
Exploring the Cold-Adaptation Mechanism of Serine Hydroxymethyltransferase by Comparative Molecular Dynamics Simulations




Abstract
:1. Introduction
2. Results
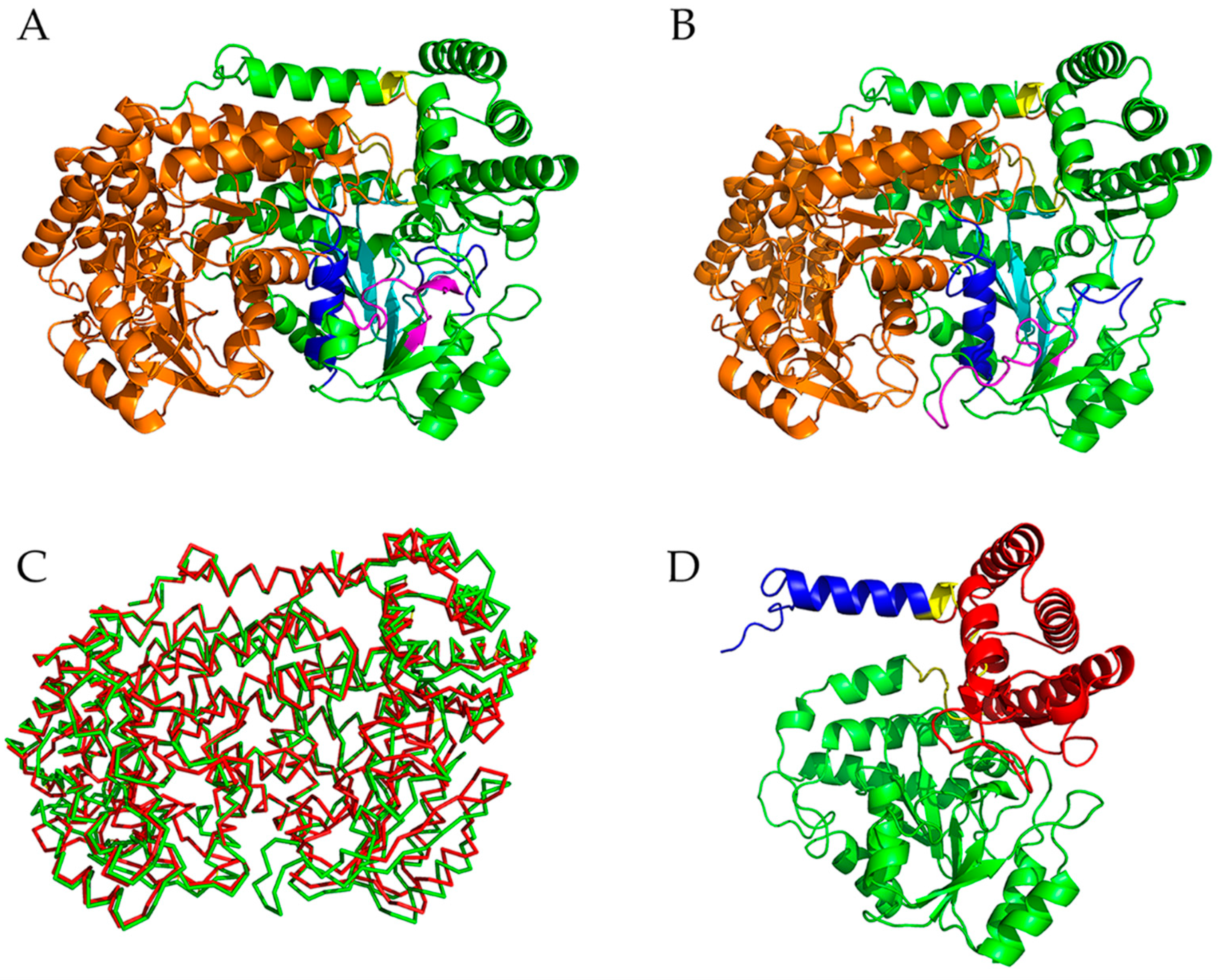
2.1. Descriptions of the Crystal Structures of mSHMT and pSHMT
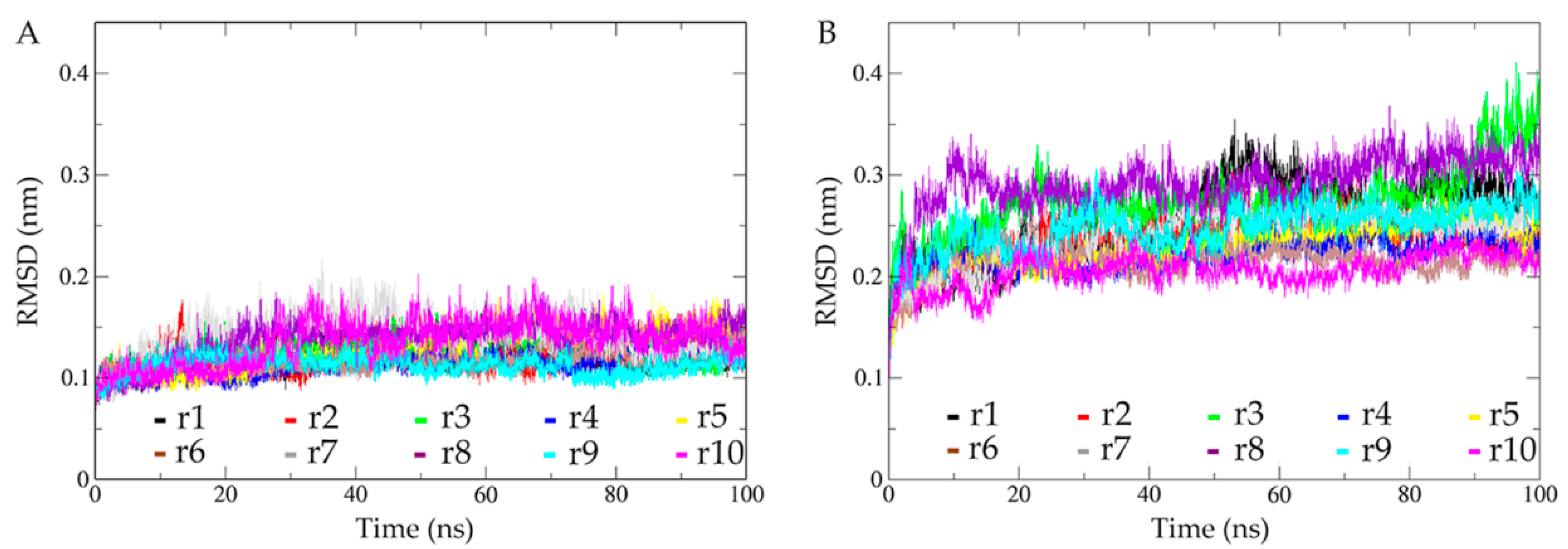
2.2. Structural Fluctuations during Simulations and Conformational Sampling Evaluation
2.3. Comparison of Structural Properties
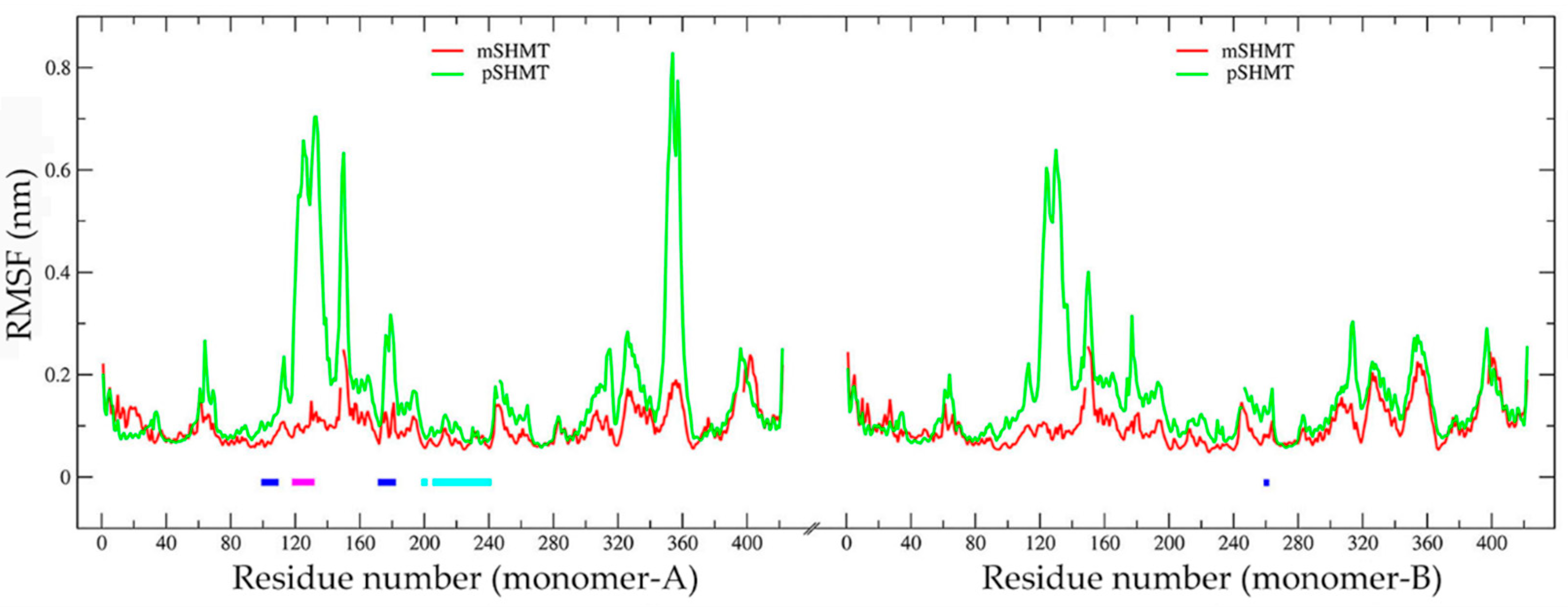
2.4. Comparison of Conformational Flexibility
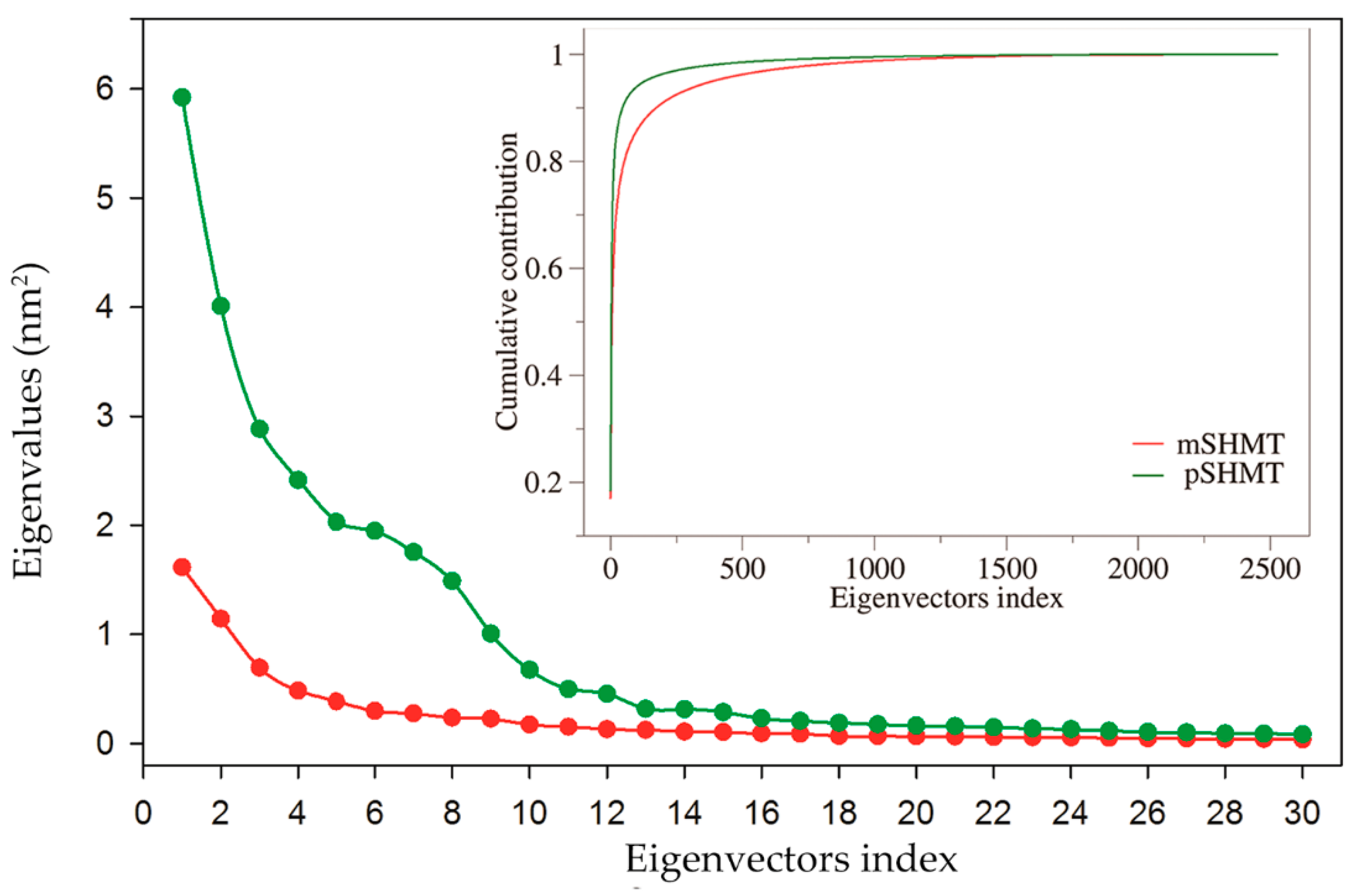
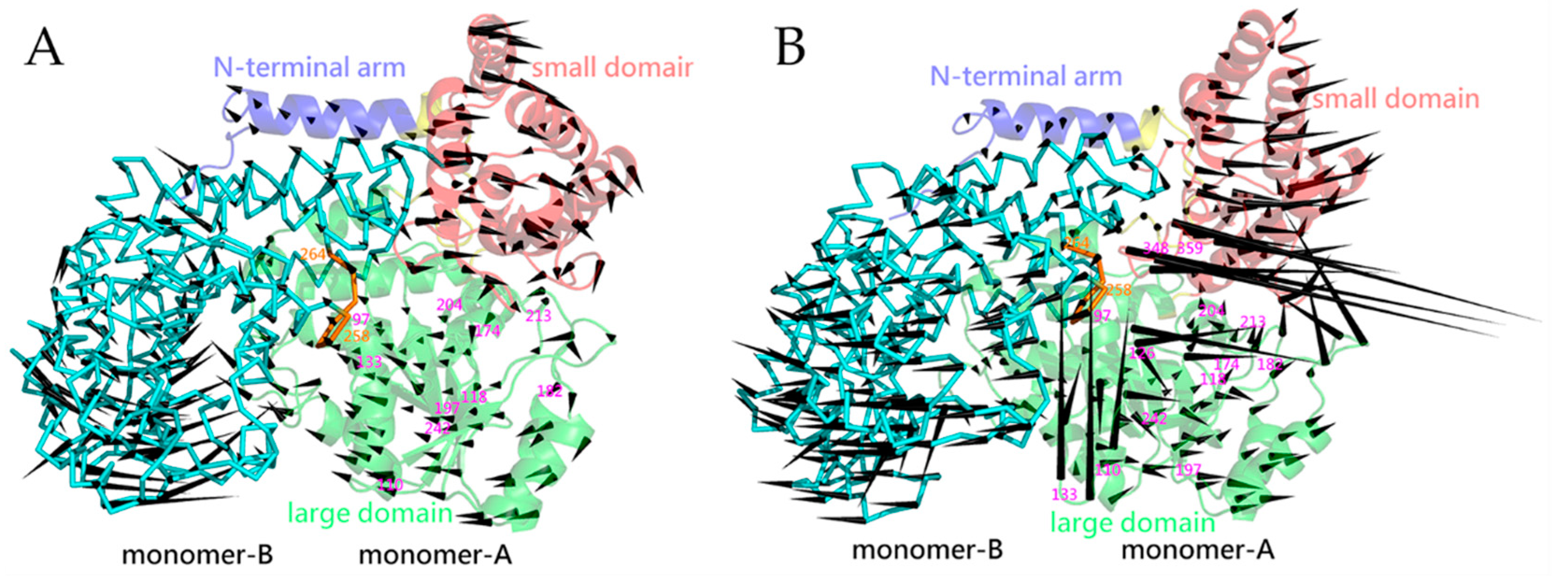
2.5. Essential Dynamics and Collective Motions
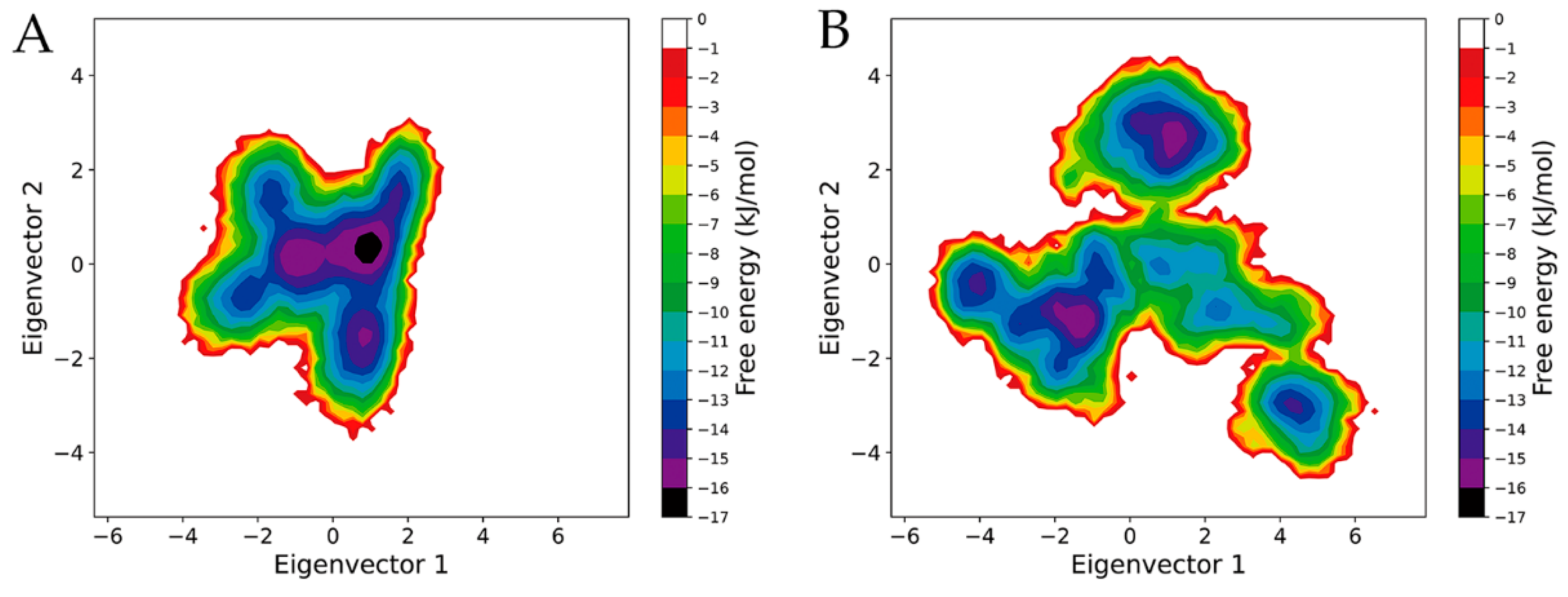
2.6. Free Energy Landscapes
3. Discussion
4. Materials and Methods
4.1. Structural Preparation
4.2. MD Simulations
4.3. Analysis Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lonhienne, T.; Gerday, C.; Feller, G. Psychrophilic enzymes: Revisiting the thermodynamic parameters of activation may explain local flexibility. Biochim. Biophys. Acta 2000, 1543, 1–10. [Google Scholar] [CrossRef]
- Somero, G.N. Proteins and temperature. Annu. Rev. Physiol. 1995, 57, 43–68. [Google Scholar] [CrossRef]
- Feller, G.; Gerday, C. Psychrophilic enzymes: Molecular basis of cold adaptation. Cell. Mol. Life Sci. 1997, 53, 830–841. [Google Scholar] [CrossRef] [Green Version]
- Papaleo, E.; Tiberti, M.; Invernizzi, G.; Pasi, M.; Ranzani, V. Molecular determinants of enzyme cold adaptation: Comparative structural and computational studies of cold- and warm-adapted enzymes. Curr. Protein Pept. Sci. 2011, 12, 657–683. [Google Scholar] [CrossRef] [PubMed]
- Feller, G.; d’Amico, D.; Gerday, C. Thermodynamic stability of a cold-active alpha-amylase from the Antarctic bacterium Alteromonas haloplanctis. Biochemistry 1999, 38, 4613–4619. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.A.; Lei, M.; Thai, V.; Kerns, S.J.; Karplus, M.; Kern, D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 2007, 450, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Papaleo, E.; Mereghetti, P.; Fantucci, P.; Grandori, R.; De Gioia, L. Free-energy landscape, principal component analysis, and structural clustering to identify representative conformations from molecular dynamics simulations: The myoglobin case. J. Mol. Graph. Model. 2009, 27, 889–899. [Google Scholar] [CrossRef]
- Li, Y.; Deng, L.; Liang, J.; Dong, G.H.; Xia, Y.L.; Fu, Y.X.; Liu, S.Q. Molecular dynamics simulations reveal distinct differences in conformational dynamics and thermodynamics between the unliganded and CD4-bound states of HIV-1 gp120. Phys. Chem. Chem. Phys. 2020, 22, 5548–5560. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Deng, L.; Yang, L.Q.; Sang, P.; Liu, S.Q. Effects of CD4 binding on conformational dynamics, molecular motions, and thermodynamics of HIV-1 gp120. Int. J. Mol. Sci. 2019, 20, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snell, K.; Baumann, U.; Byrne, P.C.; Chave, K.J.; Renwick, S.B.; Sanders, P.G.; Whitehouse, S.K. The genetic organization and protein crystallographic structure of human serine hydroxymethyltransferase. Adv. Enzyme Regul. 2000, 40, 353–403. [Google Scholar] [CrossRef]
- Appaji Rao, N.; Ambili, M.; Jala, V.R.; Subramanya, H.S.; Savithri, H.S. Structure-function relationship in serine hydroxymethyltransferase. Biochim. Biophys. Acta 2003, 1647, 24–29. [Google Scholar] [CrossRef]
- Florio, R.; di Salvo, M.L.; Vivoli, M.; Contestabile, R. Serine hydroxymethyltransferase: A model enzyme for mechanistic, structural, and evolutionary studies. Biochim. Biophys. Acta 2011, 1814, 1489–1496. [Google Scholar] [CrossRef]
- Stover, P.; Schirch, V. Serine hydroxymethyltransferase catalyzes the hydrolysis of 5,10-methenyltetrahydrofolate to 5-formyltetrahydrofolate. J. Biol. Chem. 1990, 265, 14227–14233. [Google Scholar] [CrossRef]
- Angelaccio, S. Extremophilic SHMTs: From structure to biotechnology. BioMed Res. Int. 2013, 2013, 851428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amadasi, A.; Bertoldi, M.; Contestabile, R.; Bettati, S.; Cellini, B.; di Salvo, M.L.; Borri-Voltattorni, C.; Bossa, F.; Mozzarelli, A. Pyridoxal 5′-phosphate enzymes as targets for therapeutic agents. Curr. Med. Chem. 2007, 14, 1291–1324. [Google Scholar] [CrossRef] [PubMed]
- Renwick, S.B.; Snell, K.; Baumann, U. The crystal structure of human cytosolic serine hydroxymethyltransferase: A target for cancer chemotherapy. Structure 1998, 6, 1105–1116. [Google Scholar] [CrossRef] [Green Version]
- Angelaccio, S.; Florio, R.; Consalvi, V.; Festa, G.; Pascarella, S. Serine hydroxymethyltransferase from the cold adapted microorganism Psychromonas ingrahamii: A low temperature active enzyme with broad substrate specificity. Int. J. Mol. Sci. 2012, 13, 1314–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarsdale, J.N.; Radaev, S.; Kazanina, G.; Schirch, V.; Wright, H.T. Crystal structure at 2.4 A resolution of E. coli serine hydroxymethyltransferase in complex with glycine substrate and 5-formyl tetrahydrofolate. J. Mol. Biol. 2000, 296, 155–168. [Google Scholar] [CrossRef]
- Angelaccio, S.; Dworkowski, F.; Di Bello, A.; Milano, T.; Capitani, G.; Pascarella, S. Conformational transitions driven by pyridoxal-5′-phosphate uptake in the psychrophilic serine hydroxymethyltransferase from Psychromonas ingrahamii. Proteins 2014, 82, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Siglioccolo, A.; Bossa, F.; Pascarella, S. Structural adaptation of serine hydroxymethyltransferase to low temperatures. Int. J. Biol. Macromol. 2010, 46, 37–46. [Google Scholar] [CrossRef]
- Caves, L.S.; Evanseck, J.D.; Karplus, M. Locally accessible conformations of proteins: Multiple molecular dynamics simulations of crambin. Protein Sci. 1998, 7, 649–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sang, P.; Yang, Q.; Du, X.; Yang, N.; Yang, L.Q.; Ji, X.L.; Fu, Y.X.; Meng, Z.H.; Liu, S.Q. Effect of the solvent temperatures on dynamics of serine protease proteinase K. Int. J. Mol. Sci. 2016, 17, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Deng, L.; Ai, S.M.; Sang, P.; Yang, J.; Xia, Y.L.; Zhang, Z.B.; Fu, Y.X.; Liu, S.Q. Insights into the molecular mechanism underlying CD4-dependency and neutralization sensitivity of HIV-1: A comparative molecular dynamics study on gp120s from isolates with different phenotypes. RSC Adv. 2018, 8, 14355–14368. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.L.; Sun, J.H.; Ai, S.M.; Li, Y.; Du, X.; Sang, P.; Yang, L.Q.; Fu, Y.X.; Liu, S.Q. Insights into the role of electrostatics in temperature adaptation: A comparative study of psychrophilic, mesophilic, and thermophilic subtilisin-like serine proteases. RSC Adv. 2018, 8, 29698–29713. [Google Scholar] [CrossRef] [Green Version]
- Feller, G.; Gerday, C. Psychrophilic enzymes: Hot topics in cold adaptation. Nat. Rev. Microbiol. 2003, 1, 200–208. [Google Scholar] [CrossRef]
- Chiuri, R.; Maiorano, G.; Rizzello, A.; del Mercato, L.L.; Cingolani, R.; Rinaldi, R.; Maffia, M.; Pompa, P.P. Exploring local flexibility/rigidity in psychrophilic and mesophilic carbonic anhydrases. Biophys. J. 2009, 96, 1586–1596. [Google Scholar] [CrossRef] [Green Version]
- Pasi, M.; Riccardi, L.; Fantucci, P.; De Gioia, L.; Papaleo, E. Dynamic properties of a psychrophilic alpha-amylase in comparison with a mesophilic homologue. J. Phys. Chem. B 2009, 113, 13585–13595. [Google Scholar] [CrossRef]
- Bentahir, M.; Feller, G.; Aittaleb, M.; Lamotte-Brasseur, J.; Himri, T.; Chessa, J.P.; Gerday, C. Structural, kinetic, and calorimetric characterization of the cold-active phosphoglycerate kinase from the antarctic Pseudomonas sp. TACII18. J. Biol. Chem. 2000, 275, 11147–11153. [Google Scholar] [CrossRef] [Green Version]
- Michetti, D.; Brandsdal, B.O.; Bon, D.; Isaksen, G.V.; Tiberti, M.; Papaleo, E. A comparative study of cold- and warm-adapted Endonucleases A using sequence analyses and molecular dynamics simulations. PLoS ONE 2017, 12, e0169586. [Google Scholar] [CrossRef]
- Sang, P.; Du, X.; Yang, L.Q.; Meng, Z.H.; Liu, S.Q. Molecular motions and free-energy landscape of serine proteinase K in relation to its cold-adaptation: A comparative molecular dynamics simulation study and the underlying mechanisms. RSC Adv. 2017, 7, 28580–28590. [Google Scholar] [CrossRef] [Green Version]
- Isaksen, G.V.; Åqvist, J.; Brandsdal, B.O. Enzyme surface rigidity tunes the temperature dependence of catalytic rates. Proc. Natl. Acad. Sci. USA 2016, 113, 7822–7827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pischedda, A.; Ramasamy, K.P.; Mangiagalli, M.; Chiappori, F.; Milanesi, L.; Miceli, C.; Pucciarelli, S.; Lotti, M. Antarctic marine ciliates under stress: Superoxide dismutases from the psychrophilic Euplotes focardii are cold-active yet heat tolerant enzymes. Sci. Rep. 2018, 8, 14721. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.L.; Li, Y.P.; Fu, Y.X.; Liu, S.Q. The energetic origin of different catalytic activities in temperature-adapted trypsins. ACS Omega 2020, 5, 25077–25086. [Google Scholar] [CrossRef] [PubMed]
- Isaksen, G.V.; Åqvist, J.; Brandsdal, B.O. Protein surface softness is the origin of enzyme cold-adaptation of trypsin. PLoS Comput. Biol. 2014, 10, e1003813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, K.S.; Cavicchioli, R. Cold-adapted enzymes. Annu. Rev. Biochem. 2006, 75, 403–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Åqvist, J. Cold adaptation of triosephosphate isomerase. Biochemistry 2017, 56, 4169–4176. [Google Scholar] [CrossRef] [PubMed]
- Sočan, J.; Kazemi, M.; Isaksen, G.V.; Brandsdal, B.O.; Åqvist, J. Catalytic adaptation of psychrophilic elastase. Biochemistry 2018, 57, 2984–2993. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.K.; Billeter, S.R.; Rajagopalan, P.T.; Benkovic, S.J.; Hammes-Schiffer, S. Network of coupled promoting motions in enzyme catalysis. Proc. Natl. Acad. Sci. USA 2002, 99, 2794–2799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rod, T.H.; Radkiewicz, J.L.; Brooks, C.L., III. Correlated motion and the effect of distal mutations in dihydrofolate reductase. Proc. Natl. Acad. Sci. USA 2003, 100, 6980–6985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surpeta, B.; Sequeiros-Borja, C.E.; Brezovsky, J. Dynamics, a powerful component of current and future in silico approaches for protein design and engineering. Int. J. Mol. Sci. 2020, 21, 2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.Q.; Sang, P.; Tao, Y.; Fu, Y.X.; Zhang, K.Q.; Xie, Y.H.; Liu, S.Q. Protein dynamics and motions in relation to their functions: Several case studies and the underlying mechanisms. J. Biomol. Struct. Dyn. 2014, 32, 372–393. [Google Scholar] [CrossRef] [Green Version]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Section 3.1, Hierarchical Structure of Proteins. In Molecular Cell Biology, 4th ed.; W. H. Freeman: New York, NY, USA, 2000. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK21581/ (accessed on 2 January 2021).
- Collins, K.D. Charge density-dependent strength of hydration and biological structure. Biophys. J. 1997, 72, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Qiao, B.; Jiménez-Ángeles, F.; Nguyen, T.D.; Olvera de la Cruz, M. Water follows polar and nonpolar protein surface domains. Proc. Natl. Acad. Sci. USA 2019, 116, 19274–19281. [Google Scholar] [CrossRef] [Green Version]
- Bell, G.S.; Russell, R.J.; Connaris, H.; Hough, D.W.; Danson, M.J.; Taylor, G.L. Stepwise adaptations of citrate synthase to survival at life’s extremes. From psychrophile to hyperthermophile. Eur. J. Biochem. 2002, 269, 6250–6260. [Google Scholar] [CrossRef] [PubMed]
- Chiappori, F.; Pucciarelli, S.; Merelli, I.; Ballarini, P.; Miceli, C.; Milanesi, L. Structural thermal adaptation of β-tubulins from the Antarctic psychrophilic protozoan Euplotes focardii. Proteins 2012, 80, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Rao, Z.H.; Liu, S.Q. Insight derived from molecular dynamics simulation into substrate-induced changes in protein motions of proteinase K. J. Biomol. Struct. Dyn. 2010, 28, 143–158. [Google Scholar] [CrossRef]
- D’Amico, S.; Marx, J.C.; Gerday, C.; Feller, G. Activity-stability relationships in extremophilic enzymes. J. Biol. Chem. 2003, 278, 7891–7896. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.Q.; Ji, X.L.; Liu, S.Q. The free energy landscape of protein folding and dynamics: A global view. J. Biomol. Struct. Dyn. 2013, 31, 982–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dill, K.A. Theory for the folding and stability of globular proteins. Biochemistry 1985, 24, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.H.; Tao, Y.; Liu, S.Q. 153 Wonderful roles of the entropy in protein dynamics, binding and folding. J. Biomol. Struct. Dyn. 2013, 31, 98–100. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.Q.; Ji, X.L.; Tao, Y.; Tan, D.Y.; Zhang, K.Q.; Fu, Y.X. Protein folding, binding and energy landscape: A synthesis. In Protein Engineering; Kaumaya, P., Ed.; IntechOpen: Rijeka, Croatia, 2012; pp. 207–252. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Xie, Y.; Liu, C.; Liu, S. Physicochemical bases for protein folding, dynamics, and protein-ligand binding. Sci. China Life Sci. 2014, 57, 287–302. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into protein-ligand interactions: Mechanisms, models, and methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, L. DALI and the persistence of protein shape. Protein Sci. 2020, 29, 128–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Price, D.J.; Brooks III, C.L. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comp. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. Convergence of sampling in protein simulations. Phys. Rev. E 2002, 65, 031910. [Google Scholar] [CrossRef] [Green Version]
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins 1993, 17, 412–425. [Google Scholar] [CrossRef] [PubMed]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NHB d | |||||
|---|---|---|---|---|---|
| SASA a (Å2) | NCIC b | Rg c (Å) | Intra-Protein | Protein-Solvent | |
| mSHMT | 29,103 (400) | 985,559 (4580) | 27.4 (0.13) | 693 (1.3) | 1468 (43) |
| pSHMT | 31,577 (632) | 957,507 (5087) | 28.2 (0.29) | 659 (1.4) | 1476 (34) |
| Components | mSHMT | pSHMT | ||
|---|---|---|---|---|
| Monomer-A (nm) | Monomer-B (nm) | Monomer-A (nm) | Monomer-B (nm) | |
| Floor | 0.069 (0.009) | 0.063 (0.008) | 0.085 (0.014) | 0.095 (0.017) |
| Walls | 0.085 (0.021) | 0.082 (0.019) | 0.156 (0.069) | 0.116 (0.014) |
| Roof | 0.102 (0.018) | 0.094 (0.011) | 0.539 (0.149) | 0.374 (0.182) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.-B.; Xia, Y.-L.; Dong, G.-H.; Fu, Y.-X.; Liu, S.-Q. Exploring the Cold-Adaptation Mechanism of Serine Hydroxymethyltransferase by Comparative Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 22, 1781. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041781
Zhang Z-B, Xia Y-L, Dong G-H, Fu Y-X, Liu S-Q. Exploring the Cold-Adaptation Mechanism of Serine Hydroxymethyltransferase by Comparative Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2021; 22(4):1781. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041781
Chicago/Turabian StyleZhang, Zhi-Bi, Yuan-Ling Xia, Guang-Heng Dong, Yun-Xin Fu, and Shu-Qun Liu. 2021. "Exploring the Cold-Adaptation Mechanism of Serine Hydroxymethyltransferase by Comparative Molecular Dynamics Simulations" International Journal of Molecular Sciences 22, no. 4: 1781. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041781