
A Mechanistic Model of NMDA and AMPA Receptor-Mediated Synaptic Transmission in Individual Hippocampal CA3-CA1 Synapses: A Computational Multiscale Approach

Abstract
:1. Introduction
2. Results and Discussions
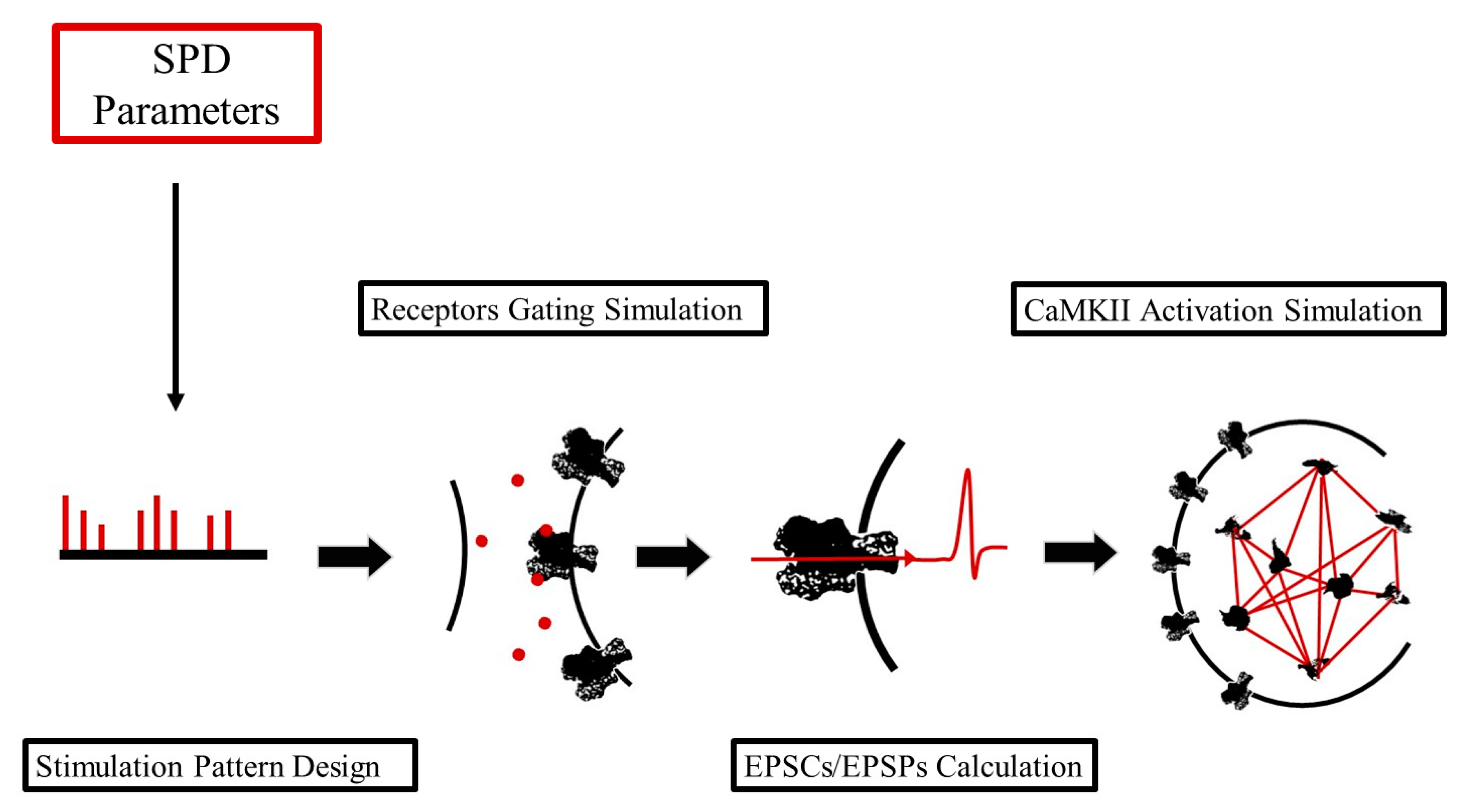
2.1. An Integrative, Python-Based Pipeline for Simulating Glutamatergic Synaptic Transmission
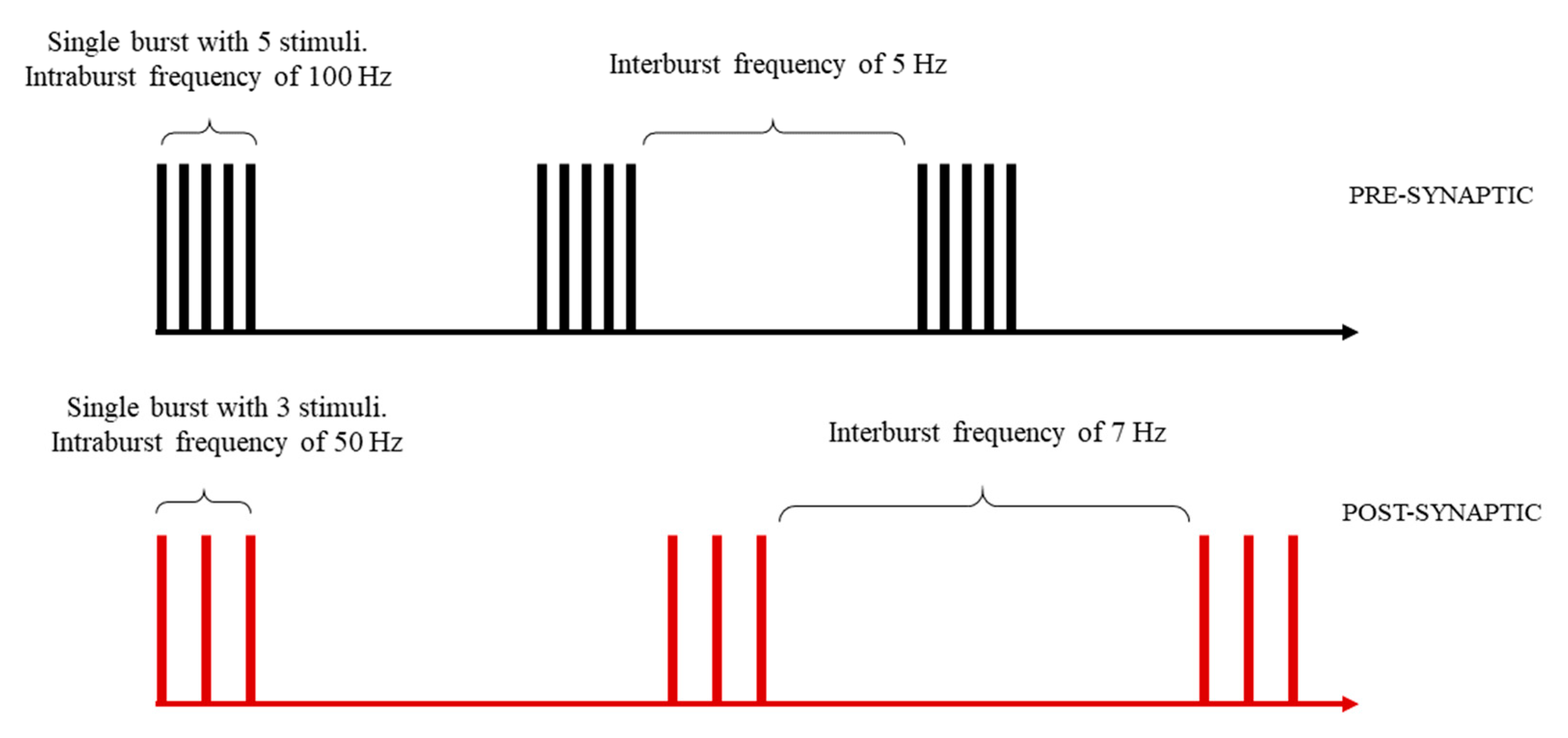
2.1.1. Stimulation Pattern Design (SPD)
2.1.2. Receptors Gating Simulation (RGS)
2.1.3. EPSCs/EPSPs Calculation (CPC)
2.1.4. CaMKII Activation Simulation (CAS)
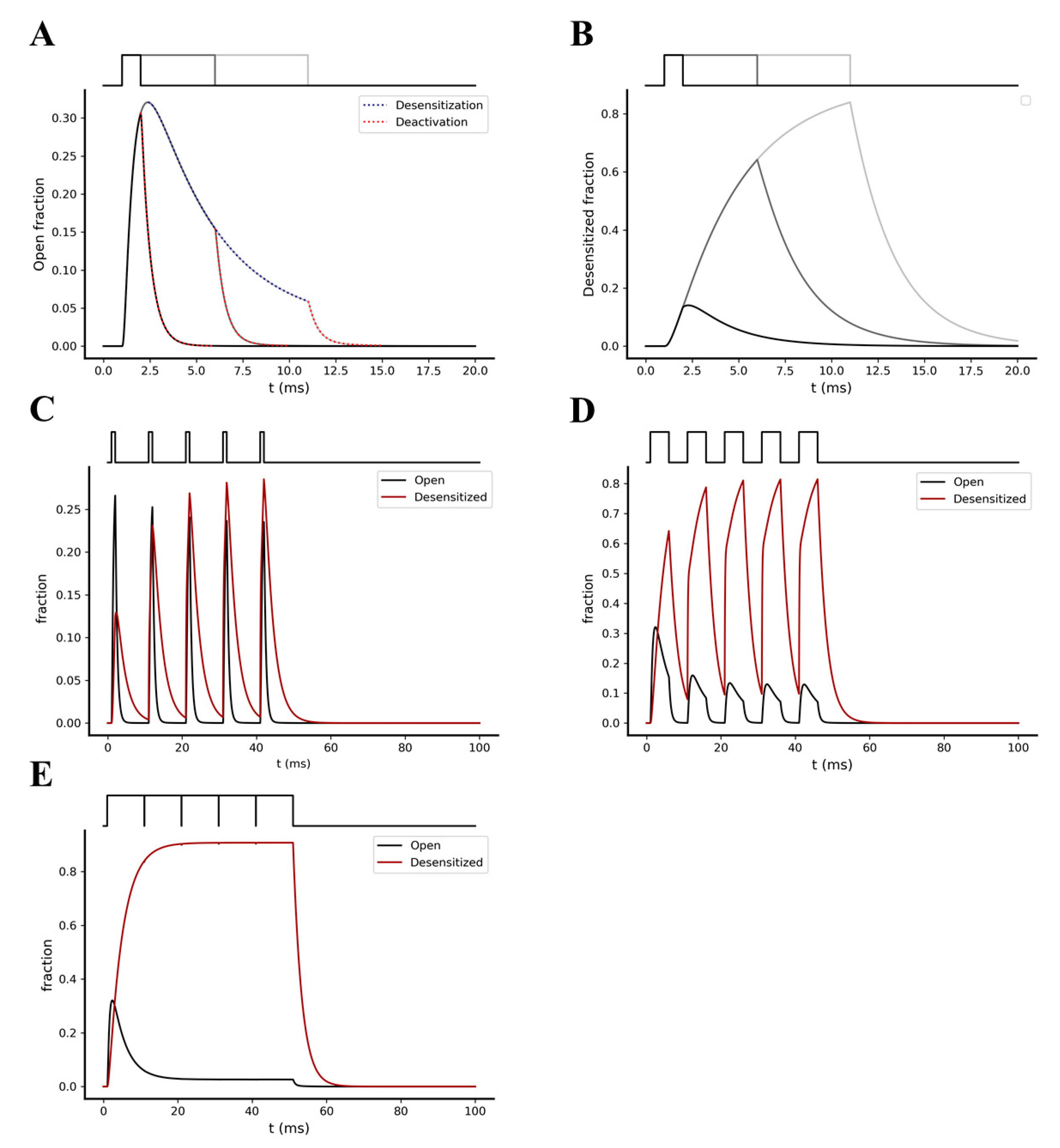
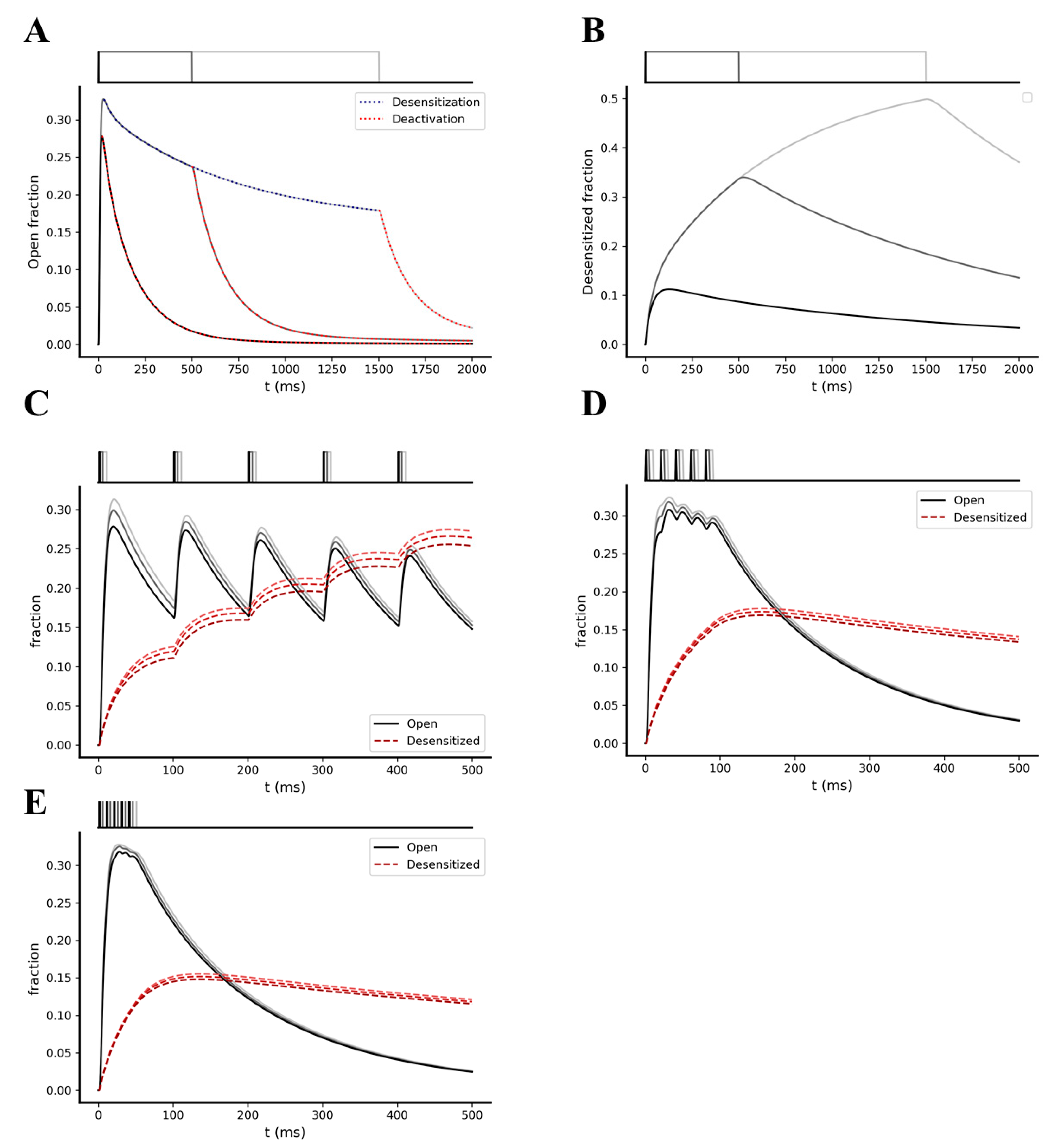
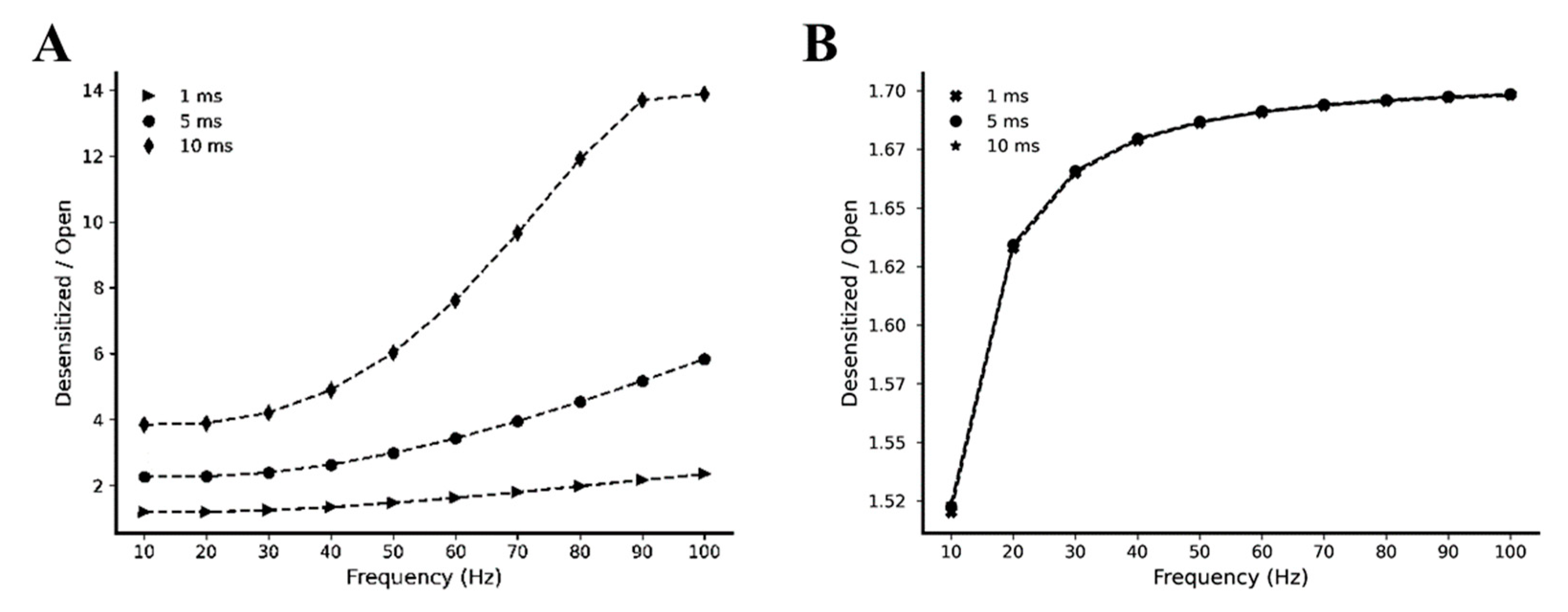
2.2. Kinetic Behavior Analysis of AMPA and NMDA Receptors under Different Pre-Synaptic Stimulation Conditions
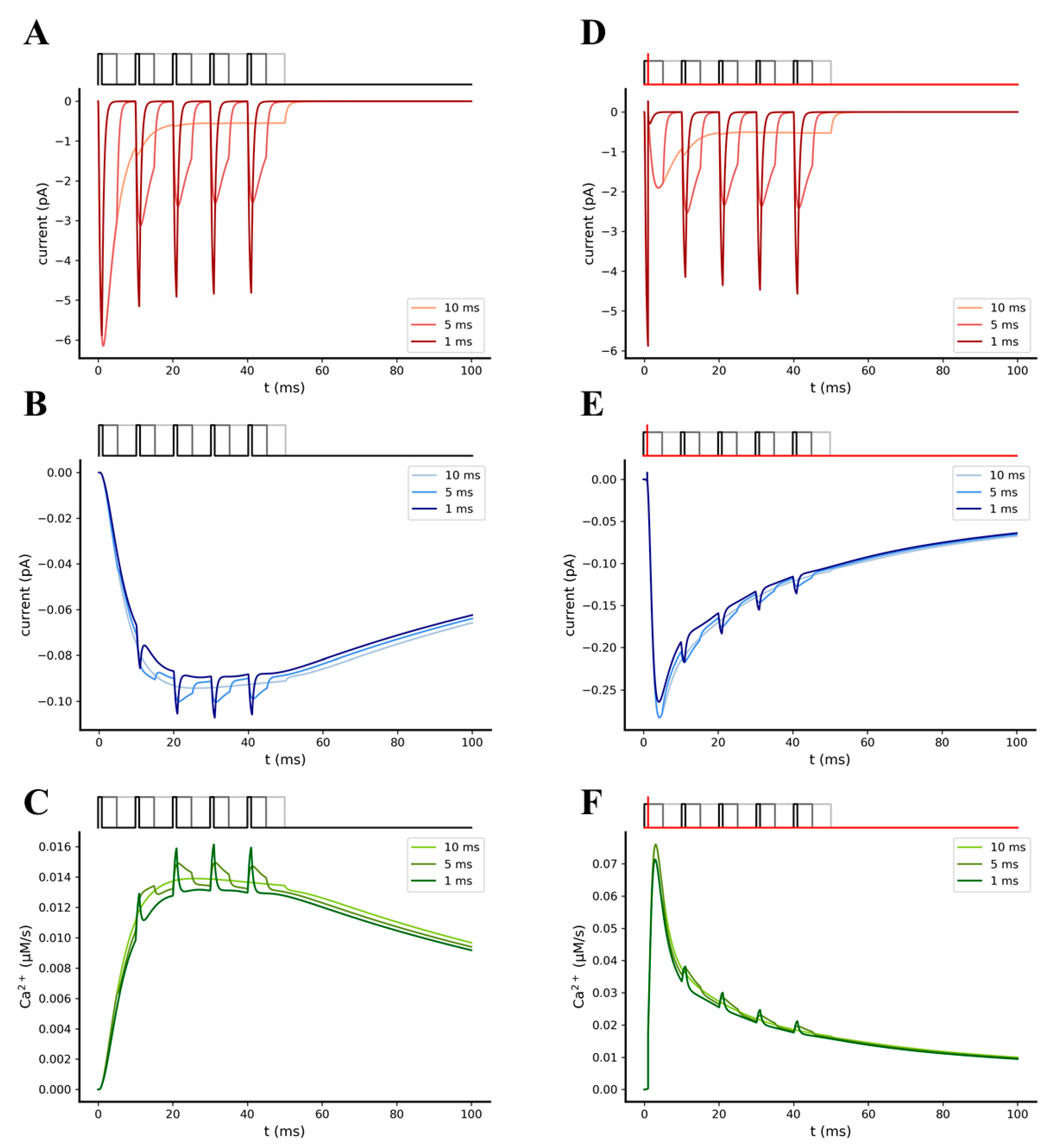
2.3. Temporal Relationship between Pre- and Post-Synaptic Stimuli Strongly Impacts Synaptic Transmission Efficiency
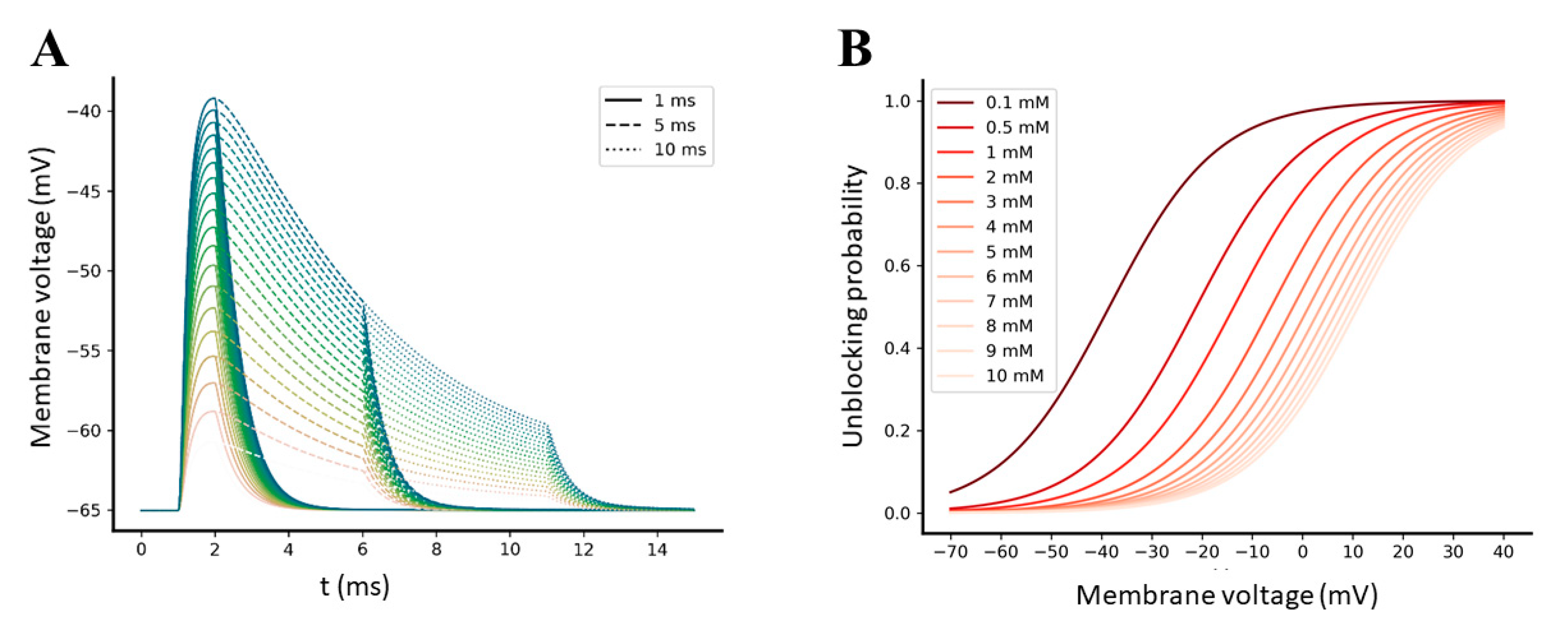
2.3.1. AMPA-Mediated EPSPs Are Not Sufficient to Efficiently Relieve the Mg2+ Block from NMDA Receptors
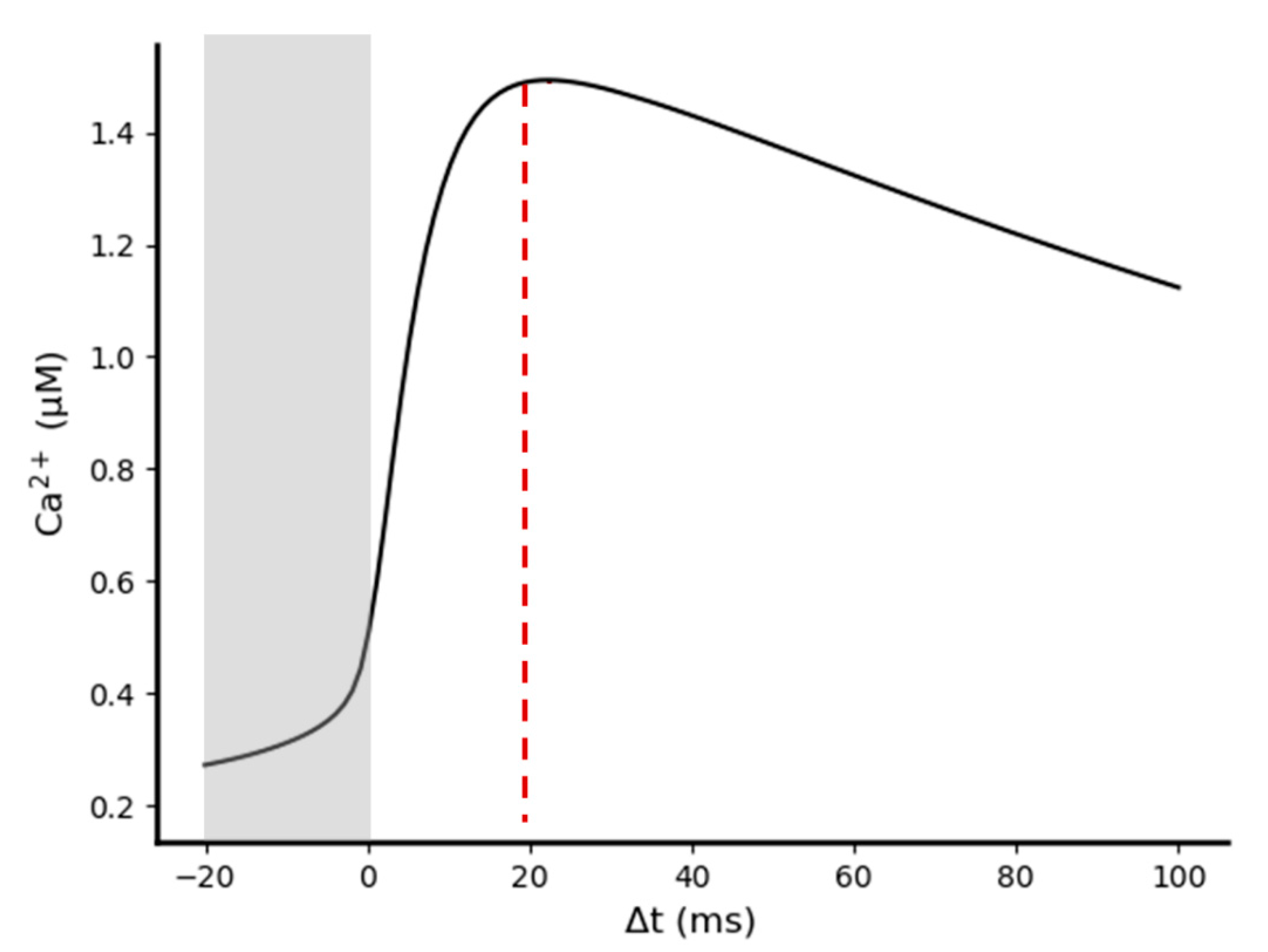
2.3.2. Synchronization between Pre- and Post-Synaptic Stimulation Significantly Increases the NMDA Receptor Contribution to Synaptic Transmission
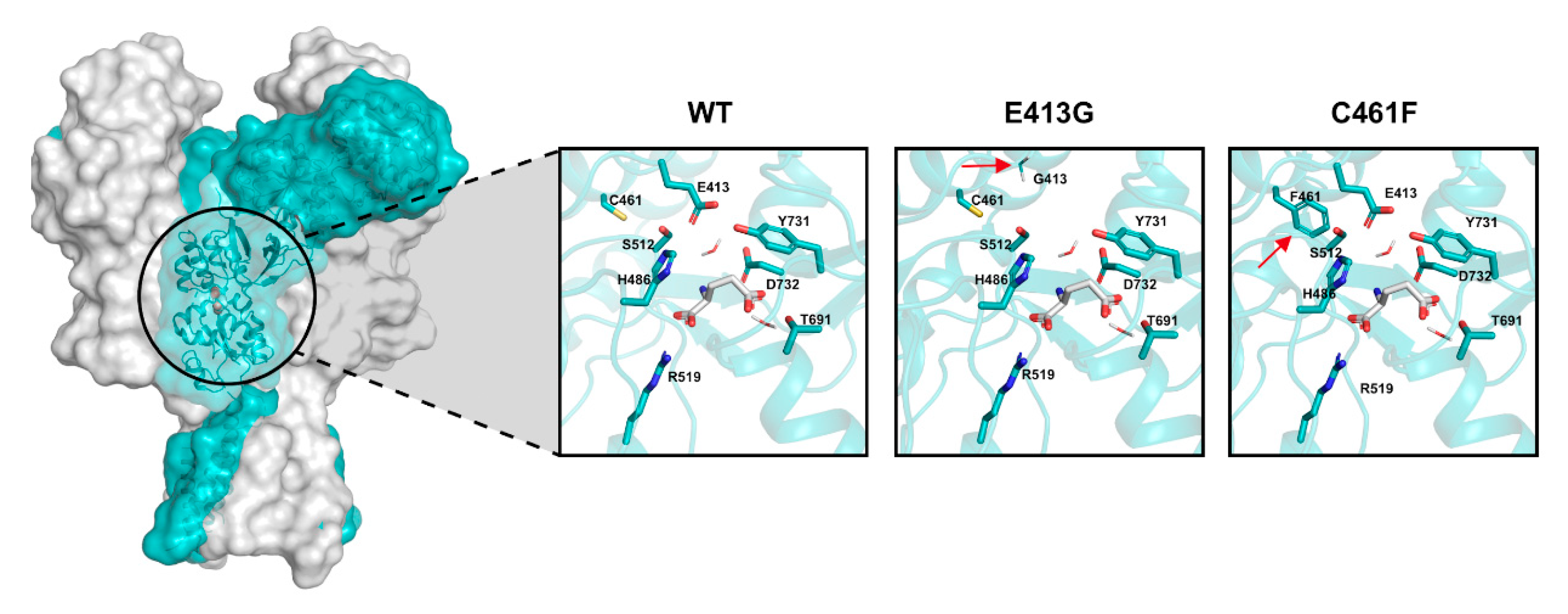
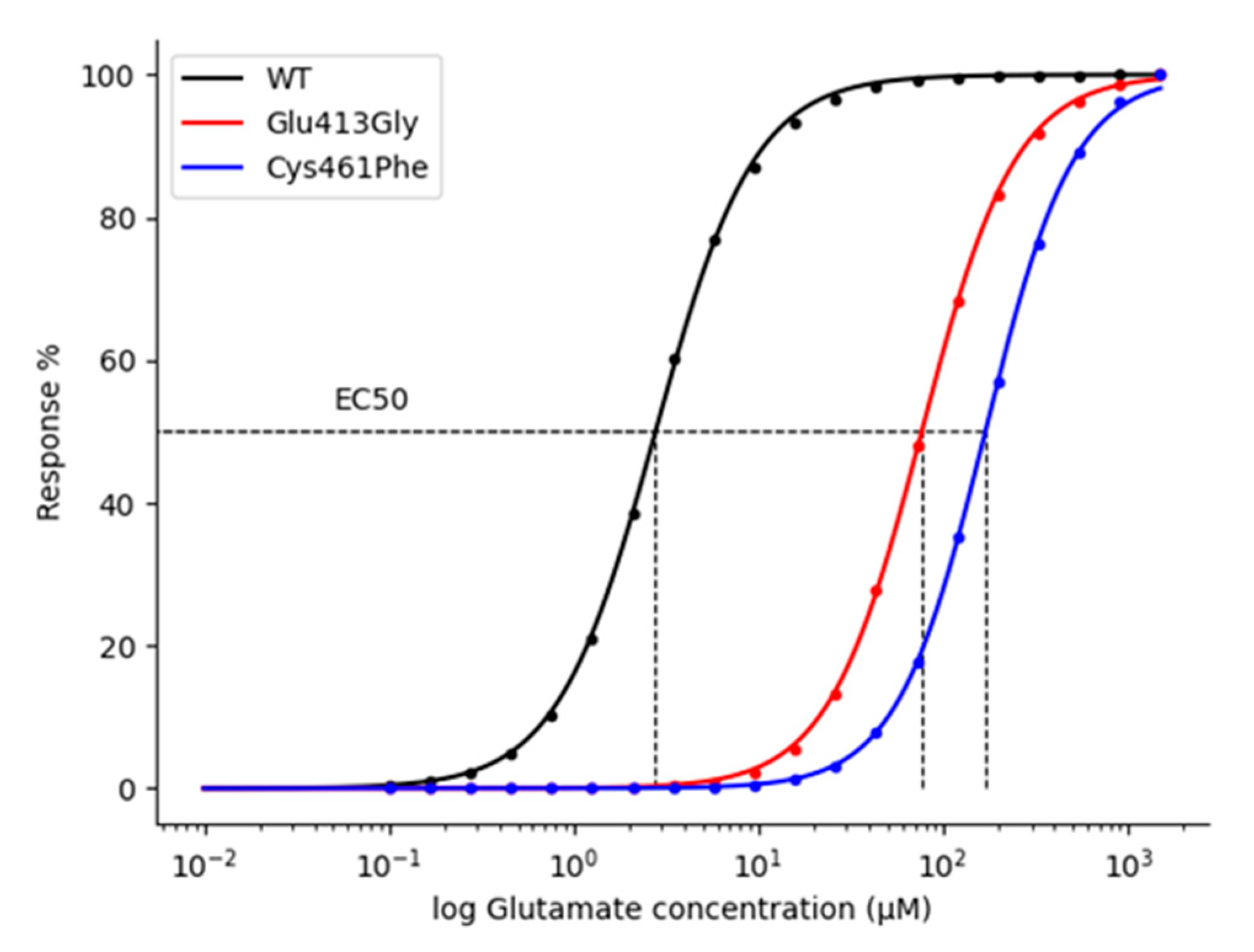
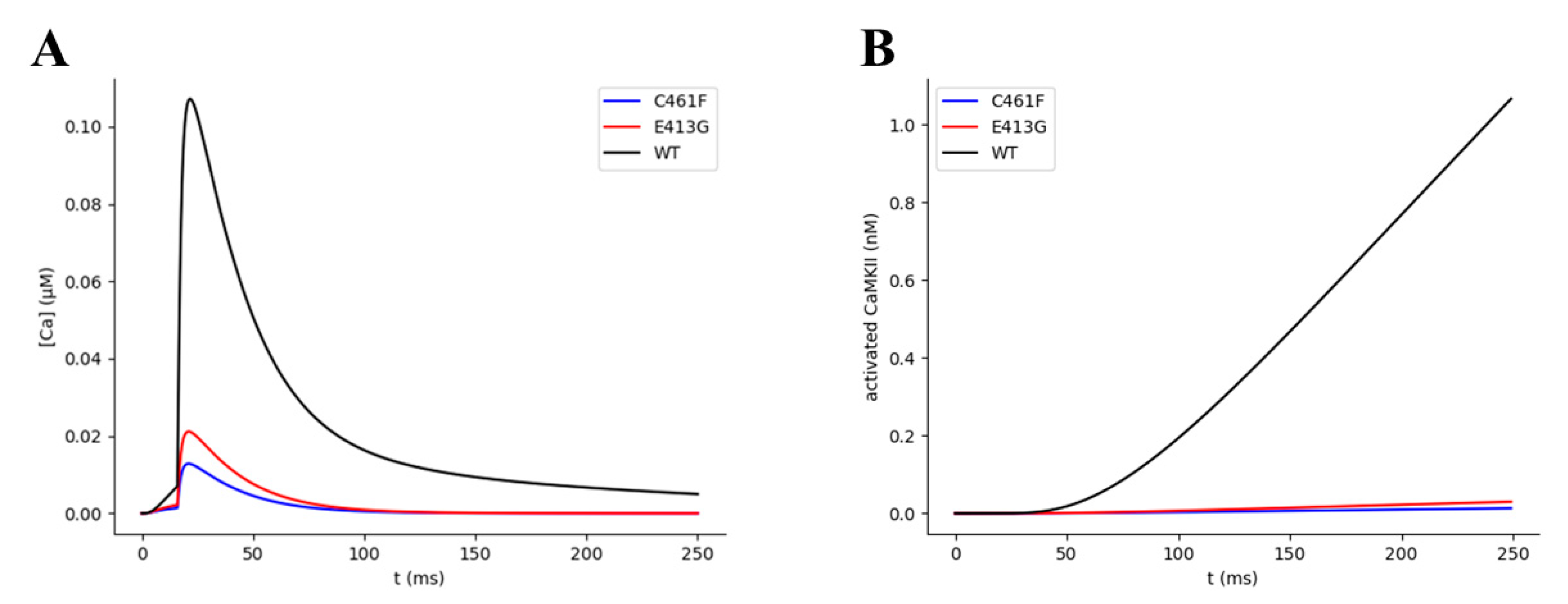
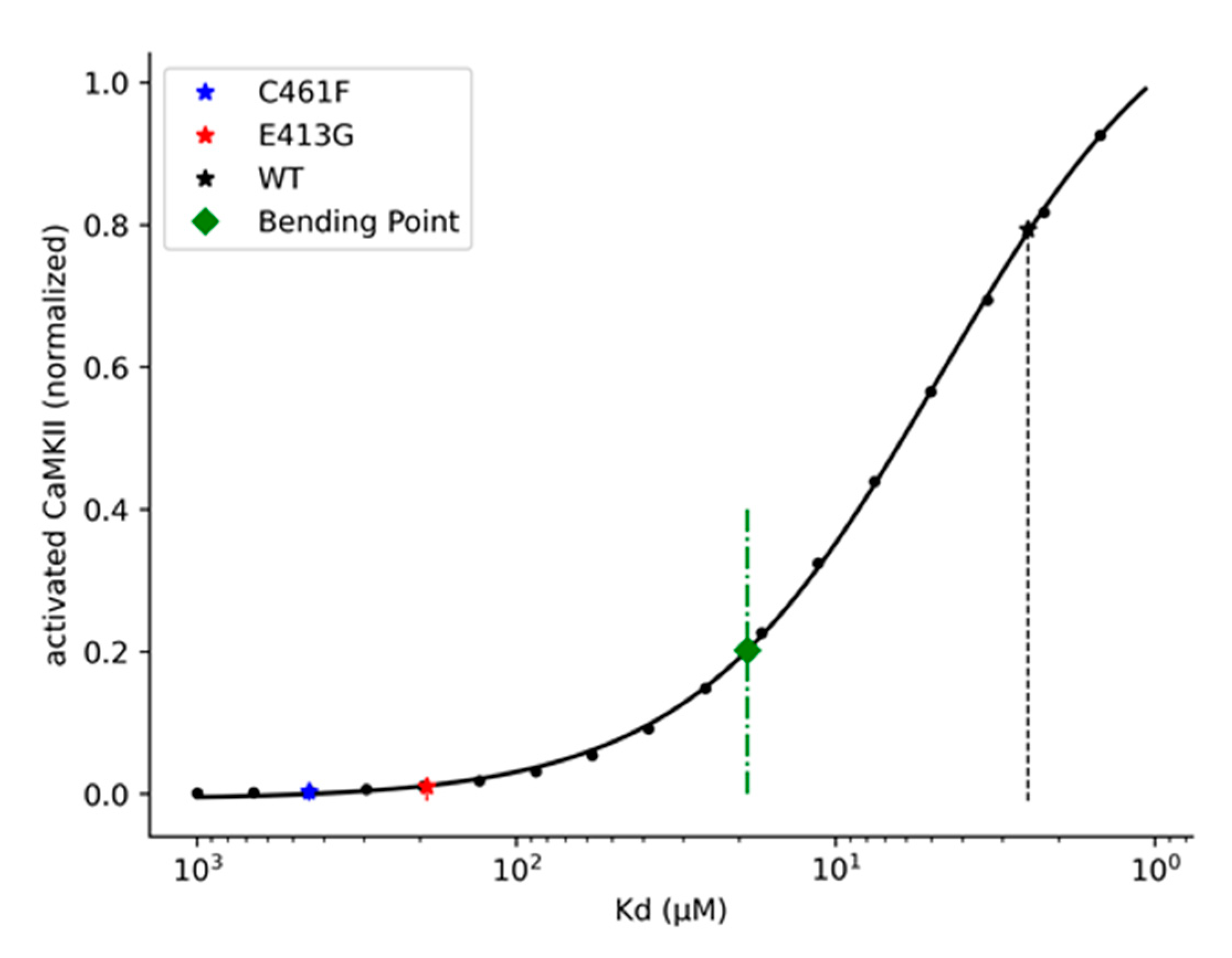
2.4. Kinetic and Pharmacological Analysis of NMDA Variants: Multiscale Integration
3. Methods
- Stimulation Pattern Design (SPD) module, where both pre- and post-synaptic stimuli can be programmed independently. This module allows us to define the inputs of the virtual synapse.
- Receptors Gating Simulation (RGS) module. This module performs a compartmentalized kinetic simulation of the virtual synaptic cleft, where a neurotransmitter released from pre-synaptic stimuli interacts with ionotropic membrane receptors expressed on the post-synaptic spine.
- EPSCs/EPSPs Calculation (CPC) module. This module analyzes the data coming from the RGS module and, calculating synaptic currents and their respective potentials, integrates pre- and post-synaptic stimuli. It constitutes a “bridge” between the extracellular and the intracellular compartments.
- CaM/CaMKII Activation Simulation (CAS) module. This module performs a compartmentalized kinetic simulation of a set of molecular reactions that takes place in the virtual post-synaptic spine, which includes the interactions between Ca2+, Calmodulin (CaM), and Ca2+/CaM-dependent Kinase II (CaMKII).
3.1. Mathematical Model Implementation
3.1.1. SPD Module
3.1.2. RGS Module
3.1.3. CPC Module
3.1.4. CAS Module
- Binding reactions between Ca2+ ions and CaM and CaM-CaMKII species;
- Dimerization reactions between Ca2+-CaM and monomeric CaMKII;
- Dimerization reactions between two Ca2+-CaM-CaMKII complexes;
- Autophosphorylation reactions of CaMKII monomers inside the 2(Ca2+-CaM-CaMKII) complexes.
3.2. Data Fitting
3.2.1. Concentration-Response Curves
3.2.2. Two-Component Exponential Function Fitting
3.2.3. Four-Parameter Logistic Function and Bending Points
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| bAP | back-propagating action potential |
| CaM | Calmodulin |
| CaMKII | Ca2+/CAM-dependent Kinase II |
| CAS | CaMKII Activation Simulation |
| CPC | Current/Potential Calculation |
| LTD | Long Term Depression |
| LTP | Long Term Potentiation |
| mCaMKII | Individual subunits of Calmodulin Kinase II |
| NMDA | N-Methyl-D-aspartic acid |
| RGS | Receptors Gating Simulation |
| STDP | Spike Timing Dependent Plasticity |
References
- Mattson, M.P. Pathways Towards and Away from Alzheimer’s Disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosjean, B.; Tsai, G.E. NMDA neurotransmission as a critical mediator of borderline personality disorder. J. Psychiatry Neurosci. 2007, 32, 103–115. [Google Scholar]
- Hsieh, H.; Boehm, J.; Sato, C.; Iwatsubo, T.; Tomita, T.; Sisodia, S.; Malinow, R. AMPA-R Removal Underlies Aβ-induced Synaptic Depression and Dendritic Spine Loss. Neuron 2006, 52, 831–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Lerma, J.; Marques, J.M. Kainate receptors in health and disease. Neuron 2013, 80, 292–311. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.J.; Grimwood, P.D.; Morris, R.G. Synaptic plasticity and memory: An evaluation of the hypothesis. Annu. Rev. Neurosci. 2000, 23, 649–711. [Google Scholar] [CrossRef] [Green Version]
- Mansvelder, H.D.; Verhoog, M.B.; Goriounova, N.A. Synaptic plasticity in human cortical circuits: Cellular mechanisms of learning and memory in the human brain? Curr. Opin. Neurobiol. 2019, 54, 186–193. [Google Scholar] [CrossRef]
- Baez, M.V.; Cercato, M.C.; Jerusalinsky, D.A. NMDA Receptor Subunits Change after Synaptic Plasticity Induction and Learning and Memory Acquisition. Neural Plast. 2018, 2018. [Google Scholar] [CrossRef]
- Kandel, E.R.; Dudai, Y.; Mayford, M.R. The Molecular and Systems Biology of Memory. Cell 2014, 157, 163–186. [Google Scholar] [CrossRef] [Green Version]
- Voss, J.L.; Bridge, D.J.; Cohen, N.J.; Walker, J.A. A closer look at the hippocampus and memory. Trends Cogn. Sci. 2017, 21, 577–588. [Google Scholar] [CrossRef]
- Lisman, J.; Buzsáki, G.; Eichenbaum, H.; Nadel, L.; Ranganath, C.; Redish, A.D. Viewpoints: How the hippocampus contributes to memory, navigation and cognition. Nat. Neurosci. 2017, 20, 1434–1447. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A. Long-Term Potentiation at CA3–CA1 Hippocampal Synapses with Special Emphasis on Aging, Disease, and Stress. Front. Aging Neurosci. 2011, 3, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsien, J.Z.; Huerta, P.T.; Tonegawa, S. The Essential Role of Hippocampal CA1 NMDA Receptor–Dependent Synaptic Plasticity in Spatial Memory. Cell 1996, 87, 1327–1338. [Google Scholar] [CrossRef] [Green Version]
- Henley, J.M.; Wilkinson, K.A. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 2016, 17, 337–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chater, T.E.; Goda, Y. The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front. Cell. Neurosci. 2014, 8, 401. [Google Scholar] [CrossRef] [PubMed]
- Blanke, M.L.; VanDongen, A.M.J. Activation Mechanisms of the NMDA Receptor. In Biology of the NMDA Receptor; Van Dongen, A.M., Ed.; Frontiers in Neuroscience; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009; ISBN 978-1-4200-4414-0. [Google Scholar]
- Vyklicky, V.; Korinek, M.; Smejkalova, T.; Balik, A.; Krausova, B.; Kaniakova, M.; Lichnerova, K.; Cerny, J.; Krusek, J.; Dittert, I.; et al. Structure, function, and pharmacology of NMDA receptor channels. Physiol. Res. 2014, 63, S191–S203. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.L.; Westbrook, G.L.; Guthrie, P.B. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 1984, 309, 261–263. [Google Scholar] [CrossRef]
- Jahr, C.E.; Stevens, C.F. A quantitative description of NMDA receptor-channel kinetic behavior. J. Neurosci. 1990, 10, 1830–1837. [Google Scholar] [CrossRef] [Green Version]
- Feldman, D.E. The spike timing dependence of plasticity. Neuron 2012, 75, 556–571. [Google Scholar] [CrossRef] [Green Version]
- Markram, H.; Gerstner, W.; Sjöström, P.J. A History of Spike-Timing-Dependent Plasticity. Front. Synaptic Neurosci. 2011, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-J.R.; Escobedo-Lozoya, Y.; Szatmari, E.M.; Yasuda, R. Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 2009, 458, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barria, A.; Muller, D.; Derkach, V.; Griffith, L.C.; Soderling, T.R. Regulatory Phosphorylation of AMPA-Type Glutamate Receptors by CaM-KII during Long-Term Potentiation. Science 1997, 276, 2042–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibarov, D.A.; Bruneau, N.; Antonov, S.M.; Szepetowski, P.; Burnashev, N.; Giniatullin, R. Functional Properties of Human NMDA Receptors Associated with Epilepsy-Related Mutations of GluN2A Subunit. Front. Cell Neurosci. 2017, 11, 155. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shipton, O.A.; Leitz, J.R.; Dworzak, J.; Acton, C.E.J.; Tunbridge, E.M.; Denk, F.; Dawson, H.N.; Vitek, M.P.; Wade-Martins, R.; Paulsen, O.; et al. Tau Protein Is Required for Amyloid β-Induced Impairment of Hippocampal Long-Term Potentiation. J. Neurosci. 2011, 31, 1688–1692. [Google Scholar] [CrossRef] [Green Version]
- Bagetta, V.; Ghiglieri, V.; Sgobio, C.; Calabresi, P.; Picconi, B. Synaptic dysfunction in Parkinson’s disease. Biochem. Soc. Trans. 2010, 38, 493–497. [Google Scholar] [CrossRef]
- Usdin, M.T.; Shelbourne, P.F.; Myers, R.M.; Madison, D.V. Impaired Synaptic Plasticity in Mice Carrying the Huntington’s Disease Mutation. Hum. Mol. Genet. 1999, 8, 839–846. [Google Scholar] [CrossRef] [Green Version]
- Murphy, K.P.S.J.; Carter, R.J.; Lione, L.A.; Mangiarini, L.; Mahal, A.; Bates, G.P.; Dunnett, S.B.; Morton, A.J. Abnormal Synaptic Plasticity and Impaired Spatial Cognition in Mice Transgenic for Exon 1 of the Human Huntington’s Disease Mutation. J. Neurosci. 2000, 20, 5115–5123. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, U.; Türkay, M.; Biegler, L.; Karasözen, B. Modeling and simulation of metabolic networks for estimation of biomass accumulation parameters. Discret. Appl. Math. 2009, 157, 2483–2493. [Google Scholar] [CrossRef] [Green Version]
- Bartol, T.M.; Keller, D.X.; Kinney, J.P.; Bajaj, C.L.; Harris, K.M.; Sejnowski, T.J.; Kennedy, M.B. Computational reconstitution of spine calcium transients from individual proteins. Front. Synaptic Neurosci. 2015, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, E.; Mergenthal, A.; Bingham, C.S.; Song, D.; Bouteiller, J.-M.; Berger, T.W. A Glutamatergic Spine Model to Enable Multi-Scale Modeling of Nonlinear Calcium Dynamics. Front. Comput. Neurosci. 2018, 12, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujol, A.; Mosca, R.; Farrés, J.; Aloy, P. Unveiling the role of network and systems biology in drug discovery. Trends Pharmacol. Sci. 2010, 31, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Shouval, H.Z.; Bear, M.F.; Cooper, L.N. A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc. Natl. Acad. Sci. USA 2002, 99, 10831–10836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, M.; Tsukada, S.; Tsuzuki, K.; Kijima, H.; Ozawa, S. Regulation of Kinetic Properties of GluR2 AMPA Receptor Channels by Alternative Splicing. J. Neurosci. 2000, 20, 2166–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amico-Ruvio, S.A.; Popescu, G.K. Stationary Gating of GluN1/GluN2B Receptors in Intact Membrane Patches. Biophys. J. 2010, 98, 1160–1169. [Google Scholar] [CrossRef] [Green Version]
- Pepke, S.; Kinzer-Ursem, T.; Mihalas, S.; Kennedy, M.B. A Dynamic Model of Interactions of Ca2+, Calmodulin, and Catalytic Subunits of Ca2+/Calmodulin-Dependent Protein Kinase II. PLoS Comput. Biol. 2010, 6, e1000675. [Google Scholar] [CrossRef] [Green Version]
- Dzubay, J.A.; Jahr, C.E. The Concentration of Synaptically Released Glutamate Outside of the Climbing Fiber–Purkinje Cell Synaptic Cleft. J. Neurosci. 1999, 19, 5265–5274. [Google Scholar] [CrossRef]
- Moussawi, K.; Riegel, A.; Nair, S.; Kalivas, P.W. Extracellular Glutamate: Functional Compartments Operate in Different Concentration Ranges. Front. Syst. Neurosci. 2011, 5, 94. [Google Scholar] [CrossRef] [Green Version]
- Robert, A.; Howe, J.R. How AMPA Receptor Desensitization Depends on Receptor Occupancy. J. Neurosci. 2003, 23, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Nusser, Z.; Lujan, R.; Laube, G.; Roberts, J.D.B.; Molnar, E.; Somogyi, P. Cell Type and Pathway Dependence of Synaptic AMPA Receptor Number and Variability in the Hippocampus. Neuron 1998, 21, 545–559. [Google Scholar] [CrossRef] [Green Version]
- Markram, H.; Lübke, J.; Frotscher, M.; Sakmann, B. Regulation of Synaptic Efficacy by Coincidence of Postsynaptic APs and EPSPs. Science 1997, 275, 213–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albensi, B.C.; Oliver, D.R.; Toupin, J.; Odero, G. Electrical stimulation protocols for hippocampal synaptic plasticity and neuronal hyper-excitability: Are they effective or relevant? Exp. Neurol. 2007, 204, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Racca, C.; Stephenson, F.A.; Streit, P.; Roberts, J.D.B.; Somogyi, P. NMDA Receptor Content of Synapses in Stratum Radiatum of the Hippocampal CA1 Area. J. Neurosci. 2000, 20, 2512–2522. [Google Scholar] [CrossRef] [PubMed]
- Lester, R.A.J.; Clements, J.D.; Westbrook, G.L.; Jahr, C.E. Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature 1990, 346, 565–567. [Google Scholar] [CrossRef] [PubMed]
- Swanger, S.A.; Chen, W.; Wells, G.; Burger, P.B.; Tankovic, A.; Bhattacharya, S.; Strong, K.L.; Hu, C.; Kusumoto, H.; Zhang, J.; et al. Mechanistic Insight into NMDA Receptor Dysregulation by Rare Variants in the GluN2A and GluN2B Agonist Binding Domains. Am. J. Hum. Genet. 2016, 99, 1261–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, G.; Yuan, H.; McDaniel, M.J.; Kusumoto, H.; Snyder, J.P.; Liotta, D.C.; Traynelis, S.F. The GluN2B-Glu413Gly NMDA receptor variant arising from a de novo GRIN2B mutation promotes ligand-unbinding and domain opening. Proteins 2018, 86, 1265–1276. [Google Scholar] [CrossRef]
- Yuan, H.; Hansen, K.B.; Vance, K.M.; Ogden, K.K.; Traynelis, S.F. Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J. Neurosci. 2009, 29, 12045–12058. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Chen, W.; Myers, S.J.; Yuan, H.; Traynelis, S.F. Human GRIN2B variants in neurodevelopmental disorders. J. Pharm. Sci. 2016, 132, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.R.; Yuan, H.; Holyoak, T.; Arajs, K.H.; Hakimi, P.; Markello, T.C.; Wolfe, L.A.; Vilboux, T.; Burton, B.K.; Fajardo, K.F.; et al. Three rare diseases in one Sib pair: RAI1, PCK1, GRIN2B mutations associated with Smith–Magenis Syndrome, cytosolic PEPCK deficiency and NMDA receptor glutamate insensitivity. Mol. Genet. Metab. 2014, 113, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.B.; Ogden, K.K.; Yuan, H.; Traynelis, S.F. Distinct functional and pharmacological properties of triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron 2014, 81, 1084–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, C.F.; Muhlich, J.L.; Bachman, J.A.; Sorger, P.K. Programming biological models in Python using PySB. Mol. Syst. Biol. 2013, 9, 646. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Stone, D.B.; Tesche, C.D. Topological dynamics in spike-timing dependent plastic model neural networks. Front. Neural Circuits 2013, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Rackham, O.J.L.; Tsaneva-Atanasova, K.; Ganesh, A.; Mellor, J.R. A Ca2+-Based Computational Model for NMDA Receptor-Dependent Synaptic Plasticity at Individual Post-Synaptic Spines in the Hippocampus. Front. Synaptic Neurosci. 2010, 2, 31. [Google Scholar] [CrossRef] [Green Version]
- Lajeunesse, F.; Kröger, H.; Timofeev, I. Regulation of AMPA and NMDA receptor-mediated EPSPs in dendritic trees of thalamocortical cells. J. Neurophysiol. 2013, 109, 13–30. [Google Scholar] [CrossRef] [Green Version]
- Pongrácz, F.; Poolos, N.P.; Kocsis, J.D.; Shepherd, G.M. A Model of NMDA Receptor-Mediated Activity in Dendrites of Hippocampal CA1 Pyramidal Neurons. J. Neurophysiol. 1992, 68, 2248–2259. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharm. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [Green Version]
- Momiyama, A.; Feldmeyer, D.; Cull-Candy, S.G. Identification of a native low-conductance NMDA channel with reduced sensitivity to Mg2+ in rat central neurones. J. Physiol. 1996, 494, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Benke, T.; Traynelis, S.F. AMPA-type glutamate receptor conductance changes and plasticity: Still a lot of noise. Neurochem. Res. 2019, 44, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Higley, M.J.; Sabatini, B.L. Calcium Signaling in Dendritic Spines. Cold Spring Harb. Perspect. Biol. 2012, 4, a005686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Maio, V.; Ventriglia, F.; Santillo, S. A model of cooperative effect of AMPA and NMDA receptors in glutamatergic synapses. Cogn. Neurodyn. 2016, 10, 315–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloodgood, B.L.; Sabatini, B.L. Nonlinear Regulation of Unitary Synaptic Signals by CaV2.3 Voltage-Sensitive Calcium Channels Located in Dendritic Spines. Neuron 2007, 53, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Malik-Sheriff, R.S.; Glont, M.; Nguyen, T.V.N.; Tiwari, K.; Roberts, M.G.; Xavier, A.; Vu, M.T.; Men, J.; Maire, M.; Kananathan, S.; et al. BioModels—15 years of sharing computational models in life science. Nucleic Acids Res. 2020, 48, D407–D415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebaugh, J.L.; McCray, P.D. Defining the linear portion of a sigmoid-shaped curve: Bend points. Pharm. Stat. 2003, 2, 167–174. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predicted Kd (μM) | Weighted Tau (ms) | ||
|---|---|---|---|
| Predicted | Exp. | ||
| Wt | 2.5 | 328 | 314–570 [47,48,52] |
| Glu413Gly | 190.5 | 29 | 20–34 [47,48] |
| Cys461Phe | 446.5 | 27 | 28 [48] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micheli, P.; Ribeiro, R.; Giorgetti, A. A Mechanistic Model of NMDA and AMPA Receptor-Mediated Synaptic Transmission in Individual Hippocampal CA3-CA1 Synapses: A Computational Multiscale Approach. Int. J. Mol. Sci. 2021, 22, 1536. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041536
Micheli P, Ribeiro R, Giorgetti A. A Mechanistic Model of NMDA and AMPA Receptor-Mediated Synaptic Transmission in Individual Hippocampal CA3-CA1 Synapses: A Computational Multiscale Approach. International Journal of Molecular Sciences. 2021; 22(4):1536. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041536
Chicago/Turabian StyleMicheli, Pietro, Rui Ribeiro, and Alejandro Giorgetti. 2021. "A Mechanistic Model of NMDA and AMPA Receptor-Mediated Synaptic Transmission in Individual Hippocampal CA3-CA1 Synapses: A Computational Multiscale Approach" International Journal of Molecular Sciences 22, no. 4: 1536. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041536