
Mass-Spectrometry-Based Functional Proteomic and Phosphoproteomic Technologies and Their Application for Analyzing Ex Vivo and In Vitro Models of Hypertrophic Cardiomyopathy

{kind=link}
Abstract
:1. Introduction
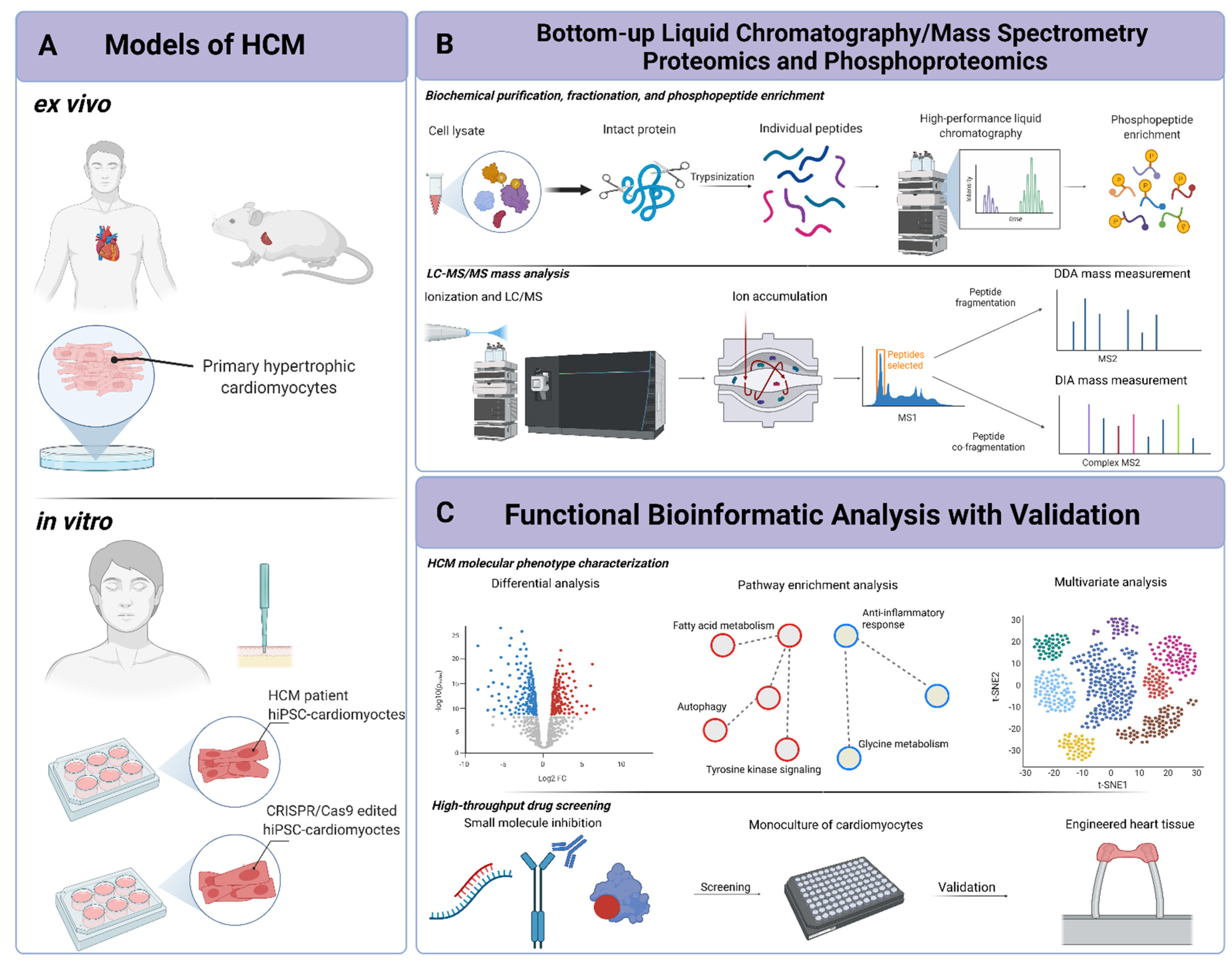
2. Mass-Spectrometry-Based Proteomics and Phosphoproteomic Technologies
2.1. Fundamentals of Mass Spectrometry
2.2. Proteomics and Phosphoproteomics
2.3. Pathway-Level Data Analysis and Functional Inference
“Minimizing type I error in proteomics”
3. Murine and Human Ex Vivo and In Vitro Models of HCM
3.1. Ex Vivo Models of HCM
3.1.1. Murine Models
3.1.2. Primary Human Tissue
“Emerging Integrative Omics: Spotlight on metabolomics”
3.2. In Vitro Models of HCM
3.3. Future Advancements in Tissue Modeling
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geske, J.B.; Ommen, S.R.; Gersh, B.J. Hypertrophic Cardiomyopathy. JACC Heart Fail. 2018, 6, 364–375. [Google Scholar] [CrossRef]
- Marian, A.J. Braunwald Eugene Hypertrophic Cardiomyopathy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Akhtar, M.; Elliott, P. The Genetics of Hypertrophic Cardiomyopathy. Glob. Cardiol. Sci. Pract. 2018, 3, 36. [Google Scholar] [CrossRef] [Green Version]
- Landstrom, A.P.; Ackerman, M.J. Mutation Type Is Not Clinically Useful in Predicting Prognosis in Hypertrophic Cardiomyopathy. Circulation 2010, 122, 2441–2450. [Google Scholar] [CrossRef] [Green Version]
- Kuzmanov, U.; Wang, E.Y.; Vanderlaan, R.; Kim, D.H.; Lee, S.-H.; Hadipour-Lakmehsari, S.; Guo, H.; Zhao, Y.; McFadden, M.; Sharma, P.; et al. Mapping signalling perturbations in myocardial fibrosis via the integrative phosphoproteomic profiling of tissue from diverse sources. Nat. Biomed. Eng. 2020, 4, 889–900. [Google Scholar] [CrossRef]
- Tucholski, T.; Cai, W.; Gregorich, Z.R.; Bayne, E.F.; Mitchell, S.D.; McIlwain, S.J.; de Lange, W.J.; Wrobbel, M.; Karp, H.; Hite, Z.; et al. Distinct Hypertrophic Cardiomyopathy Genotypes Result in Convergent Sarcomeric Proteoform Profiles Revealed by Top-down Proteomics. Proc. Natl. Acad. Sci. USA 2020, 117, 24691–24700. [Google Scholar] [CrossRef]
- Siuzdak, G. An Introduction to Mass Spectrometry Ionization: An Excerpt from the Expanding Role of Mass Spectrometry in Biotechnology, 2nd Ed.; MCC Press: San Diego, 2005. JALA J. Assoc. Lab. Autom. 2004, 9, 50–63. [Google Scholar] [CrossRef] [Green Version]
- Duong, V.-A.; Park, J.-M.; Lee, H. Review of Three-Dimensional Liquid Chromatography Platforms for Bottom-Up Proteomics. Int. J. Mol. Sci. 2020, 21, 1524. [Google Scholar] [CrossRef] [Green Version]
- Canterbury, J.D.; Merrihew, G.E.; Goodlett, D.R.; MacCoss, M.J.; Shaffer, S.A. Comparison of Data Acquisition Strategies on Quadrupole Ion Trap Instrumentation for Shotgun Proteomics. J. Am. Soc. Mass Spectrom. 2014, 25, 2048–2059. [Google Scholar] [CrossRef] [Green Version]
- Li, K.W.; Gonzalez-Lozano, M.A.; Koopmans, F.; Smit, A.B. Recent Developments in Data Independent Acquisition (DIA) Mass Spectrometry: Application of Quantitative Analysis of the Brain Proteome. Front. Mol. Neurosci. 2020, 13, 564446. [Google Scholar] [CrossRef]
- Tada, I.; Chaleckis, R.; Tsugawa, H.; Meister, I.; Zhang, P.; Lazarinis, N.; Dahlén, B.; Wheelock, C.E.; Arita, M. Correlation-Based Deconvolution (CorrDec) To Generate High-Quality MS2 Spectra from Data-Independent Acquisition in Multisample Studies. Anal. Chem. 2020, 92, 11310–11317. [Google Scholar] [CrossRef]
- Baker, A.J. Adrenergic Signaling in Heart Failure: A Balance of Toxic and Protective Effects. Pflug. Arch. Eur. J. Physiol. 2014, 466, 1139–1150. [Google Scholar] [CrossRef]
- Rolland, D.C.M.; Lim, M.S.; Elenitoba-Johnson, K.S.J. Mass Spectrometry and Proteomics in Hematology. Semin. Hematol. 2019, 56, 52–57. [Google Scholar] [CrossRef]
- Cifani, P.; Kentsis, A. High Sensitivity Quantitative Proteomics Using Automated Multidimensional Nano-Flow Chromatography and Accumulated Ion Monitoring on Quadrupole-Orbitrap-Linear Ion Trap Mass Spectrometer*. Mol. Cell. Proteom. 2017, 16, 2006–2016. [Google Scholar] [CrossRef] [Green Version]
- Bekker-Jensen, D.B.; Martínez-Val, A.; Steigerwald, S.; Rüther, P.; Fort, K.L.; Arrey, T.N.; Harder, A.; Makarov, A.; Olsen, J.V. A Compact Quadrupole-Orbitrap Mass Spectrometer with FAIMS Interface Improves Proteome Coverage in Short LC Gradients. Mol. Cell. Proteom. 2020, 19, 716–729. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Paulo, J.A.; Naverrete-Perea, J.; McAlister, G.C.; Canterbury, J.D.; Bailey, D.J.; Robitaille, A.M.; Huguet, R.; Zabrouskov, V.; Gygi, S.P.; et al. Benchmarking the Orbitrap Tribrid Eclipse for Next Generation Multiplexed Proteomics. Anal. Chem. 2020, 92, 6478–6485. [Google Scholar] [CrossRef]
- Shimada, Y.J.; Hasegawa, K.; Kochav, S.M.; Mohajer, P.; Jung, J.; Maurer, M.S.; Reilly, M.P.; Fifer, M.A. Application of Proteomics Profiling for Biomarker Discovery in Hypertrophic Cardiomyopathy. J. Cardiovasc. Trans. Res. 2019, 12, 569–579. [Google Scholar] [CrossRef]
- Sonnenschein, K.; Fiedler, J.; de Gonzalo-Calvo, D.; Xiao, K.; Pfanne, A.; Just, A.; Zwadlo, C.; Soltani, S.; Bavendiek, U.; Kraft, T.; et al. Blood-Based Protein Profiling Identifies Serum Protein c-KIT as a Novel Biomarker for Hypertrophic Cardiomyopathy. Sci. Rep. 2021, 11, 1755. [Google Scholar] [CrossRef]
- Cheung, T.K.; Lee, C.-Y.; Bayer, F.P.; McCoy, A.; Kuster, B.; Rose, C.M. Defining the Carrier Proteome Limit for Single-Cell Proteomics. Nat. Methods 2021, 18, 76–83. [Google Scholar] [CrossRef]
- Marx, V. A Dream of Single-Cell Proteomics. Nat. Methods 2019, 16, 809–812. [Google Scholar] [CrossRef] [Green Version]
- Brunner, A.-D.; Thielert, M.; Vasilopoulou, C.G.; Ammar, C.; Coscia, F.; Mund, A.; Hoerning, O.B.; Bache, N.; Apalategui, A.; Lubeck, M.; et al. Ultra-High Sensitivity Mass Spectrometry Quantifies Single-Cell Proteome Changes upon Perturbation. BioRxiv 2020. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway Enrichment Analysis and Visualization of Omics Data Using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Eckhardt, M.; Krogan, N.J. Mass Spectrometry-based Protein–Protein Interaction Networks for the Study of Human Diseases. Mol. Syst. Biol. 2021, 17, e8792. [Google Scholar] [CrossRef]
- Lualdi, M.; Fasano, M. Statistical Analysis of Proteomics Data: A Review on Feature Selection. J. Proteom. 2019, 198, 18–26. [Google Scholar] [CrossRef]
- Geisterfer-Lowrance, A.A.T.; Christe, M.; Conner, D.A.; Ingwall, J.S.; Schoen, F.J.; Seidman, C.E.; Seidman, J.G. A Mouse Model of Familial Hypertrophic Cardiomyopathy. Science 1996, 272, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.A.; Grupp, I.L.; Subramaniam, A.; Robbins, J. Cardiac Myosin Heavy Chain MRNA Expression and Myocardial Function in the Mouse Heart. Circ. Res. 1991, 68, 1742–1750. [Google Scholar] [CrossRef] [Green Version]
- Miyata, S.; Minobe, W.; Bristow, M.R.; Leinwand, L.A. Myosin Heavy Chain Isoform Expression in the Failing and Nonfailing Human Heart. Circ. Res. 2000, 86, 386–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatkin, D.; McConnell, B.K.; Mudd, J.O.; Semsarian, C.; Moskowitz, I.G.P.; Schoen, F.J.; Giewat, M.; Seidman, C.E.; Seidman, J.G. An Abnormal Ca2+ Response in Mutant Sarcomere Protein–Mediated Familial Hypertrophic Cardiomyopathy. J. Clin. Investig. 2000, 106, 1351–1359. [Google Scholar] [CrossRef] [Green Version]
- Kaltenbach, M.; Hopf, R.; Keller, M. Treatment of hypertrophic obstructive cardiomyopathy with verapamil, a calcium antagonist (author’s transl). Dtsch. Med. Wochenschr. 1976, 101, 1284–1287. [Google Scholar] [CrossRef]
- Gregor, P.; Čurila, K. Medical Treatment of Hypertrophic Cardiomyopathy—What Do We Know about It Today? Cor Vasa 2015, 57, e219–e224. [Google Scholar] [CrossRef] [Green Version]
- Ferrantini, C.; Coppini, R.; Pioner, J.M.; Gentile, F.; Tosi, B.; Mazzoni, L.; Scellini, B.; Piroddi, N.; Laurino, A.; Santini, L.; et al. Pathogenesis of Hypertrophic Cardiomyopathy Is Mutation Rather Than Disease Specific: A Comparison of the Cardiac Troponin T E163R and R92Q Mouse Models. J. Am. Heart Assoc. 2017, 6, e005407. [Google Scholar] [CrossRef] [Green Version]
- Kuster, D.W.D.; Lynch, T.L., IV; Barefield, D.Y.; Sivaguru, M.; Kuffel, G.; Zilliox, M.J.; Lee, K.H.; Craig, R.; Namakkal-Soorappan, R.; Sadayappan, S. Altered C10 Domain in Cardiac Myosin Binding Protein-C Results in Hypertrophic Cardiomyopathy. Cardiovasc. Res. 2019, 115, 1986–1997. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.J.; Makarewich, C.A.; McAnally, J.; Anderson, D.M.; Zentilin, L.; Liu, N.; Giacca, M.; Bassel-Duby, R.; Olson, E.N. A Mouse Model for Adult Cardiac-Specific Gene Deletion with CRISPR/Cas9. Proc. Natl. Acad. Sci. USA 2016, 113, 338–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.-Y.R.; Kontrogianni-Konstantopoulos, A. Proteomic Analysis of Myocardia Containing the Obscurin R4344Q Mutation Linked to Hypertrophic Cardiomyopathy. Front. Physiol. 2020, 11, 478. [Google Scholar] [CrossRef]
- Heenan, T. A Program for Standardized Training in Rodent Handling at a Large Academic Institution. Lab Anim. 2010, 39, 113–117. [Google Scholar] [CrossRef]
- Camacho, P.; Fan, H.; Liu, Z.; He, J.-Q. Small Mammalian Animal Models of Heart Disease. Am. J. Cardiovasc. Dis. 2016, 6, 70–80. [Google Scholar]
- Barefield, D.; Kumar, M.; Gorham, J.; Seidman, J.G.; Seidman, C.E.; de Tombe, P.P.; Sadayappan, S. Haploinsufficiency of MYBPC3 Exacerbates the Development of Hypertrophic Cardiomyopathy in Heterozygous Mice. J. Mol. Cell. Cardiol. 2015, 79, 234–243. [Google Scholar] [CrossRef] [Green Version]
- Santini, L.; Palandri, C.; Nediani, C.; Cerbai, E.; Coppini, R. Modelling Genetic Diseases for Drug Development: Hypertrophic Cardiomyopathy. Pharmacol. Res. 2020, 160, 105176. [Google Scholar] [CrossRef]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the Adult Human Heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef]
- Guo, Y.; Pu, W. Cardiomyocyte Maturation: New Phase in Development. Circ. Res. 2020, 126, 1086–1106. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Tang, M.; Xi, J.; Gao, L.; Zheng, Y.; Luo, H.; Hu, X.; Zhao, F.; Reppel, M.; Hescheler, J.; et al. Functional Characterization of Inward Rectifier Potassium Ion Channel in Murine Fetal Ventricular Cardiomyocytes. Cell. Physiol. Biochem. 2010, 26, 413–420. [Google Scholar] [CrossRef]
- Kuzmanov, U.; Guo, H.; Buchsbaum, D.; Cosme, J.; Abbasi, C.; Isserlin, R.; Sharma, P.; Gramolini, A.O.; Emili, A. Global Phosphoproteomic Profiling Reveals Perturbed Signaling in a Mouse Model of Dilated Cardiomyopathy. Proc. Natl. Acad. Sci. USA 2016, 113, 12592–12597. [Google Scholar] [CrossRef] [Green Version]
- Milani-Nejad, N.; Janssen, P.M.L. Small and Large Animal Models in Cardiac Contraction Research: Advantages and Disadvantages. Pharmacol. Ther. 2014, 141, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Vakrou, S.; Liu, Y.; Zhu, L.; Greenland, G.V.; Simsek, B.; Hebl, V.B.; Guan, Y.; Woldemichael, K.; Talbot, C.C.; Aon, M.A.; et al. Differences in Molecular Phenotype in Mouse and Human Hypertrophic Cardiomyopathy. Sci. Rep. 2021, 11, 13163. [Google Scholar] [CrossRef]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Fürth, D.; Qian, X.; Wärdell, E.; Custodio, J.; Reimegård, J.; Salmén, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660. [Google Scholar] [CrossRef] [PubMed]
- DeLaughter, D.M.; Bick, A.G.; Wakimoto, H.; McKean, D.; Gorham, J.M.; Kathiriya, I.S.; Hinson, J.T.; Homsy, J.; Gray, J.; Pu, W.; et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell 2016, 39, 480–490. [Google Scholar] [CrossRef] [Green Version]
- Grankvist, R.; Chireh, A.; Sandell, M.; Mukarram, A.K.; Jaff, N.; Berggren, I.; Persson, H.; Linde, C.; Arnberg, F.; Lundberg, J.; et al. Myocardial Micro-Biopsy Procedure for Molecular Characterization with Increased Precision and Reduced Trauma. Sci. Rep. 2020, 10, 8029. [Google Scholar] [CrossRef] [PubMed]
- Wojtkiewicz, M.; Berg Luecke, L.; Castro, C.; Burkovetskaya, M.; Mesidor, R.; Gundry, R.L. Bottom-up Proteomic Analysis of Human Adult Cardiac Tissue and Isolated Cardiomyocytes. J. Mol. Cell. Cardiol. 2022, 162, 20–31. [Google Scholar] [CrossRef]
- Coats Caroline, J.; Heywood Wendy, E.; Virasami, A.; Ashrafi, N.; Syrris, P.; dos Remedios, C.; Treibel Thomas, A.; Moon James, C.; Lopes Luis, R.; McGregor Christopher, G.A.; et al. Proteomic Analysis of the Myocardium in Hypertrophic Obstructive Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e001974. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, A.; Li, X.; Kao, W.-Y.; Viker, K.; Butters, K.; Masuoka, H.; Knudsen, B.; Gores, G.; Charlton, M. Lumican, an Extracellular Matrix Proteoglycan, Is a Novel Requisite for Hepatic Fibrosis. Lab. Investig. 2012, 92, 1712–1725. [Google Scholar] [CrossRef]
- Pei, J.; Schuldt, M.; Nagyova, E.; Gu, Z.; el Bouhaddani, S.; Yiangou, L.; Jansen, M.; Calis, J.J.A.; Dorsch, L.M.; Blok, C.S.; et al. Multi-Omics Integration Identifies Key Upstream Regulators of Pathomechanisms in Hypertrophic Cardiomyopathy Due to Truncating MYBPC3 Mutations. Clin. Epigenet. 2021, 13, 61. [Google Scholar] [CrossRef] [PubMed]
- Louch, W.E.; Sheehan, K.A.; Wolska, B.M. Methods in Cardiomyocyte Isolation, Culture, and Gene Transfer. J. Mol. Cell. Cardiol. 2011, 51, 288–298. [Google Scholar] [CrossRef] [Green Version]
- Li, R. Human Pediatric and Adult Ventricular Cardiomyocytes in Culture: Assessment of Phenotypic Changes with Passaging. Cardiovasc. Res. 1996, 32, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.A.; Wang, R.-S.; Shevtsov, S.; Drakos, S.G.; Arons, E.; Wever-Pinzon, O.; Huggins, G.S.; Samokhin, A.O.; Oldham, W.M.; Aguib, Y.; et al. Individualized Interactomes for Network-Based Precision Medicine in Hypertrophic Cardiomyopathy with Implications for Other Clinical Pathophenotypes. Nat. Commun. 2021, 12, 873. [Google Scholar] [CrossRef] [PubMed]
- Cambiaghi, A.; Ferrario, M.; Masseroli, M. Analysis of Metabolomic Data: Tools, Current Strategies and Future Challenges for Omics Data Integration. Brief. Bioinform. 2017, 18, 498–510. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Chou, J.; Hou, S.; Liu, X.; Yu, J.; Zhao, X.; Li, Y.; Liu, L.; Sun, C. Evaluation of Two-Step Liquid-Liquid Extraction Protocol for Untargeted Metabolic Profiling of Serum Samples to Achieve Broader Metabolome Coverage by UPLC-Q-TOF-MS. Anal. Chim. Acta 2018, 1035, 96–107. [Google Scholar] [CrossRef]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef]
- Blum, B.C.; Mousavi, F.; Emili, A. Single-Platform ‘Multi-Omic’ Profiling: Unified Mass Spectrometry and Computational Workflows for Integrative Proteomics–Metabolomics Analysis. Mol. Omics 2018, 14, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Dorr, K.M.; Conlon, F.L. Proteomic-Based Approaches to Cardiac Development and Disease. Curr. Opin. Chem. Biol. 2019, 48, 150–157. [Google Scholar] [CrossRef]
- Ahmed, R.E.; Anzai, T.; Chanthra, N.; Uosaki, H. A Brief Review of Current Maturation Methods for Human Induced Pluripotent Stem Cells-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2020, 8, 178. [Google Scholar] [CrossRef] [Green Version]
- Mosqueira, D.; Mannhardt, I.; Bhagwan, J.R.; Lis-Slimak, K.; Katili, P.; Scott, E.; Hassan, M.; Prondzynski, M.; Harmer, S.C.; Tinker, A.; et al. CRISPR/Cas9 Editing in Human Pluripotent Stem Cell-Cardiomyocytes Highlights Arrhythmias, Hypocontractility, and Energy Depletion as Potential Therapeutic Targets for Hypertrophic Cardiomyopathy. Eur. Heart J. 2018, 39, 3879–3892. [Google Scholar] [CrossRef]
- Han, L.; Li, Y.; Tchao, J.; Kaplan, A.D.; Lin, B.; Li, Y.; Mich-Basso, J.; Lis, A.; Hassan, N.; London, B.; et al. Study Familial Hypertrophic Cardiomyopathy Using Patient-Specific Induced Pluripotent Stem Cells. Cardiovasc. Res. 2014, 104, 258–269. [Google Scholar] [CrossRef]
- Koivumäki, J.T.; Naumenko, N.; Tuomainen, T.; Takalo, J.; Oksanen, M.; Puttonen, K.A.; Lehtonen, Š.; Kuusisto, J.; Laakso, M.; Koistinaho, J.; et al. Structural Immaturity of Human IPSC-Derived Cardiomyocytes: In Silico Investigation of Effects on Function and Disease Modeling. Front. Physiol. 2018, 9, 80. [Google Scholar] [CrossRef]
- van den Berg, C.W.; Okawa, S.; Chuva de Sousa Lopes, S.M.; van Iperen, L.; Passier, R.; Braam, S.R.; Tertoolen, L.G.; del Sol, A.; Davis, R.P.; Mummery, C.L. Transcriptome of Human Foetal Heart Compared with Cardiomyocytes from Pluripotent Stem Cells. Development 2015, 142, 3231–3238. [Google Scholar] [CrossRef] [Green Version]
- Correia, C.; Koshkin, A.; Duarte, P.; Hu, D.; Teixeira, A.; Domian, I.; Serra, M.; Alves, P.M. Distinct Carbon Sources Affect Structural and Functional Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Sci. Rep. 2017, 7, 8590. [Google Scholar] [CrossRef]
- Lewandowski, J.; Rozwadowska, N.; Kolanowski, T.J.; Malcher, A.; Zimna, A.; Rugowska, A.; Fiedorowicz, K.; Łabędź, W.; Kubaszewski, Ł.; Chojnacka, K.; et al. The Impact of in Vitro Cell Culture Duration on the Maturation of Human Cardiomyocytes Derived from Induced Pluripotent Stem Cells of Myogenic Origin. Cell Transplant. 2018, 27, 1047–1067. [Google Scholar] [CrossRef] [Green Version]
- Cadet, J.S.; Kamp, T.J. A Recipe for T-Tubules in Human IPS Cell-Derived Cardiomyocytes. Circ. Res. 2017, 121, 1294–1295. [Google Scholar] [CrossRef]
- Parikh, S.S.; Blackwell, D.J.; Gomez-Hurtado, N.; Frisk, M.; Wang, L.; Kim, K.; Dahl, C.P.; Fiane, A.; Tønnessen, T.; Kryshtal, D.O.; et al. Thyroid and Glucocorticoid Hormones Promote Functional T-Tubule Development in Human-Induced Pluripotent Stem Cell Derived Cardiomyocytes. Circ. Res. 2017, 121, 1323–1330. [Google Scholar] [CrossRef]
- Correia, C.; Koshkin, A.; Duarte, P.; Hu, D.; Carido, M.; Sebastião, M.J.; Gomes-Alves, P.; Elliott, D.A.; Domian, I.J.; Teixeira, A.P.; et al. 3D Aggregate Culture Improves Metabolic Maturation of Human Pluripotent Stem Cell Derived Cardiomyocytes. Biotechnol. Bioeng. 2018, 115, 630–644. [Google Scholar] [CrossRef]
- Prondzynski, M.; Krämer, E.; Laufer, S.D.; Shibamiya, A.; Pless, O.; Flenner, F.; Müller, O.J.; Münch, J.; Redwood, C.; Hansen, A.; et al. Evaluation of MYBPC3 Trans-Splicing and Gene Replacement as Therapeutic Options in Human IPSC-Derived Cardiomyocytes. Mol. Ther. Nucleic Acids 2017, 7, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Cohn, R.; Thakar, K.; Lowe, A.; Ladha, F.A.; Pettinato, A.M.; Romano, R.; Meredith, E.; Chen, Y.-S.; Atamanuk, K.; Huey, B.D.; et al. A Contraction Stress Model of Hypertrophic Cardiomyopathy Due to Sarcomere Mutations. Stem Cell Rep. 2019, 12, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A.; Yuasa, S.; Mearini, G.; Egashira, T.; Seki, T.; Kodaira, M.; Kusumoto, D.; Kuroda, Y.; Okata, S.; Suzuki, T.; et al. Endothelin-1 Induces Myofibrillar Disarray and Contractile Vector Variability in Hypertrophic Cardiomyopathy–Induced Pluripotent Stem Cell–Derived Cardiomyocytes. J. Am. Heart Assoc. 2014, 3, e001263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pioner, J.M.; Racca, A.W.; Klaiman, J.M.; Yang, K.-C.; Guan, X.; Pabon, L.; Muskheli, V.; Zaunbrecher, R.; Macadangdang, J.; Jeong, M.Y.; et al. Isolation and Mechanical Measurements of Myofibrils from Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cell Rep. 2016, 6, 885–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhagwan, J.R.; Mosqueira, D.; Chairez-Cantu, K.; Mannhardt, I.; Bodbin, S.E.; Bakar, M.; Smith, J.G.W.; Denning, C. Isogenic Models of Hypertrophic Cardiomyopathy Unveil Differential Phenotypes and Mechanism-Driven Therapeutics. J. Mol. Cell. Cardiol. 2020, 145, 43–53. [Google Scholar] [CrossRef]
- Birket, M.J.; Ribeiro, M.C.; Kosmidis, G.; Ward, D.; Leitoguinho, A.R.; van de Pol, V.; Dambrot, C.; Devalla, H.D.; Davis, R.P.; Mastroberardino, P.G.; et al. Contractile Defect Caused by Mutation in MYBPC3 Revealed under Conditions Optimized for Human PSC-Cardiomyocyte Function. Cell Rep. 2015, 13, 733–745. [Google Scholar] [CrossRef] [Green Version]
- Coppini, R.; Ferrantini, C.; Mugelli, A.; Poggesi, C.; Cerbai, E. Altered Ca2+ and Na+ Homeostasis in Human Hypertrophic Cardiomyopathy: Implications for Arrhythmogenesis. Front. Physiol. 2018, 9, 1391. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.H.; Herting, J.; Tirilomis, T.; Renner, A.; Neef, S.; Toischer, K.; Ellenberger, D.; Förster, A.; Schmitto, J.D.; Gummert, J.; et al. Ca2+/Calmodulin-Dependent Protein Kinase II and Protein Kinase A Differentially Regulate Sarcoplasmic Reticulum Ca2+ Leak in Human Cardiac Pathology. Circulation 2013, 128, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Hellen, N.; Pinto Ricardo, C.; Vauchez, K.; Whiting, G.; Wheeler, J.X.; Harding, S.E. Proteomic Analysis Reveals Temporal Changes in Protein Expression in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes In Vitro. Stem Cells Dev. 2019, 28, 565–578. [Google Scholar] [CrossRef]
- Lundy, S.D.; Zhu, W.-Z.; Regnier, M.; Laflamme, M.A. Structural and Functional Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cells Dev. 2013, 22, 1991–2002. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sheehan, R.P.; Palmer, A.C.; Everley, R.A.; Boswell, S.A.; Ron-Harel, N.; Ringel, A.E.; Holton, K.M.; Jacobson, C.A.; Erickson, A.R.; et al. Adaptation of Human IPSC-Derived Cardiomyocytes to Tyrosine Kinase Inhibitors Reduces Acute Cardiotoxicity via Metabolic Reprogramming. Cell Syst. 2019, 8, 412–426. [Google Scholar] [CrossRef]
- Humphrey, S.J.; Karayel, O.; James, D.E.; Mann, M. High-Throughput and High-Sensitivity Phosphoproteomics with the EasyPhos Platform. Nat. Protoc. 2018, 13, 1897–1916. [Google Scholar] [CrossRef] [PubMed]
- Hogrebe, A.; von Stechow, L.; Bekker-Jensen, D.B.; Weinert, B.T.; Kelstrup, C.D.; Olsen, J.V. Benchmarking Common Quantification Strategies for Large-Scale Phosphoproteomics. Nat. Commun. 2018, 9, 1045. [Google Scholar] [CrossRef] [Green Version]
- Zuppinger, C. 3D Cardiac Cell Culture: A Critical Review of Current Technologies and Applications. Front. Cardiovasc. Med. 2019, 6, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polonchuk, L.; Chabria, M.; Badi, L.; Hoflack, J.-C.; Figtree, G.; Davies, M.J.; Gentile, C. Cardiac Spheroids as Promising in Vitro Models to Study the Human Heart Microenvironment. Sci. Rep. 2017, 7, 7005. [Google Scholar] [CrossRef]
- Archer, C.R.; Sargeant, R.; Basak, J.; Pilling, J.; Barnes, J.R.; Pointon, A. Characterization and Validation of a Human 3D Cardiac Microtissue for the Assessment of Changes in Cardiac Pathology. Sci. Rep. 2018, 8, 10160. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, P.; Jackson, C.B.; Ozhathil, L.C.; Agarkova, I.; Galindo, C.L.; Sawyer, D.B.; Suter, T.M.; Zuppinger, C. 3D Co-Culture of HiPSC-Derived Cardiomyocytes with Cardiac Fibroblasts Improves Tissue-Like Features of Cardiac Spheroids. Front. Mol. Biosci. 2020, 7, 14. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moore, J.; Emili, A. Mass-Spectrometry-Based Functional Proteomic and Phosphoproteomic Technologies and Their Application for Analyzing Ex Vivo and In Vitro Models of Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 13644. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413644
Moore J, Emili A. Mass-Spectrometry-Based Functional Proteomic and Phosphoproteomic Technologies and Their Application for Analyzing Ex Vivo and In Vitro Models of Hypertrophic Cardiomyopathy. International Journal of Molecular Sciences. 2021; 22(24):13644. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413644
Chicago/Turabian StyleMoore, Jarrod, and Andrew Emili. 2021. "Mass-Spectrometry-Based Functional Proteomic and Phosphoproteomic Technologies and Their Application for Analyzing Ex Vivo and In Vitro Models of Hypertrophic Cardiomyopathy" International Journal of Molecular Sciences 22, no. 24: 13644. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms222413644