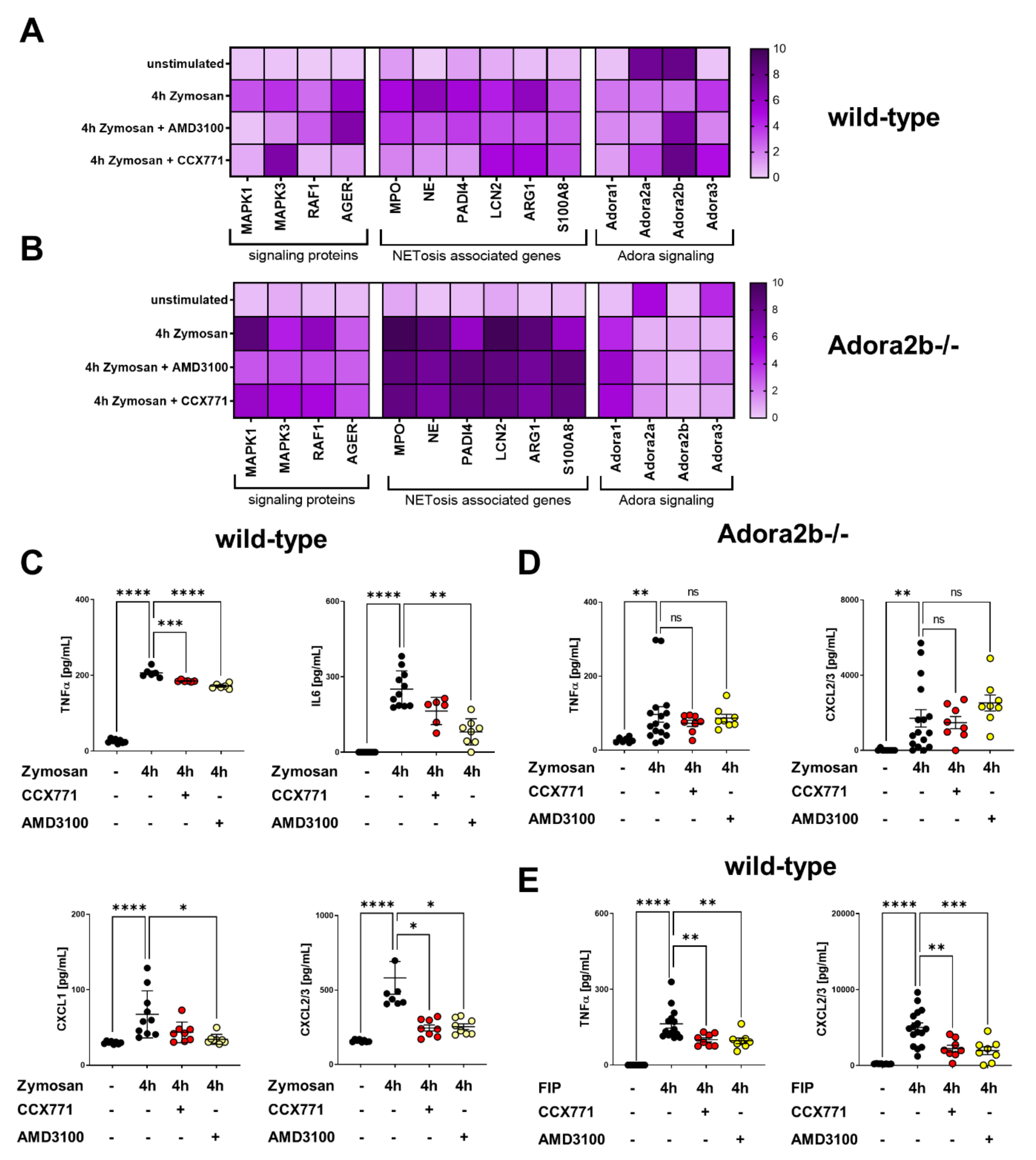
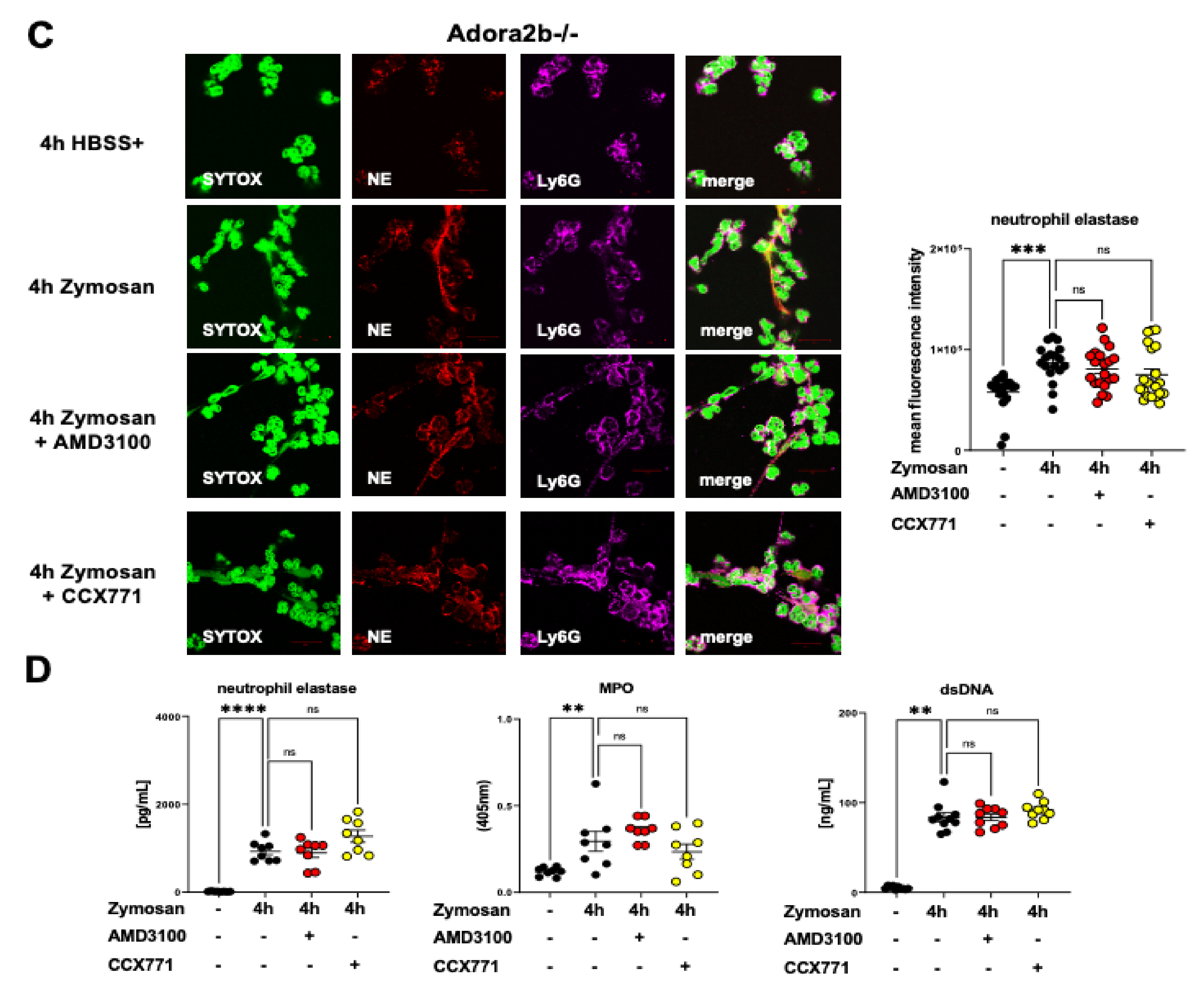
2.3. Adora2b Signaling Is Crucial for the Pharmacological Effects of CXCR4 and CXCR7 Antagonism on Inhibiting PNC Formation
2.4. Inhibition of CXCR4 and CXCR7 Dampens PNC Formation during Polymicrobial Inflammation
2.5. Specific CXCR4 and CXCR7 Inhibition Influences Intracellular Pathways and NETosis-Related Gene Expression
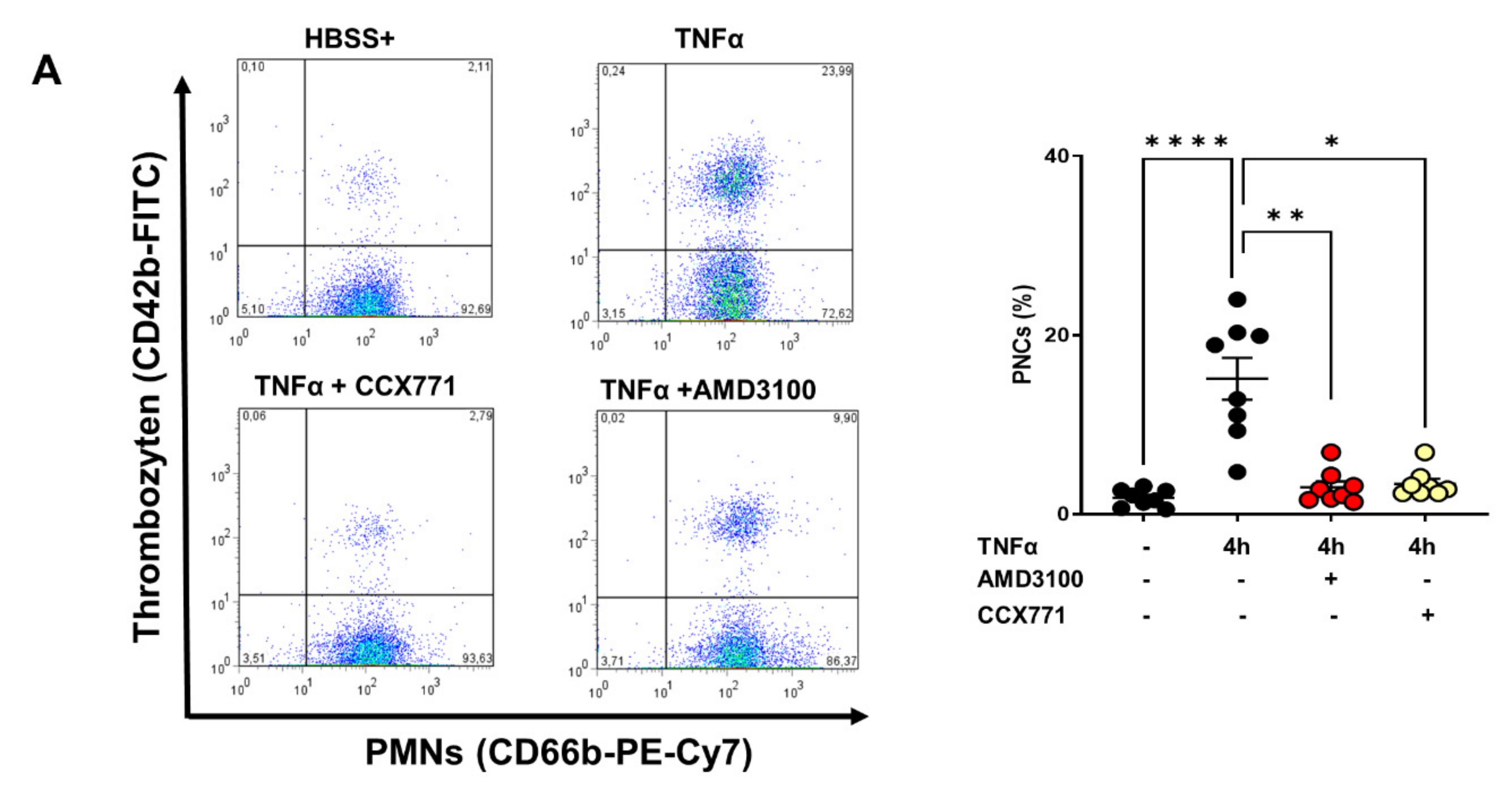
2.6. Effects of CXCR4 and CXCR7 Antagonism on Human PNC Formation and PNC Migratory Behavior
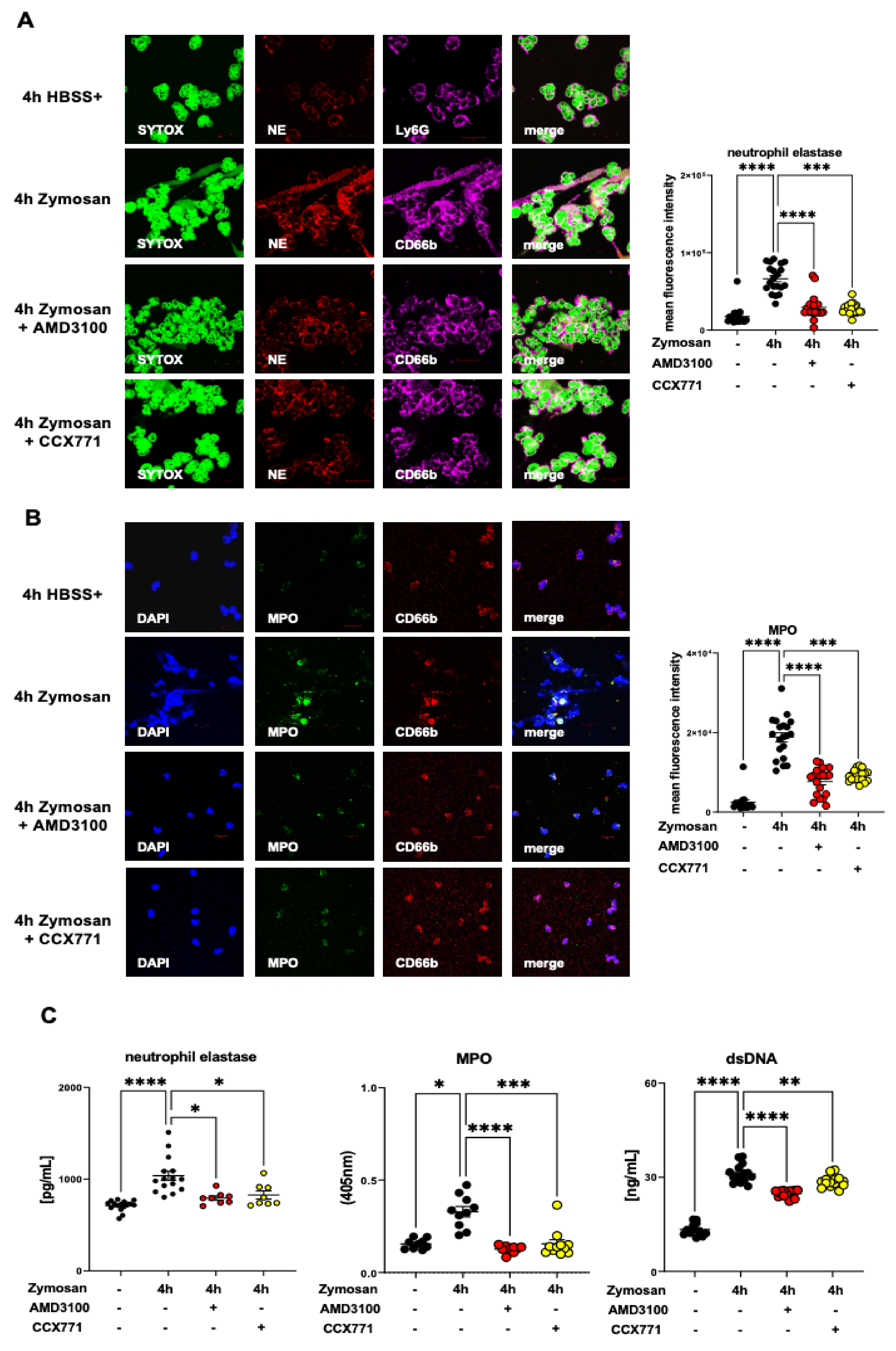
2.7. CXCR4 and CXCR7 Antagonism Affects the Formation of NETs Ex Vivo
2.8. Effects of CXCR4 and CXCR7 Inhibition on Human NET Formation In Vitro
3. Discussion
4. Material and Methods
4.1. Animals
4.2. Reagents
4.3. Zymosan-Induced Peritonitis and Sepsis
4.4. Fecal-Induced Peritonitis and Sepsis
4.5. Platelet–Neutrophil Complex Formation In Vivo and In Vitro
4.6. NET Quantification and Myeloperoxidase Release
4.7. Immunohistochemical Detection of Platelet–Neutrophil Complexes
4.8. Immunofluorescence Experiments
4.9. RT-PCR
4.10. Cytokine and Chemokine Concentration
4.11. Software and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Adora2b | Adenosine receptor A2B |
| AGER | Advanced glycosylation end product-specific receptor |
| ACKR3 | Atypical chemokine receptor 3 |
| APC | Allophycocyanin |
| CXCL | C-X-C motif chemokine ligand |
| DAPI | 4‘,6-diamidin-2-phenylindol |
| ELISA | Enzyme-linked immunosorbent assay |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| FITC | Fluorescein isothiocyanate |
| FSC | Forward scatter |
| HBSS | Hank´s balanced salt solution |
| HMGB1 | High-mobility-group-box 1 |
| IL | Interleukin |
| LCN2 | Lipocalin 2 |
| MAPK | Mitogen-activated protein kinase |
| MPO | Myeloperoxidase |
| NE | Neutrophil elastase |
| NET | Neutrophil extracellular traps |
| PADI4 | Peptidyl arginine deiminase type 4 |
| PE | Phycoerythrin |
| PE/Cy7 | Phycoeryhtrin/cyanin 7 |
| PerCP | Peridinin-chlorophyll-protein |
| PMNs | Polymorphonuclear neutrophils |
| PNCs | Platelet–neutrophil complexes |
| PBS- | Phosphate-buffered solution without calcium |
| RAF1 | Rapidly accelerated fibrosarcoma 1 |
| SDF-1 | Stromal cell-derived factor-1 |
| SSC | Sideward scatter |
| TNF-α | Tumor necrosis factor alpha |
References
- Nasa, P.; Juneja, D.; Singh, O.; Dang, R.; Arora, V. Severe sepsis and its impact on outcome in elderly and very elderly patients admitted in intensive care unit. J. Intensive Care Med. 2012, 27, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Coopersmith, C.M.; De Backer, D.; Deutschman, C.S.; Ferrer, R.; Lat, I.; Machado, F.R.; Martin, G.S.; Martin-Loeches, I.; Nunnally, M.E.; Antonelli, M.; et al. Surviving Sepsis Campaign: Research Priorities for Sepsis and Septic Shock. Crit. Care Med. 2018, 46, 1334–1356. [Google Scholar] [CrossRef]
- Cahilog, Z.; Zhao, H.; Wu, L.; Alam, A.; Eguchi, S.; Weng, H.; Ma, D. The Role of Neutrophil NETosis in Organ Injury: Novel Inflammatory Cell Death Mechanisms. Inflammation 2020, 43, 2021–2032. [Google Scholar] [CrossRef]
- Johansson, D.; Shannon, O.; Rasmussen, M. Platelet and neutrophil responses to Gram positive pathogens in patients with bacteremic infection. PLoS ONE 2011, 6, e26928. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, M.; Rath, D.; Gawaz, M. Role of chemokine receptors CXCR4 and CXCR7 for platelet function. Biochem. Soc. Trans. 2015, 43, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Konrad, F.M.; Meichssner, N.; Bury, A.; Ngamsri, K.C.; Reutershan, J. Inhibition of SDF-1 receptors CXCR4 and CXCR7 attenuates acute pulmonary inflammation via the adenosine A2B-receptor on blood cells. Cell Death Dis. 2017, 8, e2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngamsri, K.C.; Jans, C.; Putri, R.A.; Schindler, K.; Gamper-Tsigaras, J.; Eggstein, C.; Kohler, D.; Konrad, F.M. Inhibition of CXCR4 and CXCR7 Is Protective in Acute Peritoneal Inflammation. Front. Immunol. 2020, 11, 407. [Google Scholar] [CrossRef]
- Ngamsri, K.C.; Muller, A.; Bosmuller, H.; Gamper-Tsigaras, J.; Reutershan, J.; Konrad, F.M. The Pivotal Role of CXCR7 in Stabilization of the Pulmonary Epithelial Barrier in Acute Pulmonary Inflammation. J. Immunol. 2017, 198, 2403–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, M.; von Ungern-Sternberg, S.N.; Seizer, P.; Schlegel, F.; Buttcher, M.; Sindhu, N.A.; Muller, S.; Mack, A.; Gawaz, M. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4-CXCR7. Cell Death Dis. 2015, 6, e1989. [Google Scholar] [CrossRef] [Green Version]
- Alampour-Rajabi, S.; El Bounkari, O.; Rot, A.; Muller-Newen, G.; Bachelerie, F.; Gawaz, M.; Weber, C.; Schober, A.; Bernhagen, J. MIF interacts with CXCR7 to promote receptor internalization, ERK1/2 and ZAP-70 signaling, and lymphocyte chemotaxis. FASEB J. 2015, 29, 4497–4511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eltzschig, H.K. Extracellular adenosine signaling in molecular medicine. J. Mol. Med. 2013, 91, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Aherne, C.M.; Saeedi, B.; Collins, C.B.; Masterson, J.C.; McNamee, E.N.; Perrenoud, L.; Rapp, C.R.; Curtis, V.F.; Bayless, A.; Fletcher, A.; et al. Epithelial-specific A2B adenosine receptor signaling protects the colonic epithelial barrier during acute colitis. Mucosal. Immunol. 2015, 8, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
- Eckle, T.; Hughes, K.; Ehrentraut, H.; Brodsky, K.S.; Rosenberger, P.; Choi, D.S.; Ravid, K.; Weng, T.; Xia, Y.; Blackburn, M.R.; et al. Crosstalk between the equilibrative nucleoside transporter ENT2 and alveolar Adora2b adenosine receptors dampens acute lung injury. FASEB J. 2013, 27, 3078–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eltzschig, H.K.; Bonney, S.K.; Eckle, T. Attenuating myocardial ischemia by targeting A2B adenosine receptors. Trends. Mol. Med. 2013, 19, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Konrad, F.M.; Witte, E.; Vollmer, I.; Stark, S.; Reutershan, J. Adenosine receptor A2b on hematopoietic cells mediates LPS-induced migration of PMNs into the lung interstitium. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2012, 303, L425–L438. [Google Scholar] [CrossRef] [PubMed]
- Eckle, T.; Hartmann, K.; Bonney, S.; Reithel, S.; Mittelbronn, M.; Walker, L.A.; Lowes, B.D.; Han, J.; Borchers, C.H.; Buttrick, P.M.; et al. Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat. Med. 2012, 18, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Rosenberger, P.; Schwab, J.M.; Mirakaj, V.; Masekowsky, E.; Mager, A.; Morote-Garcia, J.C.; Unertl, K.; Eltzschig, H.K. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat. Immunol. 2009, 10, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Joffre, J.; Hellman, J.; Ince, C.; Ait-Oufella, H. Endothelial Responses in Sepsis. Am. J. Respir. Crit. Care. Med. 2020, 202, 361–370. [Google Scholar] [CrossRef]
- Wong, E.; Xu, F.; Joffre, J.; Nguyen, N.; Wilhelmsen, K.; Hellman, J. ERK1/2 Has Divergent Roles in LPS-Induced Microvascular Endothelial Cell Cytokine Production and Permeability. Shock 2021, 55, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Deng, M.; Liu, X.; Ai, W.; Tang, Q.; Hu, J. TLR4 activation induces nontolerant inflammatory response in endothelial cells. Inflammation 2011, 34, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; van der Poll, T. Coagulation and sepsis. Thromb. Res. 2017, 149, 38–44. [Google Scholar] [CrossRef]
- Levi, M.; Schultz, M.; van der Poll, T. Sepsis and thrombosis. Semin. Thromb. Hemost. 2013, 39, 559–566. [Google Scholar] [CrossRef]
- Rossaint, J.; Margraf, A.; Zarbock, A. Role of Platelets in Leukocyte Recruitment and Resolution of Inflammation. Front. Immunol. 2018, 9, 2712. [Google Scholar] [CrossRef]
- Sreeramkumar, V.; Adrover, J.M.; Ballesteros, I.; Cuartero, M.I.; Rossaint, J.; Bilbao, I.; Nacher, M.; Pitaval, C.; Radovanovic, I.; Fukui, Y.; et al. Neutrophils scan for activated platelets to initiate inflammation. Science 2014, 346, 1234–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granja, T.F.; Kohler, D.; Schad, J.; de Oliveira, C.B.; Konrad, F.; Hoch-Gutbrod, M.; Streienberger, A.; Rosenberger, P.; Straub, A. Adenosine Receptor Adora2b Plays a Mechanistic Role in the Protective Effect of the Volatile Anesthetic Sevoflurane during Liver Ischemia/Reperfusion. Anesthesiology 2016, 125, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Remijsen, Q.; Vanden Berghe, T.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, D.A.S.; Prestes, E.B.; Gama, A.M.S.; Silva, L.S.; Pinheiro, A.A.S.; Ribeiro, J.M.C.; Campos, R.M.P.; Pimentel-Coelho, P.M.; De Souza, H.S.; Dicko, A.; et al. CXCR4 and MIF are required for neutrophil extracellular trap release triggered by Plasmodium-infected erythrocytes. PLoS Pathog. 2020, 16, e1008230. [Google Scholar] [CrossRef]
- Ali, R.A.; Gandhi, A.A.; Meng, H.; Yalavarthi, S.; Vreede, A.P.; Estes, S.K.; Palmer, O.R.; Bockenstedt, P.L.; Pinsky, D.J.; Greve, J.M.; et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat. Commun. 2019, 10, 1916. [Google Scholar] [CrossRef]
- Zucoloto, A.Z.; Jenne, C.N. Platelet-Neutrophil Interplay: Insights Into Neutrophil Extracellular Trap (NET)-Driven Coagulation in Infection. Front. Cardiovasc. Med. 2019, 6, 85. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yu, X.; He, Y.; Zhang, L.; Huang, X.; Xu, X.; Chen, M.; Chen, X.; Wang, L. Activation of A2aR attenuates bleomycin-induced pulmonary fibrosis via the SDF-1/CXCR4 axis-related pathway. Am. J. Transl. Res. 2017, 9, 4125–4136. [Google Scholar] [PubMed]
- Jackson, S.P.; Darbousset, R.; Schoenwaelder, S.M. Thromboinflammation: Challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 2019, 133, 906–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivetic, A.; Hoskins Green, H.L.; Hart, S.J. L-selectin: A Major Regulator of Leukocyte Adhesion, Migration and Signaling. Front. Immunol. 2019, 10, 1068. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Caro, T.; Lendner, M.; Daugschies, A.; Hermosilla, C.; Taubert, A. NADPH oxidase, MPO, NE, ERK1/2, p38 MAPK and Ca2+ influx are essential for Cryptosporidium parvum-induced NET formation. Dev. Comp. Immunol. 2015, 52, 245–254. [Google Scholar] [CrossRef]
- Muraro, S.P.; De Souza, G.F.; Gallo, S.W.; Da Silva, B.K.; De Oliveira, S.D.; Vinolo, M.A.R.; Saraiva, E.M.; Porto, B.N. Respiratory Syncytial Virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci. Rep. 2018, 8, 14166. [Google Scholar] [CrossRef]
- Baston-Bust, D.M.; Schanz, A.; Boddeker, S.J.; Altergot-Ahmad, O.; Krussel, J.S.; Rein, D.; Hess, A.P. CXCL1 expression in human decidua in vitro is mediated via the MAPK signalling cascade. Cytokine 2013, 64, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Risco, A.; del Fresno, C.; Mambol, A.; Alsina-Beauchamp, D.; MacKenzie, K.F.; Yang, H.T.; Barber, D.F.; Morcelle, C.; Arthur, J.S.; Ley, S.C.; et al. p38gamma and p38delta kinases regulate the Toll-like receptor 4 (TLR4)-induced cytokine production by controlling ERK1/2 protein kinase pathway activation. Proc. Natl. Acad. Sci. USA 2012, 109, 11200–11205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, M.J.; Radic, M. Neutrophil extracellular traps: Double-edged swords of innate immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, V.; Laube, B.; Abu Abed, U.; Goosmann, C.; Zychlinsky, A. Neutrophil extracellular traps: How to generate and visualize them. J. Vis. Exp. 2010, 36, 1724. [Google Scholar] [CrossRef] [Green Version]
- Eash, K.J.; Greenbaum, A.M.; Gopalan, P.K.; Link, D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Investig. 2010, 120, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Levoye, A.; Balabanian, K.; Baleux, F.; Bachelerie, F.; Lagane, B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 2009, 113, 6085–6093. [Google Scholar] [CrossRef] [Green Version]
- Devi, S.; Wang, Y.; Chew, W.K.; Lima, R.; A-González, N.; Mattar, C.N.; Chong, S.Z.; Schlitzer, A.; Bakocevic, N.; Chew, S.; et al. Neutrophil mobilization via plerixafor-mediated CXCR4 inhibition arises from lung demargination and blockade of neutrophil homing to the bone marrow. J. Exp. Med. 2013, 210, 2321–2336. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Xia, Y.; Zuo, K.; Wang, Y.; Zhang, S.; Kuang, D.; Duan, Y.; Zhao, X.; Wang, G. Crosstalk between SDF-1/CXCR4 and SDF-1/CXCR7 in cardiac stem cell migration. Sci. Rep. 2015, 5, 16813. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Liu, Y.; Wang, C.; Zhang, L.; Crocker, L.; Shen, J. Atorvastatin inhibits CXCR7 induction to reduce macrophage migration. Biochem. Pharmacol. 2014, 89, 99–108. [Google Scholar] [CrossRef]
- Feng, Y.F.; Guo, H.; Yuan, F.; Shen, M.Q. Lipopolysaccharide Promotes Choroidal Neovascularization by Up-Regulation of CXCR4 and CXCR7 Expression in Choroid Endothelial Cell. PLoS ONE 2015, 10, e0136175. [Google Scholar] [CrossRef]
- Houard, X.; Touat, Z.; Ollivier, V.; Louedec, L.; Philippe, M.; Sebbag, U.; Meilhac, O.; Rossignol, P.; Michel, J.B. Mediators of neutrophil recruitment in human abdominal aortic aneurysms. Cardiovasc. Res. 2009, 82, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Ngamsri, K.C.; Bohne, J.; Simelitidis, M.S.; Gamper-Tsigaras, J.; Zhang, Y.; Ehnert, S.; Konrad, F.M. CX3CR1 Depletion Promotes the Formation of Platelet-Neutrophil Complexes and Aggravates Acute Peritonitis. Shock 2021, 56, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Witte, A.; Rohlfing, A.K.; Dannenmann, B.; Dicenta, V.; Nasri, M.; Kolb, K.; Sudmann, J.; Castor, T.; Rath, D.; Borst, O.; et al. The chemokine CXCL14 mediates platelet function and migration via direct interaction with CXCR4. Cardiovasc. Res. 2021, 117, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.C.; Makena, P.S.; Gorantla, V.; Sinclair, S.E.; Waters, C.M. CXCR4 regulates migration of lung alveolar epithelial cells through activation of Rac1 and matrix metalloproteinase-2. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L846–L856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Lu, R.; Wang, S.; Chen, H.; Wang, F.; Liu, K. Effects of SDF-1/CXCR4 on Acute Lung Injury Induced by Cardiopulmonary Bypass. Inflammation 2017, 40, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, M.; Williams, D.W.; Calderon, T.M.; Anastos, K.; Morgello, S.; Berman, J.W. Frontline Science: CXCR7 mediates CD14(+)CD16(+) monocyte transmigration across the blood brain barrier: A potential therapeutic target for NeuroAIDS. J. Leukoc. Biol. 2017, 102, 1173–1185. [Google Scholar] [CrossRef] [Green Version]
- Kontos, C.; El Bounkari, O.; Krammer, C.; Sinitski, D.; Hille, K.; Zan, C.; Yan, G.; Wang, S.; Gao, Y.; Brandhofer, M.; et al. Designed CXCR4 mimic acts as a soluble chemokine receptor that blocks atherogenic inflammation by agonist-specific targeting. Nat. Commun. 2020, 11, 5981. [Google Scholar] [CrossRef]
- Page, C.; Pitchford, S. Neutrophil and platelet complexes and their relevance to neutrophil recruitment and activation. Int. Immunopharmacol. 2013, 17, 1176–1184. [Google Scholar] [CrossRef]
- Cleary, S.J.; Hobbs, C.; Amison, R.T.; Arnold, S.; O’Shaughnessy, B.G.; Lefrancais, E.; Mallavia, B.; Looney, M.R.; Page, C.P.; Pitchford, S.C. LPS-induced Lung Platelet Recruitment Occurs Independently from Neutrophils, PSGL-1, and P-Selectin. Am. J. Respir. Cell Mol. Biol. 2019, 61, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Pircher, J.; Engelmann, B.; Massberg, S.; Schulz, C. Platelet-Neutrophil Crosstalk in Atherothrombosis. Thromb. Haemost. 2019, 119, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Slotta, J.E.; Braun, O.O.; Menger, M.D.; Thorlacius, H. Capture of platelets to the endothelium of the femoral vein is mediated by CD62P and CD162. Platelets 2009, 20, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.I.; Siddiqui, F.M.; Goldstein, J.N.; Cox, M.; Xian, Y.; Matsouaka, R.A.; Heidenreich, P.A.; Peterson, E.D.; Bhatt, D.L.; Fonarow, G.C.; et al. Association Between Previous Use of Antiplatelet Therapy and Intracerebral Hemorrhage Outcomes. Stroke 2017, 48, 1810–1817. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Berg, N.K.; Mills, T.; Zhang, K.; Eltzschig, H.K.; Yuan, X. Adenosine at the Interphase of Hypoxia and Inflammation in Lung Injury. Front. Immunol. 2020, 11, 604944. [Google Scholar] [CrossRef] [PubMed]
- Kiers, D.; Wielockx, B.; Peters, E.; van Eijk, L.T.; Gerretsen, J.; John, A.; Janssen, E.; Groeneveld, R.; Peters, M.; Damen, L.; et al. Short-Term Hypoxia Dampens Inflammation in vivo via Enhanced Adenosine Release and Adenosine 2B Receptor Stimulation. EBioMedicine 2018, 33, 144–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reutershan, J.; Vollmer, I.; Stark, S.; Wagner, R.; Ngamsri, K.C.; Eltzschig, H.K. Adenosine and inflammation: CD39 and CD73 are critical mediators in LPS-induced PMN trafficking into the lungs. FASEB J. 2009, 23, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Pieterse, E.; Rother, N.; Yanginlar, C.; Hilbrands, L.B.; van der Vlag, J. Neutrophils Discriminate between Lipopolysaccharides of Different Bacterial Sources and Selectively Release Neutrophil Extracellular Traps. Front. Immunol. 2016, 7, 484. [Google Scholar] [CrossRef] [Green Version]
- McDonald, B.; Davis, R.P.; Kim, S.J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porto, B.N.; Stein, R.T. Neutrophil Extracellular Traps in Pulmonary Diseases: Too Much of a Good Thing? Front. Immunol. 2016, 7, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.W.; Lee, H.; Lee, H.K.; Kim, I.D.; Lee, J.K. Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain. Acta Neuropathol. Commun. 2019, 7, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Radermecker, C.; Sabatel, C.; Vanwinge, C.; Ruscitti, C.; Marechal, P.; Perin, F.; Schyns, J.; Rocks, N.; Toussaint, M.; Cataldo, D.; et al. Locally instructed CXCR4(hi) neutrophils trigger environment-driven allergic asthma through the release of neutrophil extracellular traps. Nat. Immunol. 2019, 20, 1444–1455. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, M.; Shan, Q.; D’Ortona, S.; Maurer, R.; Mitchell, R.; Olesen, H.; Thiel, S.; Huebner, J.; Gadjeva, M. Cystic fibrosis sputum DNA has NETosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. J. Innate Immun. 2014, 6, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Rivera, C.; Khaznadar, S.S.; Shwin, K.W.; Irizarry-Caro, J.A.; O’Neil, L.J.; Liu, Y.; Jacobson, K.A.; Ombrello, A.K.; Stone, D.L.; Tsai, W.L.; et al. Deficiency of adenosine deaminase 2 triggers adenosine-mediated NETosis and TNF production in patients with DADA2. Blood 2019, 134, 395–406. [Google Scholar] [CrossRef]
- Yan, Z.; Luo, H.; Xie, B.; Tian, T.; Li, S.; Chen, Z.; Liu, J.; Zhao, X.; Zhang, L.; Deng, Y.; et al. Targeting adaptor protein SLP76 of RAGE as a therapeutic approach for lethal sepsis. Nat. Commun. 2021, 12, 308. [Google Scholar] [CrossRef]
- Smith, J.A.; Mayeux, P.R.; Schnellmann, R.G. Delayed Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase Inhibition by Trametinib Attenuates Systemic Inflammatory Responses and Multiple Organ Injury in Murine Sepsis. Crit. Care Med. 2016, 44, e711–e720. [Google Scholar] [CrossRef] [Green Version]
- Kuhns, D.B.; Priel, D.A.L.; Chu, J.; Zarember, K.A. Isolation and Functional Analysis of Human Neutrophils. Curr. Protoc. Immunol. 2015, 111, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Weissmuller, T.; Campbell, E.L.; Rosenberger, P.; Scully, M.; Beck, P.L.; Furuta, G.T.; Colgan, S.P. PMNs facilitate translocation of platelets across human and mouse epithelium and together alter fluid homeostasis via epithelial cell-expressed ecto-NTPDases. J. Clin. Investig. 2008, 118, 3682–3692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swamydas, M.; Lionakis, M.S. Isolation, purification and labeling of mouse bone marrow neutrophils for functional studies and adoptive transfer experiments. J. Vis. Exp. 2013, 77, e50586. [Google Scholar] [CrossRef] [PubMed]
- Aurbach, K.; Spindler, M.; Haining, E.J.; Bender, M.; Pleines, I. Blood collection, platelet isolation and measurement of platelet count and size in mice-a practical guide. Platelets 2019, 30, 698–707. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngamsri, K.-C.; Putri, R.A.; Jans, C.; Schindler, K.; Fuhr, A.; Zhang, Y.; Gamper-Tsigaras, J.; Ehnert, S.; Konrad, F.M. CXCR4 and CXCR7 Inhibition Ameliorates the Formation of Platelet–Neutrophil Complexes and Neutrophil Extracellular Traps through Adora2b Signaling. Int. J. Mol. Sci. 2021, 22, 13576. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413576
Ngamsri K-C, Putri RA, Jans C, Schindler K, Fuhr A, Zhang Y, Gamper-Tsigaras J, Ehnert S, Konrad FM. CXCR4 and CXCR7 Inhibition Ameliorates the Formation of Platelet–Neutrophil Complexes and Neutrophil Extracellular Traps through Adora2b Signaling. International Journal of Molecular Sciences. 2021; 22(24):13576. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413576
Chicago/Turabian StyleNgamsri, Kristian-Christos, Rizki A. Putri, Christoph Jans, Katharina Schindler, Anika Fuhr, Yi Zhang, Jutta Gamper-Tsigaras, Sabrina Ehnert, and Franziska M. Konrad. 2021. "CXCR4 and CXCR7 Inhibition Ameliorates the Formation of Platelet–Neutrophil Complexes and Neutrophil Extracellular Traps through Adora2b Signaling" International Journal of Molecular Sciences 22, no. 24: 13576. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413576