
Mass Spectrometry-Based Redox and Protein Profiling of Failing Human Hearts

 ,
,  , , and
, , and 
Abstract
:
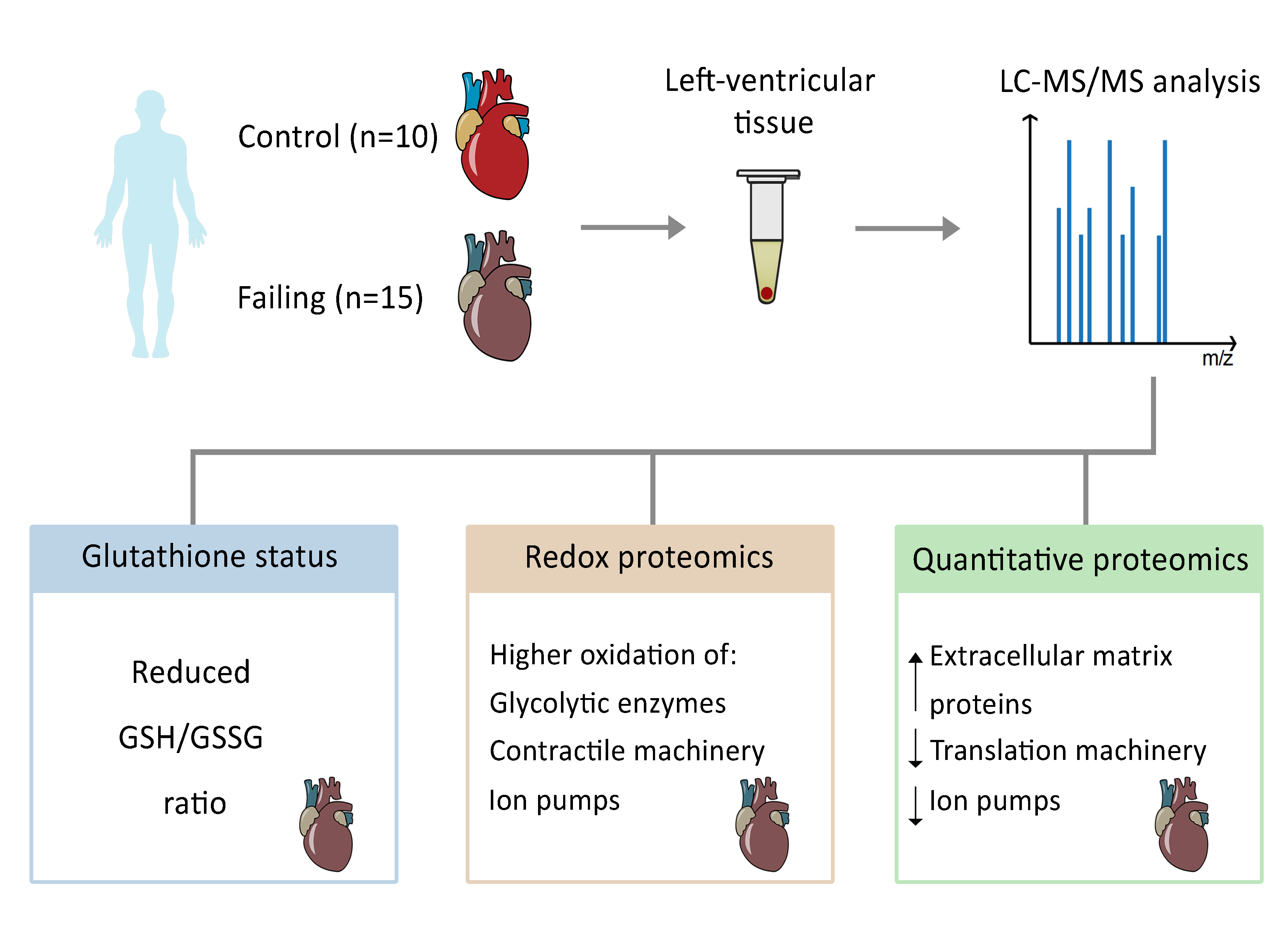{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The GSH/GSSG Ratio is Reduced in Failing Hearts
2.2. Redox Proteomics Reveal Higher Oxidation of Glucose Metabolic Enzymes and Proteins of the Contractile Machinery
2.3. Protein Expression Patterns in Failing Hearts Demonstrate a Prominent Increase of Proteins Responsible for Extracellular-Matrix (ECM) Remodeling
2.4. Reduced Expression of Ion Regulators and Members of Translation and Protein-Folding Machinery in Failing Hearts
3. Discussion
4. Materials and Methods
4.1. Tissue Specimens
4.2. Study Approval and Ethical Aspects
4.3. Cell Culture
4.4. Glutathione Measurement Sample Preparation
4.5. Proteomics Sample Preparation
4.6. LC-MS/MS Parameters (Proteomics)
4.7. Proteomics Data Processing
4.7.1. Patient Samples
4.7.2. Cell Culture Samples
4.8. Enrichment and Protein–Protein Interaction Analysis
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid. Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef] [PubMed]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature: Part 2 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Niemann, B.; Rohrbach, S.; Miller, M.R.; Newby, D.E.; Fuster, V.; Kovacic, J.C. Oxidative Stress and Cardiovascular Risk: Obesity, Diabetes, Smoking, and Pollution: Part 3 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 230–251. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Damy, T.; Kirsch, M.; Khouzami, L.; Caramelle, P.; Le Corvoisier, P.; Roudot-Thoraval, F.; Dubois-Randé, J.-L.; Hittinger, L.; Pavoine, C.; Pecker, F. Glutathione deficiency in cardiac patients is related to the functional status and structural cardiac abnormalities. PLoS ONE 2009, 4, e4871. [Google Scholar] [CrossRef] [PubMed]
- Michelhaugh, S.A.; Januzzi, J.L. Finding a Needle in a Haystack: Proteomics in Heart Failure. JACC Basic Transl. Sci. 2020, 5, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xia, Y.; Liu, X.; Wang, Y.; Chen, Z.; Xie, J.; Qian, J.; Shen, H.; Yang, P. In-depth proteomic profiling of left ventricular tissues in human end-stage dilated cardiomyopathy. Oncotarget 2017, 8, 48321–48332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Xia, Y.; Chen, Z.; Chen, A.; Wu, Y.; Jia, J.; Sun, A.; Zou, Y.; Qian, J.; Ge, J. Cardiac Proteome Profiling in Ischemic and Dilated Cardiomyopathy Mouse Models. Front. Physiol. 2019, 10, 750. [Google Scholar] [CrossRef]
- Lynch, T.L.; Sivaguru, M.; Velayutham, M.; Cardounel, A.J.; Michels, M.; Barefield, D.; Govindan, S.; dos Remedios, C.; van der Velden, J.; Sadayappan, S. Oxidative Stress in Dilated Cardiomyopathy Caused by MYBPC3 Mutation. Oxid. Med. Cell. Longev. 2015, 2015, 424751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verrastro, I.; Pasha, S.; Jensen, K.T.; Pitt, A.R.; Spickett, C.M. Mass spectrometry-based methods for identifying oxidized proteins in disease: Advances and challenges. Biomolecules 2015, 5, 378–411. [Google Scholar] [CrossRef] [Green Version]
- Tomin, T.; Schittmayer, M.; Honeder, S.; Heininger, C.; Birner-Gruenberger, R. Irreversible oxidative post-translational modifications in heart disease. Expert Rev. Proteom. 2019, 16, 681–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Carroll, K.S. Activity-Based Sensing for Site-Specific Proteomic Analysis of Cysteine Oxidation. Acc. Chem. Res. 2020, 53, 20–31. [Google Scholar] [CrossRef]
- McDonagh, B. Detection of ROS Induced Proteomic Signatures by Mass Spectrometry. Front. Physiol. 2017, 8, 470. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Carroll, K.S.; Liebler, D.C. The Expanding Landscape of the Thiol Redox Proteome. Mol. Cell. Proteom. 2016, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomin, T.; Schittmayer, M.; Birner-Gruenberger, R. Addressing Glutathione Redox Status in Clinical Samples by Two-Step Alkylation with N-ethylmaleimide Isotopologues. Metabolites 2020, 10, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- Ashfaq, S.; Abramson, J.L.; Jones, D.P.; Rhodes, S.D.; Weintraub, W.S.; Hooper, W.C.; Vaccarino, V.; Harrison, D.G.; Quyyumi, A.A. The relationship between plasma levels of oxidized and reduced thiols and early atherosclerosis in healthy adults. J. Am. Coll. Cardiol. 2006, 47, 1005–1011. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.S.; Ghasemzadeh, N.; Eapen, D.J.; Sher, S.; Arshad, S.; Ko, Y.-A.; Veledar, E.; Samady, H.; Zafari, A.M.; Sperling, L.; et al. Novel Biomarker of Oxidative Stress Is Associated with Risk of Death in Patients with Coronary Artery Disease. Circulation 2016, 133, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.I.; van Eyk, J.E. Chasing cysteine oxidative modifications: Proteomic tools for characterizing cysteine redox status. Circ. Cardiovasc. Genet. 2012, 5, 591. [Google Scholar] [CrossRef] [Green Version]
- Davidson, M.M.; Nesti, C.; Palenzuela, L.; Walker, W.F.; Hernandez, E.; Protas, L.; Hirano, M.; Isaac, N.D. Novel cell lines derived from adult human ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2005, 39, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Alegre-Cebollada, J.; Kosuri, P.; Giganti, D.; Eckels, E.; Rivas-Pardo, J.A.; Hamdani, N.; Warren, C.M.; Solaro, R.J.; Linke, W.A.; Fernández, J.M. S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell 2014, 156, 1235–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giganti, D.; Yan, K.; Badilla, C.L.; Fernandez, J.M.; Alegre-Cebollada, J. Disulfide isomerization reactions in titin immunoglobulin domains enable a mode of protein elasticity. Nat. Commun. 2018, 9, 185. [Google Scholar] [CrossRef] [PubMed]
- Labeit, S.; Kolmerer, B. Titins: Giant proteins in charge of muscle ultrastructure and elasticity. Science 1995, 270, 293–296. [Google Scholar] [CrossRef]
- Rienks, M.; Papageorgiou, A.-P.; Frangogiannis, N.G.; Heymans, S. Myocardial extracellular matrix: An ever-changing and diverse entity. Circ. Res. 2014, 114, 872–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabotuwana, N.; Murtha, L.; Hardy, S.; Boyle, A. Fibulin-3 in Cardiac Fibrosis. Heart Lung Circ. 2017, 26, S133. [Google Scholar] [CrossRef] [Green Version]
- Barallobre-Barreiro, J.; Didangelos, A.; Schoendube, F.A.; Drozdov, I.; Yin, X.; Fernández-Caggiano, M.; Willeit, P.; Puntmann, V.O.; Aldama-López, G.; Shah, A.M.; et al. Proteomics analysis of cardiac extracellular matrix remodeling in a porcine model of ischemia/reperfusion injury. Circulation 2012, 125, 789–802. [Google Scholar] [CrossRef]
- Blackburn, P.R.; Xu, Z.; Tumelty, K.E.; Zhao, R.W.; Monis, W.J.; Harris, K.G.; Gass, J.M.; Cousin, M.A.; Boczek, N.J.; Mitkov, M.V.; et al. Bi-allelic Alterations in AEBP1 Lead to Defective Collagen Assembly and Connective Tissue Structure Resulting in a Variant of Ehlers-Danlos Syndrome. Am. J. Hum. Genet. 2018, 102, 696–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Hui, H.; Li, Z.; Pan, J.; Jiang, X.; Wei, T.; Cui, H.; Li, L.; Yuan, X.; Sun, T.; et al. Pigment epithelium-derived factor attenuates myocardial fibrosis via inhibiting Endothelial-to-Mesenchymal Transition in rats with acute myocardial infarction. Sci. Rep. 2017, 7, 41932. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Horiguchi, M.; Zilberberg, L.; Dabovic, B.; Hadjiolova, K.; Rifkin, D.B. Latent TGF-β-binding proteins. Matrix Biol. 2015, 47, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Petretto, E.; Sarwar, R.; Grieve, I.; Lu, H.; Kumaran, M.K.; Muckett, P.J.; Mangion, J.; Schroen, B.; Benson, M.; Punjabi, P.P.; et al. Integrated genomic approaches implicate osteoglycin (Ogn) in the regulation of left ventricular mass. Nat. Genet. 2008, 40, 546–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marro, J.; Pfefferli, C.; de Preux Charles, A.-S.; Bise, T.; Jaźwińska, A. Collagen XII Contributes to Epicardial and Connective Tissues in the Zebrafish Heart during Ontogenesis and Regeneration. PLoS ONE 2016, 11, e0165497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torella, D.; Ellison, G.M.; Torella, M.; Vicinanza, C.; Aquila, I.; Iaconetti, C.; Scalise, M.; Marino, F.; Henning, B.J.; Lewis, F.C.; et al. Carbonic anhydrase activation is associated with worsened pathological remodeling in human ischemic diabetic cardiomyopathy. J. Am. Heart Assoc. 2014, 3, e000434. [Google Scholar] [CrossRef] [Green Version]
- Hammadah, M.; Fan, Y.; Wu, Y.; Hazen, S.L.; Tang, W.H.W. Prognostic value of elevated serum ceruloplasmin levels in patients with heart failure. J. Card. Fail. 2014, 20, 946–952. [Google Scholar] [CrossRef] [Green Version]
- Schwinger, R. The Na, K-ATPase in the failing human heart. Cardiovasc. Res. 2003, 57, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zou, J.; Littlejohn, R.; Liu, J.; Su, H. Neddylation, an Emerging Mechanism Regulating Cardiac Development and Function. Front. Physiol. 2020, 11, 612927. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Shin, J.; Rajan, S.; Yoon, H.S. Structural basis of nucleic acid recognition by FK506-binding protein 25 (FKBP25), a nuclear immunophilin. Nucleic Acids Res. 2016, 44, 2909–2925. [Google Scholar] [CrossRef] [Green Version]
- Lopez, E.; Berna-Erro, A.; Salido, G.M.; Rosado, J.A.; Redondo, P.C. FKBP25 and FKBP38 regulate non-capacitative calcium entry through TRPC6. Biochim. Biophys. Acta 2015, 1853, 2684–2696. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Wu, G.; Yang, L.; Lu, Y.-P.; Liu, X.-X.; Han, F.; Deng, Y.-P.; Fu, X.-C.; Liu, Q.-B.; Lu, Y.-M. Elucidation of the FKBP25-60S Ribosomal Protein L7a Stress Response Signaling During Ischemic Injury. Cell. Physiol. Biochem. 2018, 47, 2018–2030. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yücel, D.; Aydoğdu, S.; Cehreli, S.; Saydam, G.; Canatan, H.; Seneş, M.; Ciğdem Topkaya, B.; Nebioğlu, S. Increased oxidative stress in dilated cardiomyopathic heart failure. Clin. Chem. 1998, 44, 148–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, D.H.; Wang, Z.V. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J. Am. Heart Assoc. 2019, 8, e012673. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullarky, E.; Cantley, L.C. Diverting Glycolysis to Combat Oxidative Stress. In Innovative Medicine: Basic Research and Development; Springer: Tokyo, Japan, 2015; ISBN 9784431556503. [Google Scholar]
- López-Grueso, M.J.; González-Ojeda, R.; Requejo-Aguilar, R.; McDonagh, B.; Fuentes-Almagro, C.A.; Muntané, J.; Bárcena, J.A.; Padilla, C.A. Thioredoxin and glutaredoxin regulate metabolism through different multiplex thiol switches. Redox Biol. 2019, 21, 101049. [Google Scholar] [CrossRef]
- Singh, S.; Lämmle, S.; Giese, H.; Kämmerer, S.; Meyer-Roxlau, S.; Alfar, E.A.; Dihazi, H.; Guan, K.; El-Armouche, A.; Richter, F. The reduced activity of PP-1α under redox stress condition is a consequence of GSH-mediated transient disulfide formation. Sci. Rep. 2018, 8, 17711. [Google Scholar] [CrossRef]
- Behring, J.B.; Kumar, V.; Whelan, S.A.; Chauhan, P.; Siwik, D.A.; Costello, C.E.; Colucci, W.S.; Cohen, R.A.; McComb, M.E.; Bachschmid, M.M. Does reversible cysteine oxidation link the Western diet to cardiac dysfunction? FASEB J. 2014, 28, 1975–1987. [Google Scholar] [CrossRef] [Green Version]
- Krüger, M.; Kötter, S. Titin, a Central Mediator for Hypertrophic Signaling, Exercise-Induced Mechanosignaling and Skeletal Muscle Remodeling. Front. Physiol. 2016, 7, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuello, F.; Wittig, I.; Lorenz, K.; Eaton, P. Oxidation of cardiac myofilament proteins: Priming for dysfunction? Mol. Asp. Med. 2018, 63, 47–58. [Google Scholar] [CrossRef]
- Hertelendi, Z.; Tóth, A.; Borbély, A.; Galajda, Z.; van der Velden, J.; Stienen, G.J.M.; Edes, I.; Papp, Z. Oxidation of myofilament protein sulfhydryl groups reduces the contractile force and its Ca2+ sensitivity in human cardiomyocytes. Antioxid. Redox Signal. 2008, 10, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- LeWinter, M.M.; Granzier, H.L. Cardiac titin and heart disease. J. Cardiovasc. Pharmacol. 2014, 63, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146. [Google Scholar] [CrossRef]
- De Boer, R.A.; de Keulenaer, G.; Bauersachs, J.; Brutsaert, D.; Cleland, J.G.; Diez, J.; Du, X.-J.; Ford, P.; Heinzel, F.R.; Lipson, K.E.; et al. Towards better definition, quantification and treatment of fibrosis in heart failure. A scientific roadmap by the Committee of Translational Research of the Heart Failure Association (HFA) of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 272–285. [Google Scholar] [CrossRef] [Green Version]
- Lindsey, M.L.; Jung, M.; Hall, M.E.; DeLeon-Pennell, K.Y. Proteomic analysis of the cardiac extracellular matrix: Clinical research applications. Expert Rev. Proteom. 2018, 15, 105–112. [Google Scholar] [CrossRef]
- Van der Reest, J.; Lilla, S.; Zheng, L.; Zanivan, S.; Gottlieb, E. Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat. Commun. 2018, 9, 1581. [Google Scholar] [CrossRef] [PubMed]
- Murtha, L.A.; Mabotuwana, N.R.; Hardy, S.A.; Boyle, A.J. Fibulin-3 as a Potential Therapeutic Target for Cardiac Fibrosis. J. Card. Fail. 2017, 23, S28. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Øie, E.; Vinge, L.E.; Yndestad, A.; Andersen, G.G.Ø.; Andersson, Y.; Attramadal, T.; Attramadal, H. Induction of myocardial biglycan in heart failure in rats—An extracellular matrix component targeted by AT(1) receptor antagonism. Cardiovasc. Res. 2003, 60, 557–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, D.; Mersmann, J.; Melchior, A.; Freudenberger, T.; Petrik, C.; Schaefer, L.; Lüllmann-Rauch, R.; Lettau, O.; Jacoby, C.; Schrader, J.; et al. Biglycan is required for adaptive remodeling after myocardial infarction. Circulation 2008, 117, 1269–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.-B.; Huang, R.; Yang, K.; Xu, M.; Fan, D.; Liu, M.-X.; Huang, S.-H.; Liu, L.-B.; Wu, H.-M.; Tang, Q.-Z. Identification of differentially expressed genes and preliminary validations in cardiac pathological remodeling induced by transverse aortic constriction. Int. J. Mol. Med. 2019, 44, 1447–1461. [Google Scholar] [CrossRef] [Green Version]
- Petrushanko, I.Y.; Yakushev, S.; Mitkevich, V.A.; Kamanina, Y.V.; Ziganshin, R.H.; Meng, X.; Anashkina, A.A.; Makhro, A.; Lopina, O.D.; Gassmann, M.; et al. S-glutathionylation of the Na,K-ATPase catalytic α subunit is a determinant of the enzyme redox sensitivity. J. Biol. Chem. 2012, 287, 32195–32205. [Google Scholar] [CrossRef] [Green Version]
- Zeitz, M.J.; Smyth, J.W. Translating Translation to Mechanisms of Cardiac Hypertrophy. J. Cardiovasc. Dev. Dis. 2020, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, L.J.; Reader, J.S.; Tzima, E. Mechanical Regulation of Protein Translation in the Cardiovascular System. Front. Cell. Dev. Biol. 2020, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Forte, M.; Frati, G.; Sadoshima, J. New Insights into the Role of mTOR Signaling in the Cardiovascular System. Circ. Res. 2018, 122, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Zhai, P.; Shao, D.; Maejima, Y.; Robbins, J.; Volpe, M.; Condorelli, G.; Sadoshima, J. Rheb is a critical regulator of autophagy during myocardial ischemia: Pathophysiological implications in obesity and metabolic syndrome. Circulation 2012, 125, 1134–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Contu, R.; Latronico, M.V.G.; Zhang, J.; Zhang, J.L.; Rizzi, R.; Catalucci, D.; Miyamoto, S.; Huang, K.; Ceci, M.; et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J. Clin. Investig. 2010, 120, 2805–2816. [Google Scholar] [CrossRef] [PubMed]
- World Medical Association. Declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA 2013, 310, 2191–2194. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Schumacher, M.; Scherer, A.; Sanoudou, D.; Megherbi, D.; Davison, T.; Shi, T.; Tong, W.; Shi, L.; Hong, H.; et al. A comparison of batch effect removal methods for enhancement of prediction performance using MAQC-II microarray gene expression data. Pharmacogenom. J. 2010, 10, 278–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygaard, V.; Rødland, E.A.; Hovig, E. Methods that remove batch effects while retaining group differences may lead to exaggerated confidence in downstream analyses. Biostatistics 2016, 17, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomin, T.; Schittmayer, M.; Sedej, S.; Bugger, H.; Gollmer, J.; Honeder, S.; Darnhofer, B.; Liesinger, L.; Zuckermann, A.; Rainer, P.P.; et al. Mass Spectrometry-Based Redox and Protein Profiling of Failing Human Hearts. Int. J. Mol. Sci. 2021, 22, 1787. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041787
Tomin T, Schittmayer M, Sedej S, Bugger H, Gollmer J, Honeder S, Darnhofer B, Liesinger L, Zuckermann A, Rainer PP, et al. Mass Spectrometry-Based Redox and Protein Profiling of Failing Human Hearts. International Journal of Molecular Sciences. 2021; 22(4):1787. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041787
Chicago/Turabian StyleTomin, Tamara, Matthias Schittmayer, Simon Sedej, Heiko Bugger, Johannes Gollmer, Sophie Honeder, Barbara Darnhofer, Laura Liesinger, Andreas Zuckermann, Peter P. Rainer, and et al. 2021. "Mass Spectrometry-Based Redox and Protein Profiling of Failing Human Hearts" International Journal of Molecular Sciences 22, no. 4: 1787. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041787