
Endoplasmic Reticulum Stress Signaling and the Pathogenesis of Hepatocarcinoma

Department of Pathology, Northwestern University Feinberg School of Medicine, Chicago, IL 60611, USA
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(4), 1799; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041799
Submission received: 20 January 2021
/
Revised: 5 February 2021
/
Accepted: 8 February 2021
/
Published: 11 February 2021
(This article belongs to the Special Issue Transcription Factors in Cancer)
Abstract
:Hepatocellular carcinoma (HCC), also known as hepatoma, is a primary malignancy of the liver and the third leading cause of cancer mortality globally. Although much attention has focused on HCC, its pathogenesis remains largely obscure. The endoplasmic reticulum (ER) is a cellular organelle important for regulating protein synthesis, folding, modification and trafficking, and lipid metabolism. ER stress occurs when ER homeostasis is disturbed by numerous environmental, physiological, and pathological challenges. In response to ER stress due to misfolded/unfolded protein accumulation, unfolded protein response (UPR) is activated to maintain ER function for cell survival or, in cases of excessively severe ER stress, initiation of apoptosis. The liver is especially susceptible to ER stress given its protein synthesis and detoxification functions. Experimental data suggest that ER stress and unfolded protein response are involved in HCC development, aggressiveness and response to treatment. Herein, we highlight recent findings and provide an overview of the evidence linking ER stress to the pathogenesis of HCC.
1. Introduction
The endoplasmic reticulum (ER) is a multifunctional organelle within which protein folding, lipid biosynthesis, and calcium storage occur [1,2,3,4]. The ER in hepatocytes has a remarkable capacity to adapt to extracellular and intracellular challenges [5,6,7]. However, in humans, numerous disturbances impair hepatocytes’ ability to handle protein folding and proteostasis, which disrupts ER homeostasis and leads to misfolded protein accumulation, ER stress and unfolded protein response (UPR) [8,9,10,11,12]. ER stress and subsequent adaptive UPR activation are important in the pathogenesis of chronic liver disease, ranging from steatosis and nonalcoholic fatty liver disease (NAFLD) to hepatocellular carcinoma (HCC) [13,14,15,16,17]. The key interaction between the UPR and metabolic liver disease has been comprehensively discussed in previous reviews [18,19,20]. This review compiles the recent findings supporting the functional impact of ER stress signaling on the pathogenesis of HCC.
2. ER Stress and UPR
The UPR plays a critical role in mediating cellular adaptation to ER stress [21,22]. UPR acts in concert to increase ER content, expands the ER protein folding capacity, degrades misfolded proteins and reduces the load of new proteins to the ER [23,24,25,26]. Activation of the UPR depends on the ER transmembrane proteins and sensors including inositol-requiring enzyme 1 (IRE1α), protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6) and cyclic adenosine monophosphate (cAMP)-responsive element-binding protein H (CREBH) [27,28,29,30] (Figure 1). Both PERK and IRE1α are type I transmembrane proteins with similar ER luminal domain structures and a cytosolic Serine/threonine kinase domain, whereas ATF6α is a type II transmembrane protein that contains a cytosolic cyclic AMP response element-binding protein (CREB)–ATF basic leucine zipper domain [31,32,33,34]. IRE1α, PERK, ATF6 and CREBH are ultimately responsible for the activation of a set of transcription factors (TF), including spliced X-box binding protein 1 (XBP1s), activating transcription factor 4 (ATF4), CCAAT enhancer-binding protein (C/EBP) homologous protein (CHOP), nuclear factor κB (NF-κB) and activator protein 1 (AP-1), through a complicated and nonparallel process [35,36,37,38,39].
Hepatocytes, like other secretory cells, have a well-developed ER [18,40]. Due to their high protein and lipid synthesis, hepatic UPR is activated by rhythmic or transient physiological conditions (e.g., feeding-fasting cycles) to regulate lipid and glucose homeostasis [5,41,42,43,44]. However, the UPR is also chronically activated by irreversible or chronic stress (e.g., viral infection and obesity) and causes hepatic dysfunction, leading to the pathogenesis of the HCC, including viral hepatitis, alcoholic fatty liver disease, non-alcoholic fatty liver disease (NAFLD) and alpha1-antitrypsin deficiency [13,14,15,16,17].
2.1. Serine/Threonine-Protein Kinase/Endoribonuclease IRE1α and Transcription Factor Xbp1s
IRE1 is the most conserved ER stress sensor, with two isoforms identified in humans: IRE1α and IRE1β [28]. IRE1α is a ubiquitously expressed and dual function transmembrane protein with serine/threonine kinase and endoribonuclease (RNase) activities on its cytosolic tail [28,45]. Upon binding with ER chaperone Hsp47 and dissociation from BiP, IRE1α oligomerizes, trans-autophosphorylates and activates its RNase activity [46,47]. The mRNA encoding XBP1 undergoes a cytosolic unconventional splicing reaction by IRE1α and the tRNA ligase RTCB at a stem-loop structure that results in the translation of an active transcription factor, termed spliced XBP1 (XBP1s) [48]. Transcriptional targets of XBP1s include genes that encode functions in ER protein folding, secretion, ER-associated degradation (ERAD), ER biogenesis and lipid synthesis [28,41,49].
In mice, genetic ablation of either IRE1α or XBP1 results in embryonic lethality with major liver defects, which indicate that IRE1α/Xbp1s pathway is involved in hepatic physiological functions other than its involvement in ER stress response [50,51]. XBP1 deficiency leads to diminished hepatocyte development and prominent apoptosis [51]. One of the specific target genes of XBP1s in the liver is identified as αFP, which may be a regulator of hepatocyte growth [51]. IRE1α knockout results in a reduction in vascular endothelial growth factor and fetal liver hypoplasia49. Liver conditional knockout studies have helped to understand the important role of IRE1α/Xbp1s pathway in liver physiology [52]. XBP1 deficiency in the liver results in a profound compromise of de novo hepatic lipid synthesis, thereby leading to decreased serum triglycerides (TG), cholesterol and free fatty acids (FFAs) without causing hepatic steatosis [52]. As a transcription factor, XBP1s directly regulates the expression of a subset of lipogenic genes including stearoyl-CoA desaturase 1 (SCD1), Acetyl-CoA carboxylase 2(ACC2) and Diacylglycerol O-acyltransferase 2 (DGAT2) [52]. Moreover, IRE1α-mediated mRNA decay lowers plasma lipids through silencing of lipid metabolism genes [53].
2.2. The Kinase PERK and Transcription Factor ATF4 and CHOP
PERK is a type 1 transmembrane protein and a member of the eukaryotic initiation factor 2α (eIF2α) kinase family that is typically activated through recruitment of chaperone BiP away from PERK, leading to oligomerization and activation of the cytosolic kinase domain [54]. Protein structural analysis also shows that PERK’s luminal domain can recognize and selectively interact with misfolded protein and thereby trigger oligomerization and activation [55,56]. PERK is activated to phosphorylate eIF2α at Ser51 (p-eIF2α), which briefly attenuates initiation of mRNA translation, to reduce the ER protein folding load [29]. Although it reduces global protein synthesis, eIF2α selective upregulates the translation of a subset of mRNAs, the best-studied of which is the transcription factor ATF4 [57]. ATF4 transcriptionally upregulates several UPR-target genes that mediate antioxidant responses and amino acid synthesis and transport [58,59]. ATF4 also upregulates transcription factor CHOP, which forms heterodimers with ATF4 to mediate pro-apoptotic signaling activation [58]. Moreover, ATF4 and CHOP induce transcription of growth arrest and DNA-damage-inducible protein 34 (GADD34) to direct p-eIF2α dephosphorylation and restart global mRNA translation, which is essential if cells are to survive an acute ER stress challenge [60].
Germline ablation of PERK leads to 30–40% prenatal mortality and at least 23% postnatal mortality [61]. The surviving newborn Perk−/− mice are of normal size but exhibit severe postnatal growth retardation [61]. Consistently, homozygotes of Atf4−/− pups generally die during the first hour after birth, although excess mortality occurs throughout the first 3 weeks of life [62]. Liver-specific knockout studies show that PERK/ATF4/CHOP signaling also regulates lipid homeostasis [63]. Triglyceride levels are increased in livers of the PERK liver-specific knockout mice through regulating Very Low-Density Lipoprotein Receptor (VLDLR) expression upon tunicamycin treatment [64]. However, ATF4 overexpression induces early onset of hyperlipidemia and hepatic steatosis in zebrafish [65]. Aged female Chop−/− mice reportedly gain much more weight with increased adiposity and hepatic steatosis [66]. CHOP mediates insulin resistance by modulating adipose tissue macrophage polarity [67].
2.3. Transcription Factor ATF6α
ATF6 is a type II ER transmembrane protein, with a cytoplasmic N-terminus that contains a basic leucine zipper (bZIP) domain that functions as a transcription factor following regulated intramembrane proteolysis by site-1 and site-2 proteases (S1P and S2P) in ER-stressed cells [30]. Two Atf6 genes, Atf6α and Atf6β, are expressed ubiquitously, with no obvious phenotype in mice lacking either individual isoform [68,69]. Interestingly, the combined deletion of Atf6α and Atf6β causes a very early embryonic lethality, suggesting these genes provide essential complementary functions in early embryonic development [70]. Transcriptional induction of ER chaperone and ERAD genes such as BiP, glucose-regulated protein 94 (GRP94) and E3 ubiquitin-protein ligase HRD1 is mostly mediated by ATF6α binding to the cis-acting ER Stress Response Element (ERSE) consensus sequence [71,72].
Intriguely, pharmacological challenge of ATF6α deficiency mice, but not ATF6β-null mice, with ER stress leading to aggravated hepatosteatosis and death in the liver [73]. In hepatocytes, ATF6α interacts with peroxisome proliferator-activated receptor α (PPARα) to enhance its transcriptional activity, upregulating hepatic fatty acid oxidation [74]. Moreover, ATF6α suppresses SREBP2 transactivation in hepatocytes to downregulate lipogenesis [75]. ATF6α also reduces hepatic glucose output by disrupting the CREB-CRTC2 interaction and thereby inhibiting CRTC2 occupancy over gluconeogenic genes [76].
2.4. Transcription Factor CREBH
CREBH, encoded by the CREB3L3 gene, is also an ER-residing transcription factor that is robustly expressed in the liver [31]. CREBH contains an ER transmembrane domain, a transcription-activation domain and a basic leucine zipper (bZIP) domain [31,43]. Similar to ATF6, in response to ER stress, CREBH is activated by intramembrane proteolysis and translocates from the ER to the Golgi apparatus, where it is cleaved by the S1P and S2P, releasing bZIP domain to induce transcriptional activity [31,77].
CREBH is a transcription factor that is key in the regulation of hepatic lipid accumulation [78,79]. Hepatic CREBH bZIP domain overexpression significantly decreases the TG levels in plasma and increases hepatic TG levels compared to WT controls [80]. Consistently, the levels of plasma TG in the CREBH null mice are dramatically increased compared with those of the WT mice [80]. Under nutrient starvation, a condition that stimulates lipolysis, the levels of ketone, 3-hydroxybutyric acid, a product of FA oxidation in the plasma, are slightly reduced in the CREBH-null mice, compared with control mice, suggesting that CREBH deletion leads to a defect in TG lipolysis, resulting in higher levels of plasma TG [80]. CREBH regulates hepatic lipid accumulation by directly modulating key metabolic enzyme expression [81]. CREBH directly regulates the transcriptional activation of apolipoprotein, APOC2, APOA4, APOA5 and cell death activator CIDE-3/FSP27 by binding the CRE binding motifs in their gene promoters [81,82]. Moreover, CREBH regulates hepatokine FGF21 expression and subsequently regulates glucose and lipid metabolism [43,83]. Lysine 294 ubiquitination and acetylation of CREBH regulate its stability and transcription activity [43,84].
3. Pathogenesis of Hepatocellular Carcinoma
Hepatocellular carcinoma (HCC) is a primary malignancy of the liver and the third leading cause of cancer mortality worldwide [85,86]. HCC is typically diagnosed at advanced stages, with a median survival following diagnosis of approximately 6 to 20 months [85]. HCC is now the third leading cause of cancer deaths worldwide, with over 500,000 people affected [86]. The incidence of HCC is highest in Asia and Africa, where the endemic high prevalence of hepatitis B and hepatitis C strongly predisposes to the development of chronic liver disease and subsequent development of HCC [86,87]. In the United States, the current 5-year survival of HCC patients is only 10% [88]. More importantly, despite the advances in the treatment of many other types of tumors, the advanced HCC five-year survival rate has not been significantly improved during the last 30 years [89]. Major risk factors for HCC include chronic HBV (hepatitis B virus) and HCV (hepatitis C virus) infections, chronic alcohol consumption and non-alcoholic fatty liver disease (NAFLD) (Figure 2) (Table 1) [85,86]. Other risk factors for the development of HCC are alpha1-antitrypsin deficiency, Wilson’s disease, hereditary hemochromatosis, primary biliary cirrhosis and autoimmune hepatitis (Figure 2) [90]. The epidemiologic distribution of these risk factors varies according to geographic location and host-specific factors [90,91].
3.1. Viral Hepatitis
Chronic hepatitis B virus (HBV) and hepatitis C virus (HCV) infections are the most important causes of HCC, accounting for about 80% of cases worldwide [92]. Chronic HBV infection accounts for approximately 80% of virus-associated HCC cases and virtually all of childhood HCC, especially in Africa and East Asia, while HCV infection, involved in about 20% of the total HCC cases, seems to be mainly related to HCC development in Western Europe and North America [91]. Prospective cohort studies have shown a significant increase in the risk of developing HCC among persons chronically infected with HBV or HCV [91,93]. Between 70 and 90% of the HBV-infected individuals who develop HCC have cirrhosis secondary to chronic necroinflammation [91,93]. HCV also increases the risk of HCC by inducing fibrosis and, eventually, cirrhosis [91].
In HBV-infected hepatocytes, large amounts of HBV-expressing proteins are synthesized and folded in ER, leading to ER dysfunction and resulting in ER stress [94]. Clinically, ground-glass hepatocyte (GGH), whose mutant surface proteins (preS1 and preS2 mutants) accumulate in the ER, represents a histological hallmark of chronic hepatitis B virus infection (Table 1) [95]. Many studies report that pre-S deletion has positive relationships with HCC [96]. Pre-S surface antigen variant induces PERK-ATF4-CHOP signal activation, ER-derived oxidative stress and apoptosis [97]. Moreover, transit or stable expression of HBx, a regulatory X protein, induces ATF6 and IRE1α-XBP1s pathway activation [98,99]. Moreover, CREBH is activated by HBx binding, leading to a synergistic effect on the expression of AP-1 target genes through c-Jun induction [100]. HBx-mediated activation of these pathways of UPR probably promotes HBV replication and expression in liver cells [99,100].
HCV replication also disrupts normal ER functions and modulates ER stress signaling activation (Table 1) [101,102]. XBP1 expression is elevated in cells carrying HCV subgenomic replicons, but XBP1 trans-activating activity is repressed [103]. This prevents the IRE1α -XBP1 transcriptional induction of EDEM (ER degradation-enhancing α-mannosidase-like protein), which is a type II ER transmembrane protein that binds directly to and enhances the degradation of misfolded proteins through the ERAD pathway [103]. Consequently, misfolded proteins are stable in cells expressing HCV replicons [103]. In addition, Hepatitis C virus core protein activates PERK and ATF6 UPR pathway, leading to autophagy induction by increasing MAP1LC3B and ATG12 expression [104]. HCV induces transforming growth factor β1 (TGFβ1) through activation of IRE1α -JNK and CREBH pathway [101,105].
3.2. Alcoholic Fatty Liver Disease (ALD)
Alcoholic beverages are classified as a human carcinogen and act synergistically with pre-existing underlying chronic liver disease to further increase the risk of HCC [115]. Heavy drinkers (50 g/day) of ethanol exhibit a 1.4-fold risk for HCC, and chronic alcohol use of more than 80 g/day for longer than 10 years increases the risk of HCC by 5-fold [116,117]. Genetic variations in the alcohol metabolizing enzymes, such as alcohol dehydrogenase 1C (ADH1C) and aldehyde dehydrogenase (ALDH2), which modulate the resulting amount of the carcinogenic aldehyde, are thought of as potential inherited markers of HCC [118]. Generation of tissue-damaging reactive oxygen species (ROS) from alcohol oxidation has been identified as an important mechanism in hepatocarcinogenesis [119]. ROS promotes HCC development via damaging cellular macromolecules and forming lipid peroxides [120,121]. It also stimulates the production of cytokines, inflammation, cell proliferation and upregulates angiogenesis [122].
Alcohol is mainly metabolized in the liver, and ethanol metabolism directly impairs ER structure and function in hepatocytes [123]. More evidence shows that alcohol consumption causes hepatic ER stress along with steatosis, inflammation and apoptosis [123]. Moreover, reducing ER stress has been shown to decrease alcoholic liver injury [123]. Interestingly, alcohol feeding only induces IRE1α-Xbp1s pathway activation in the pancreas but not in the liver [124]. However, alanine aminotransferase (ALT) level is elevated in XBP1-deficient mice fed ethanol [124]. Hepatic gene expression profiling of ethanol-fed mice demonstrates upregulated mRNA abundance of UPR target genes after 4 weeks of ethanol feeding [125]. CHOP deficient mice have a remarkable absence of hepatocellular apoptosis in response to alcohol feeding but no protection against hyperhomocysteinemia, ER stress and fatty liver [106]. This suggests a role for CHOP-mediated hepatocyte apoptosis in ethanol-induced liver injury. Moreover, PERK-ATF4-induced upregulation of nicotinamide methyltransferase (NNMT) contributes to alcohol-related fatty liver development [107]. In Zebrafish, blocking ATF6 larvae prevents alcohol-induced steatosis, and ATF6 overexpression induces genes that drive lipogenesis, increases lipid production and causes steatosis [108]. CREBH-knockdown mice are protected against alcohol-induced perturbation of bile acid homeostasis [109].
3.3. Non-Alcoholic Fatty Liver Disease (NAFLD) and Nonalcoholic Steatohepatitis (NASH)
In recent decades, NAFLD has become the most common liver disease in developed countries [126,127,128]. NAFLD ranges from simple steatosis in the absence of excessive alcohol intake to non-alcoholic NASH with or without cirrhosis [126,127,128]. The risk of HCC is higher in NAFLD patients than that observed in the general clinical population [126,129]. Most HCC cases in NAFLD develop in patients with cirrhosis [129]. Among patients with NAFLD cirrhosis, HCC risk ranges from 1.6 to 23.7 per 1000 persons per year based on other demographic characteristics [129].
ER stress and dysregulation of the UPR proteins have been implicated in human NAFLD [129,130]. Moreover, studies from animal models of NAFLD support the important role of the three UPR pathways in the development and progression of NAFLD. Hepatocyte-specific Ire1α−/− mice develop severe hepatic steatosis and insulin resistance under high-fat diets (HFDs) [110]. IRE1α deficiency increases the abundance of a subset of miRNA clusters in the steatotic livers of HFD-fed mice and patients [110]. Hepatocyte Xbp1 deficiency increases liver injury in mice fed with HFDs/high-carbohydrate diets (HCD) [131]. ATF4 liver-specific knockout mice are protected from HFD or HCD-induced liver steatosis [111]. Mechanistically, ATF4 deficiency suppresses HCD-induced SCD1 and HFD induced cytochrome P450, family 2, subfamily, polypeptide 1 (CYP2E1) expression [111,112]. Overexpression of dominant-negative ATF6 increases susceptibility to development hepatic steatosis and insulin resistance in HFD or HCD [74]. After having been fed an HFD, a massive accumulation of hepatic lipid metabolites and significant increases in plasma TG levels are observed in the CREBH knockout mice. Along with the hypertriglyceridemia phenotype, CREBH-deficient mice display significantly reduced body-weight gain, diminished abdominal fat and increased nonalcoholic steatohepatitis activities [80].
UPR also drives key features of progressive NASH including inflammation and cell death under long-term NAFLD development [132]. Upon ER stress, activated IREα activates JNKs and NF-κB, which are implicated in the transcriptional activation of pro-inflammatory pathways [113]. In NASH, NF-κB has been identified as a central link between hepatic injury and fibrosis and even favoring progression to HCC [133]. Genetic ablation of NF-κB regulators in mouse models leads to spontaneous liver injury, fibrosis and HCC [134,135]. Moreover, liver-specific CREBH KO mice display severe hepatitis in MCD diet feeding without an increase in liver lipid contents [136].
3.4. Alpha1-Antitrypsin Deficiency (AATD)
Alpha-1-antitrypsin (A1AT) is a protein mainly produced in the liver that protects the lungs from damage caused by activated enzymes [137]. AATD is the most common genetic cause of metabolic liver disease in children and the most frequent inherited indication for liver failure and transplantation in the pediatric population [138]. However, only approximately 10% of homozygosity for AATD, usually of the genotype PiZZ, have marginal deviations in liver test results [139,140]. Generally, AATD adults show clinical manifestations of chronic liver disease during middle or old age [140]. Moreover, heterozygous carriage of the AATD PiMZ variant increases the risk to develop liver cirrhosis [141]. Autopsy studies conducted on PiZZ patients show that over one-third of elderly patients have developed cirrhosis and primary liver cancer [142,143]. Furthermore, AATD is a risk factor for HBV/HCV infection that promotes advanced liver disease and consequently hepatocellular carcinoma [144].
Hereditary deficiency of A1AT is a consequence of the accumulation of polymers of A1AT mutants in the endoplasmic reticulum of hepatocytes [144]. Accumulation of unfolded Z alpha1-antitrypsin inside the endoplasmic reticulum (ER) induces ER proteotoxic stress and increases the incidence of liver injury, fibrosis/cirrhosis and carcinogenesis [145]. Interestingly, although A1AT mutants can accumulate dramatically within the ER and show increased interaction with the chaperone BiP, many fail to constitutively activate the UPR by itself [146]. However, the cells expressing A1AT Z or H334D mutants exhibit hypersensitivity to both pharmacological and physiological ER stressors [147]. Recently, gene array profiling shows that CHOP is upregulated in the liver of PiZ transgenic mice [114,148]. Increased CHOP levels are also detected in diseased livers of children homozygous for the Z allele [114]. CHOP and c-JUN upregulate A1AT expression and play an important role in hepatic disease by increasing the burden of proteotoxic A1AT Z mutants, particularly in the pediatric population [114].
4. Targeting ER Stress for HCC Treatment
The ER stress and UPR play important roles in the regulation of cell fate and have become novel signaling targets for HCC treatment. IRE1α in stellate cells is activated by co-culturing with HCC cells [149]. Inhibiting IRE1α-endonuclease activity decreases tumor burden in a mouse model for hepatocellular carcinoma [150]. The expression of XBP1s is increased in HCC cell lines and tissues [151]. Overexpression of XBP1s promotes epithelial–mesenchymal transition (EMT) and metastasis of HCC cells [151]. Moreover, the PERK pathway is robustly activated in HCC [152]. PERK inhibitor (GSK2656157) administration induces ER-stress-mediated cell death and HCC regression [152]. Melatonin increases the sensitivity of HCC to Sorafenib through the PERK-ATF4-Beclin1 pathway [153]. No reported studies have targeted ATF6 and CREBH in HCC treatment, probably because of the lack of availability of specific chemical inhibtors.
5. Conclusions
Chronic UPR/ER stress contributes to the pathogenesis of HCC and disease development [149,154]. Multiple risk factors are commonly associated with carcinogenesis of HCC such as HBV/HCV viral infections, excessive alcoholic consumption, obesity and NAFLD [13,14,15,16,17,155]. However, the precise contribution of each of the factors to ER stress induction is not clear, and their importance to HCC may depend on ER stress duration, presence or absence of genetic factors, cross-talks with other pathogenic pathways, and liver disease stages. Emerging evidence indicates a direct involvement of long-term ER stress in liver hepatocellular tumorigenesis [149]. However, the causative or resultant role of ER stress in HCC development and the precise contribution of each individual UPR pathway are incompletely understood. Moreover, hepatocarcinogenesis is a complex multistep process involving multiple different signaling cascades’ activation. Telomere shortening, oncogenic copy number variants, single-nucleotide variants and epigenetic modifications are also well-established molecular mechanisms for HCC tumorigenesis [156]. The molecular crosstalk between these factors and ER stress needs to be further investigated (Figure 3). It is of vital importance to define the cellular and molecular pathogenesis that driven HCC as current treatments for HCC, such as the tyrosine kinase inhibitor sorafenib, are ineffective therapies, as they only extend median survival by a few weeks to months in a subset of patients [157]. Relieving ER stress may serve as a new specific and effective strategy for preventing or treating HCC.
Author Contributions
Writing—original draft preparation, J.W.; writing—review and editing, D.F. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Institutes of Health (NIH) R01CA257520; R01DK120330 and R01CA232347.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
All figures were created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Halperin, L.; Jung, J.; Michalak, M. The many functions of the endoplasmic reticulum chaperones and folding enzymes. IUBMB Life 2014, 66, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beretta, M.; Santos, C.X.; Molenaar, C.; Hafstad, A.D.; Miller, C.C.; Revazian, A.; Betteridge, K.; Schröder, K.; Streckfuß-Bömeke, K.; Doroshow, J.H.; et al. Nox4 regulates InsP(3) receptor-dependent Ca(2+) release into mitochondria to promote cell survival. EMBO J. 2020, 39, e103530. [Google Scholar] [CrossRef] [PubMed]
- Sasako, T.; Ohsugi, M.; Kubota, N.; Itoh, S.; Okazaki, Y.; Terai, A.; Kubota, T.; Yamashita, S.; Nakatsukasa, K.; Kamura, T.; et al. Hepatic Sdf2l1 controls feeding-induced ER stress and regulates metabolism. Nat. Commun. 2019, 10, 947. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, K. Endoplasmic Reticulum Stress-Associated Lipid Droplet Formation and Type II Diabetes. Biochem. Res. Int. 2012, 2012, 247275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.J.; Malhi, H. The unfolded protein response and hepatic lipid metabolism in non alcoholic fatty liver disease. Pharmacol. Ther. 2019, 203, 107401. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Dandekar, A.; Mendez, R.; Zhang, K. Cross talk between ER stress, oxidative stress, and inflammation in health and disease. Methods Mol. Biol. 2015, 1292, 205–214. [Google Scholar] [PubMed]
- Zhang, K. Endoplasmic reticulum stress response and transcriptional reprogramming. Front. Genet. 2014, 5, 460. [Google Scholar] [PubMed] [Green Version]
- Sabath, N.; Levy-Adam, F.; Younis, A.; Rozales, K.; Meller, A.; Hadar, S.; Soueid-Baumgarten, S.; Shalgi, R. Cellular proteostasis decline in human senescence. Proc. Natl. Acad. Sci. USA 2020, 117, 31902–31913. [Google Scholar] [CrossRef] [PubMed]
- Platko, K.; Lebeau, P.F.; Byun, J.H.; Poon, S.V.; Day, E.A.; MacDonald, M.E.; Holzapfel, N.; Mejia-Benitez, A.; Maclean, K.N.; Krepinsky, J.C.; et al. GDF10 blocks hepatic PPARγ activation to protect against diet-induced liver injury. Mol. Metab. 2019, 27, 62–74. [Google Scholar] [CrossRef]
- Wei, J.; Yuan, Y.; Chen, L.; Xu, Y.; Zhang, Y.; Wang, Y.; Yang, Y.; Peek, C.B.; Diebold, L.; Yang, Y.; et al. ER-associated ubiquitin ligase HRD1 programs liver metabolism by targeting multiple metabolic enzymes. Nat. Commun. 2018, 9, 3659. [Google Scholar] [CrossRef]
- Ji, C. New Insights into the Pathogenesis of Alcohol-Induced ER Stress and Liver Diseases. Int. J. Hepatol. 2014, 2014, 513787. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Green, R.M. Endoplasmic reticulum stress and liver diseases. Liver Res. 2019, 3, 55–64. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Mohan, S.; Brown, L.; Ayyappan, P. Endoplasmic reticulum stress: A master regulator of metabolic syndrome. Eur. J. Pharmacol. 2019, 860, 172553. [Google Scholar] [CrossRef]
- Maiers, J.L.; Malhi, H. Endoplasmic Reticulum Stress in Metabolic Liver Diseases and Hepatic Fibrosis. Semin. Liver Dis. 2019, 39, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Zhang, C.; Zhang, K. Measurement of ER stress response and inflammation in the mouse model of nonalcoholic fatty liver disease. Methods Enzymol. 2011, 489, 329–348. [Google Scholar] [PubMed]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [Green Version]
- Kapuy, O.; Márton, M.; Bánhegyi, G.; Vinod, P.K. Multiple system-level feedback loops control life-and-death decisions in endoplasmic reticulum stress. FEBS Lett. 2020, 594, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Li, T.; Liu, Y.; Wang, X.; Zhang, J.; Wang, X.; Shi, G.; Lou, J.; Wang, L.; Wang, C.C.; et al. Phosphorylation switches protein disulfide isomerase activity to maintain proteostasis and attenuate ER stress. EMBO J. 2020, 39, e103841. [Google Scholar] [CrossRef]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell. Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fang, D. Endoplasmic reticulum-associated degradation and beyond: The multitasking roles for HRD1 in immune regulation and autoimmunity. J. Autoimmun. 2020, 109, 102423. [Google Scholar] [CrossRef] [PubMed]
- Rainbolt, T.K.; Frydman, J. Dynamics and clustering of IRE1α during ER stress. Proc. Natl. Acad. Sci. USA 2020, 117, 3352–3354. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 2006, 124, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belyy, V.; Tran, N.H.; Walter, P. Quantitative microscopy reveals dynamics and fate of clustered IRE1α. Proc. Natl. Acad. Sci. USA 2020, 117, 1533–1542. [Google Scholar] [CrossRef] [Green Version]
- Dufey, E.; Bravo-San Pedro, J.M.; Eggers, C.; González-Quiroz, M.; Urra, H.; Sagredo, A.I.; Sepulveda, D.; Pihán, P.; Carreras-Sureda, A.; Hazari, Y.; et al. Genotoxic stress triggers the activation of IRE1α-dependent RNA decay to modulate the DNA damage response. Nat. Commun. 2020, 11, 2401. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.B.; Snyder, J.T.; Alonso, L.C. Atf6α impacts cell number by influencing survival, death and proliferation. Mol. Metab. 2019, 27s, S69–S80. [Google Scholar] [CrossRef] [PubMed]
- Duvigneau, J.C.; Luís, A.; Gorman, A.M.; Samali, A.; Kaltenecker, D.; Moriggl, R.; Kozlov, A.V. Crosstalk between inflammatory mediators and endoplasmic reticulum stress in liver diseases. Cytokine 2019, 124, 154577. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Huang, L.; Gong, J.; Shi, C.; Wang, Z.; Ye, B.; Xuan, A.; He, X.; Long, D.; Zhu, X.; et al. NF-κB pathway link with ER stress-induced autophagy and apoptosis in cervical tumor cells. Cell Death Discov. 2017, 3, 17059. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Trillo-Tinoco, J.; Sierra, R.A.; Anadon, C.; Dai, W.; Mohamed, E.; Cen, L.; Costich, T.L.; Magliocco, A.; Marchion, D.; et al. ER stress-induced mediator C/EBP homologous protein thwarts effector T cell activity in tumors through T-bet repression. Nat. Commun. 2019, 10, 1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasudevan, D.; Neuman, S.D.; Yang, A.; Lough, L.; Brown, B.; Bashirullah, A.; Cardozo, T.; Ryoo, H.D. Translational induction of ATF4 during integrated stress response requires noncanonical initiation factors eIF2D and DENR. Nat. Commun. 2020, 11, 4677. [Google Scholar] [CrossRef] [PubMed]
- Kasetti, R.B.; Patel, P.D.; Maddineni, P.; Patil, S.; Kiehlbauch, C.; Millar, J.C.; Searby, C.C.; Raghunathan, V.; Sheffield, V.C.; Zode, G.S. ATF4 leads to glaucoma by promoting protein synthesis and ER client protein load. Nat. Commun. 2020, 11, 5594. [Google Scholar] [CrossRef] [PubMed]
- Schulze, R.J.; Schott, M.B.; Casey, C.A.; Tuma, P.L.; McNiven, M.A. The cell biology of the hepatocyte: A membrane trafficking machine. J. Cell. Biol. 2019, 218, 2096–2112. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wang, Z.V.; Tao, C.; Gao, N.; Holland, W.L.; Ferdous, A.; Repa, J.J.; Liang, G.; Ye, J.; Lehrman, M.A.; et al. The Xbp1s/GalE axis links ER stress to postprandial hepatic metabolism. J. Clin. Investig. 2013, 123, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Maillo, C.; Martín, J.; Sebastián, D.; Hernández-Alvarez, M.; García-Rocha, M.; Reina, O.; Zorzano, A.; Fernandez, M.; Méndez, R. Circadian- and UPR-dependent control of CPEB4 mediates a translational response to counteract hepatic steatosis under ER stress. Nat. Cell Biol. 2017, 19, 94–105. [Google Scholar] [CrossRef]
- Wei, J.; Chen, L.; Li, F.; Yuan, Y.; Wang, Y.; Xia, W.; Zhang, Y.; Xu, Y.; Yang, Z.; Gao, B.; et al. HRD1-ERAD controls production of the hepatokine FGF21 through CREBH polyubiquitination. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Chen, L.; Wei, J.; Zhu, H.; Pan, H.; Fang, D. Energy supplementation rescues growth restriction and female infertility of mice with hepatic HRD1 ablation. Am. J. Transl. Res. 2020, 12, 2018–2027. [Google Scholar] [PubMed]
- Sun, S.; Shi, G.; Sha, H.; Ji, Y.; Han, X.; Shu, X.; Ma, H.; Inoue, T.; Gao, B.; Kim, H.; et al. IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 2015, 17, 1546–1555. [Google Scholar] [CrossRef] [Green Version]
- Sepulveda, D.; Rojas-Rivera, D.; Rodríguez, D.A.; Groenendyk, J.; Köhler, A.; Lebeaupin, C.; Ito, S.; Urra, H.; Carreras-Sureda, A.; Hazari, Y.; et al. Interactome Screening Identifies the ER Luminal Chaperone Hsp47 as a Regulator of the Unfolded Protein Response Transducer IRE1α. Mol. Cell 2018, 69, 238–252.e7. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Nagata, K. Roles of the endoplasmic reticulum-resident, collagen-specific molecular chaperone Hsp47 in vertebrate cells and human disease. J. Biol. Chem. 2019, 294, 2133–2141. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Liang, F.X.; Wang, X. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol. Cell 2014, 55, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Melo-Cardenas, J.; Zhang, Y.; Gau, I.; Wei, J.; Montauti, E.; Zhang, Y.; Gao, B.; Jin, H.; Sun, Z.; et al. The E3 ligase Hrd1 stabilizes Tregs by antagonizing inflammatory cytokine-induced ER stress response. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wong, H.N.; Song, B.; Miller, C.N.; Scheuner, D.; Kaufman, R.J. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J. Clin. Investig. 2005, 115, 268–281. [Google Scholar] [CrossRef] [Green Version]
- Reimold, A.M.; Etkin, A.; Clauss, I.; Perkins, A.; Friend, D.S.; Zhang, J.; Horton, H.F.; Scott, A.; Orkin, S.H.; Byrne, M.C.; et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000, 14, 152–157. [Google Scholar]
- Lee, A.H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, J.S.; Hur, K.Y.; Tarrio, M.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Lichtman, A.H.; Iwawaki, T.; Glimcher, L.H.; et al. Silencing of lipid metabolism genes through IRE1α-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012, 16, 487–499. [Google Scholar] [CrossRef] [Green Version]
- McQuiston, A.; Diehl, J.A. Recent insights into PERK-dependent signaling from the stressed endoplasmic reticulum. F1000Research 2017, 6, 1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Li, J.; Sha, B. The ER stress sensor PERK luminal domain functions as a molecular chaperone to interact with misfolded proteins. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72 Pt 12, 1290–1297. [Google Scholar] [CrossRef]
- Wang, P.; Li, J.; Tao, J.; Sha, B. The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. J. Biol. Chem. 2018, 293, 4110–4121. [Google Scholar] [CrossRef] [Green Version]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucl. Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell. Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Dey, S.; Sayers, C.M.; Verginadis, I.I.; Lehman, S.L.; Cheng, Y.; Cerniglia, G.J.; Tuttle, S.W.; Feldman, M.D.; Zhang, P.J.; Fuchs, S.Y.; et al. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Investig. 2015, 125, 2592–2608. [Google Scholar] [CrossRef] [Green Version]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell. Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol. Cell 2001, 7, 1153–1163. [Google Scholar] [CrossRef]
- Zhang, P.; McGrath, B.; Li, S.; Frank, A.; Zambito, F.; Reinert, J.; Gannon, M.; Ma, K.; McNaughton, K.; Cavener, D.R. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell. Biol. 2002, 22, 3864–3874. [Google Scholar] [CrossRef] [Green Version]
- Fusakio, M.E.; Willy, J.A.; Wang, Y.; Mirek, E.T.; Al Baghdadi, R.J.; Adams, C.M.; Anthony, T.G.; Wek, R.C. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol. Biol. Cell 2016, 27, 1536–1551. [Google Scholar] [CrossRef] [PubMed]
- Jo, H.; Choe, S.S.; Shin, K.C.; Jang, H.; Lee, J.H.; Seong, J.K.; Back, S.H.; Kim, J.B. Endoplasmic reticulum stress induces hepatic steatosis via increased expression of the hepatic very low-density lipoprotein receptor. Hepatology 2013, 57, 1366–1377. [Google Scholar] [CrossRef]
- Yeh, K.Y.; Lai, C.Y.; Lin, C.Y.; Hsu, C.C.; Lo, C.P.; Her, G.M. ATF4 overexpression induces early onset of hyperlipidaemia and hepatic steatosis and enhances adipogenesis in zebrafish. Sci. Rep. 2017, 7, 16362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhi, H.; Kropp, E.M.; Clavo, V.F.; Kobrossi, C.R.; Han, J.; Mauer, A.S.; Yong, J.; Kaufman, R.J. C/EBP homologous protein-induced macrophage apoptosis protects mice from steatohepatitis. J. Biol. Chem. 2013, 288, 18624–18642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Gao, J.; Ishigaki, Y.; Kondo, K.; Sawada, S.; Izumi, T.; Uno, K.; Kaneko, K.; Tsukita, S.; Takahashi, K.; et al. ER Stress Protein CHOP Mediates Insulin Resistance by Modulating Adipose Tissue Macrophage Polarity. Cell. Rep. 2017, 18, 2045–2057. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Forouhan, M.; Mori, K.; Boot-Handford, R.P. Paradoxical roles of ATF6α and ATF6β in modulating disease severity caused by mutations in collagen X. Matrix Biol. J. Int. Soc. Matrix Biol. 2018, 70, 50–71. [Google Scholar] [CrossRef]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.; Kaufman, R.J. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Snapp, E.L.; Lippincott-Schwartz, J.; Prywes, R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol. Cell. Biol. 2005, 25, 921–932. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, M.; Yasui, S.; Niinuma, Y.; Arai, K.; Omura, T.; Okuma, Y.; Nomura, Y. A different pathway in the endoplasmic reticulum stress-induced expression of human HRD1 and SEL1 genes. FEBS Lett. 2007, 581, 5355–5360. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Takahara, K.; Oyadomari, S.; Okada, T.; Sato, T.; Harada, A.; Mori, K. Induction of liver steatosis and lipid droplet formation in ATF6alpha-knockout mice burdened with pharmacological endoplasmic reticulum stress. Mol. Biol. Cell 2010, 21, 2975–2986. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, F.; Gong, Q.; Cui, A.; Zhuo, S.; Hu, Z.; Han, Y.; Gao, J.; Sun, Y.; Liu, Z.; et al. Hepatic ATF6 Increases Fatty Acid Oxidation to Attenuate Hepatic Steatosis in Mice Through Peroxisome Proliferator-Activated Receptor α. Diabetes 2016, 65, 1904–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Lu, M.; Mori, K.; Luo, S.; Lee, A.S.; Zhu, Y.; Shyy, J.Y. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 2004, 23, 950–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Vera, L.; Fischer, W.H.; Montminy, M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature 2009, 460, 534–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Williams, D.; Qiu, Y.; Song, Z.; Yang, Z.; Kimler, V.; Goldberg, A.; Zhang, R.; Yang, Z.; Chen, X.; et al. Regulation of hepatic autophagy by stress-sensing transcription factor CREBH. FASEB J. Off. Publ. Federat. Am. Soc. Exp. Biol. 2019, 33, 7896–7914. [Google Scholar] [CrossRef]
- Zheng, Z.; Kim, H.; Qiu, Y.; Chen, X.; Mendez, R.; Dandekar, A.; Zhang, X.; Zhang, C.; Liu, A.C.; Yin, L.; et al. CREBH Couples Circadian Clock With Hepatic Lipid Metabolism. Diabetes 2016, 65, 3369–3383. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Zhao, M.; Cheng, X.; Shen, J.; Khound, R.; Zhang, K.; Su, Q. CREBH mediates metabolic inflammation to hepatic VLDL overproduction and hyperlipoproteinemia. J. Mol. Med. 2017, 95, 839–849. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, G.; Zheng, Z.; Maddipati, K.R.; Zhang, X.; Dyson, G.; Williams, P.; Duncan, S.A.; Kaufman, R.J.; Zhang, K. Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology 2012, 55, 1070–1082. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Giannikopoulos, P.; Duncan, S.A.; Wang, J.; Johansen, C.T.; Brown, J.D.; Plutzky, J.; Hegele, R.A.; Glimcher, L.H.; Lee, A.H. The transcription factor cyclic AMP-responsive element-binding protein H regulates triglyceride metabolism. Nat. Med. 2011, 17, 812–815. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Park, J.G.; So, J.S.; Hur, K.Y.; Lee, A.H. Transcriptional regulation of apolipoprotein A-IV by the transcription factor CREBH. J. Lipid Res. 2014, 55, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Mendez, R.; Zheng, Z.; Chang, L.; Cai, J.; Zhang, R.; Zhang, K. Liver-enriched transcription factor CREBH interacts with peroxisome proliferator-activated receptor α to regulate metabolic hormone FGF21. Endocrinology 2014, 155, 769–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Mendez, R.; Chen, X.; Fang, D.; Zhang, K. Lysine Acetylation of CREBH Regulates Fasting-Induced Hepatic Lipid Metabolism. Mol. Cell. Biol. 2015, 35, 4121–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, K.A.; London, W.T. The global epidemiology of hepatocellular carcinoma: Present and future. Clin. Liver Dis. 2011, 15, 223–243, vii–x. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordenstedt, H.; White, D.L.; El-Serag, H.B. The changing pattern of epidemiology in hepatocellular carcinoma. Digest. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2010, 42 (Suppl. 3), S206–S214. [Google Scholar] [CrossRef] [Green Version]
- Golabi, P.; Fazel, S.; Otgonsuren, M.; Sayiner, M.; Locklear, C.T.; Younossi, Z.M. Mortality assessment of patients with hepatocellular carcinoma according to underlying disease and treatment modalities. Medicine 2017, 96, e5904. [Google Scholar] [CrossRef]
- Colagrande, S.; Inghilesi, A.L.; Aburas, S.; Taliani, G.G.; Nardi, C.; Marra, F. Challenges of advanced hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 7645–7659. [Google Scholar] [CrossRef] [PubMed]
- Hamed, M.A.; Ali, S.A. Non-viral factors contributing to hepatocellular carcinoma. World J. Hepatol. 2013, 5, 311–322. [Google Scholar] [CrossRef] [Green Version]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petruzziello, A. Epidemiology of Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV) Related Hepatocellular Carcinoma. Open Virol. J. 2018, 12, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.W.; Lee, J.S.; Ahn, S.H. Hepatitis B Virus Cure: Targets and Future Therapies. Int. J. Mol. Sci. 2020, 22, 213. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kyaw, Y.Y.; Cheong, J. Functional interaction of endoplasmic reticulum stress and hepatitis B virus in the pathogenesis of liver diseases. World J. Gastroenterol. 2017, 23, 7657–7665. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Wu, H.C.; Chen, C.F.; Fausto, N.; Lei, H.Y.; Su, I.J. Different types of ground glass hepatocytes in chronic hepatitis B virus infection contain specific pre-S mutants that may induce endoplasmic reticulum stress. Am. J. Pathol. 2003, 163, 2441–2449. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Teng, Z.; Zhu, Y.; Zhao, A.Z.; Sun, C. Associations between pre-S deletion mutation of hepatitis B virus and risk of hepatocellular carcinoma in the Asian population: A meta-analysis. Med. Sci. Monitor Int. Med. J. Exp. Clin. Res. 2015, 21, 1072–1077. [Google Scholar]
- Choi, Y.M.; Lee, S.Y.; Kim, B.J. Naturally Occurring Hepatitis B Virus Mutations Leading to Endoplasmic Reticulum Stress and Their Contribution to the Progression of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 597. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.K.; Cheong, K.J.; Kim, H.Y.; Cheong, J. Endoplasmic reticulum stress induced by hepatitis B virus X protein enhances cyclo-oxygenase 2 expression via activating transcription factor 4. Biochem. J. 2011, 435, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gao, B.; Ye, L.; Han, X.; Wang, W.; Kong, L.; Fang, X.; Zeng, Y.; Zheng, H.; Li, S.; et al. Hepatitis B virus X protein (HBx) activates ATF6 and IRE1-XBP1 pathways of unfolded protein response. Virus Res. 2007, 124, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.K.; Kim, S.Y.; Kyaw, Y.Y.; Win, A.A.; Koo, S.H.; Kim, H.H.; Cheong, J. HBx induces the proliferation of hepatocellular carcinoma cells via AP1 over-expressed as a result of ER stress. Biochem. J. 2015, 466, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Chusri, P.; Kumthip, K.; Hong, J.; Zhu, C.; Duan, X.; Jilg, N.; Fusco, D.N.; Brisac, C.; Schaefer, E.A.; Cai, D.; et al. HCV induces transforming growth factor β1 through activation of endoplasmic reticulum stress and the unfolded protein response. Sci. Rep. 2016, 6, 22487. [Google Scholar] [CrossRef]
- Goto, K.; Roca Suarez, A.A.; Wrensch, F.; Baumert, T.F.; Lupberger, J. Hepatitis C Virus and Hepatocellular Carcinoma: When the Host Loses Its Grip. Int. J. Mol. Sci. 2020, 21, 57. [Google Scholar] [CrossRef]
- Tardif, K.D.; Mori, K.; Kaufman, R.J.; Siddiqui, A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 2004, 279, 17158–17164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Wang, J.; Zhao, Z. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 2014, 10, 766–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chida, T.; Ito, M.; Nakashima, K.; Kanegae, Y.; Aoshima, T.; Takabayashi, S.; Kawata, K.; Nakagawa, Y.; Yamamoto, M.; Shimano, H.; et al. Critical role of CREBH-mediated induction of transforming growth factor β2 by hepatitis C virus infection in fibrogenic responses in hepatic stellate cells. Hepatology 2017, 66, 1430–1443. [Google Scholar] [CrossRef]
- Ji, C.; Mehrian-Shai, R.; Chan, C.; Hsu, Y.H.; Kaplowitz, N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol. Clin. Exp. Res. 2005, 29, 1496–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Q.; Chen, Y.; Wang, J.; Hao, L.; Huang, C.; Griffiths, A.; Sun, Z.; Zhou, Z.; Song, Z. ER stress-induced upregulation of NNMT contributes to alcohol-related fatty liver development. J. Hepatol. 2020, 73, 783–793. [Google Scholar] [CrossRef]
- Howarth, D.L.; Lindtner, C.; Vacaru, A.M.; Sachidanandam, R.; Tsedensodnom, O.; Vasilkova, T.; Buettner, C.; Sadler, K.C. Activating transcription factor 6 is necessary and sufficient for alcoholic fatty liver disease in zebrafish. PLoS Genet. 2014, 10, e1004335. [Google Scholar] [CrossRef]
- Chanda, D.; Kim, Y.H.; Li, T.; Misra, J.; Kim, D.K.; Kim, J.R.; Kwon, J.; Jeong, W.I.; Ahn, S.H.; Park, T.S.; et al. Hepatic cannabinoid receptor type 1 mediates alcohol-induced regulation of bile acid enzyme genes expression via CREBH. PLoS ONE 2013, 8, e68845. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.M.; Qiu, Y.; Yang, Z.; Kim, H.; Qian, Q.; Sun, Q.; Zhang, C.; Yin, L.; Fang, D.; Back, S.H.; et al. IRE1α prevents hepatic steatosis by processing and promoting the degradation of select microRNAs. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Meng, Q.; Xiao, F.; Chen, S.; Du, Y.; Yu, J.; Wang, C.; Guo, F. ATF4 deficiency protects mice from high-carbohydrate-diet-induced liver steatosis. Biochem. J. 2011, 438, 283–289. [Google Scholar] [CrossRef]
- Wang, C.; Li, H.; Meng, Q.; Du, Y.; Xiao, F.; Zhang, Q.; Yu, J.; Li, K.; Chen, S.; Huang, Z.; et al. ATF4 deficiency protects hepatocytes from oxidative stress via inhibiting CYP2E1 expression. J. Cell. Mol. Med. 2014, 18, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.B.; Mercado, E.L.; Hoffmann, A.; Niwa, M. ER stress activates NF-κB by integrating functions of basal IKK activity, IRE1 and PERK. PLoS ONE 2012, 7, e45078. [Google Scholar] [CrossRef] [Green Version]
- Attanasio, S.; Ferriero, R.; Gernoux, G.; De Cegli, R.; Carissimo, A.; Nusco, E.; Campione, S.; Teckman, J.; Mueller, C.; Piccolo, P.; et al. CHOP and c-JUN up-regulate the mutant Z α(1)-antitrypsin, exacerbating its aggregation and liver proteotoxicity. J. Biol. Chem. 2020, 295, 13213–13223. [Google Scholar] [CrossRef] [PubMed]
- Testino, G.; Leone, S.; Borro, P. Alcohol and hepatocellular carcinoma: A review and a point of view. World J. Gastroenterol. 2014, 20, 15943–15954. [Google Scholar] [CrossRef] [PubMed]
- McKillop, I.H.; Schrum, L.W.; Thompson, K.J. Role of alcohol in the development and progression of hepatocellular carcinoma. Hepatic Oncol. 2016, 3, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.R.; Mandayam, S.; Jamal, M.M. Alcohol and hepatocellular carcinoma. Gastroenterology 2004, 127 (Suppl. 1), S87–S96. [Google Scholar] [CrossRef]
- Edenberg, H.J. The genetics of alcohol metabolism: Role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol. Res. Health J. Natl. Inst. Alcohol. Abuse Alcohol. 2007, 30, 5–13. [Google Scholar]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol. Res. Health J. Natl. Inst. Alcohol. Abuse Alcohol. 2006, 29, 245–254. [Google Scholar]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinogenes. 2006, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Nowak, A.J.; Relja, B. The Impact of Acute or Chronic Alcohol Intake on the NF-κB Signaling Pathway in Alcohol-Related Liver Disease. Int. J. Mol. Sci. 2020, 21, 9407. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol. Res. Curr. Rev. 2017, 38, 147–161. [Google Scholar]
- Lugea, A.; Tischler, D.; Nguyen, J.; Gong, J.; Gukovsky, I.; French, S.W.; Gorelick, F.S.; Pandol, S.J. Adaptive unfolded protein response attenuates alcohol-induced pancreatic damage. Gastroenterology 2011, 140, 987–997. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Xiao, Y.; Yu, J.; Xia, T.; Liu, B.; Guo, Y.; Deng, J.; Chen, S.; Wang, C.; Guo, F. Liver-specific Gene Inactivation of the Transcription Factor ATF4 Alleviates Alcoholic Liver Steatosis in Mice. J. Biol. Chem. 2016, 291, 18536–18546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhamija, E.; Paul, S.B.; Kedia, S. Non-alcoholic fatty liver disease associated with hepatocellular carcinoma: An increasing concern. Ind. J. Med. Res. 2019, 149, 9–17. [Google Scholar]
- Bence, K.K.; Birnbaum, M.J. Metabolic drivers of non-alcoholic fatty liver disease. Mol. Metab. 2020, 101143. [Google Scholar] [CrossRef]
- Berardo, C.; Di Pasqua, L.G.; Cagna, M.; Richelmi, P.; Vairetti, M.; Ferrigno, A. Nonalcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: Current Issues and Future Perspectives in Preclinical and Clinical Research. Int. J. Mol. Sci. 2020, 21, 9646. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837.e2. [Google Scholar] [CrossRef] [Green Version]
- Henkel, A.; Green, R.M. The unfolded protein response in fatty liver disease. Semin. Liver Dis. 2013, 33, 321–329. [Google Scholar] [PubMed] [Green Version]
- Liu, X.; Henkel, A.S.; LeCuyer, B.E.; Schipma, M.J.; Anderson, K.A.; Green, R.M. Hepatocyte X-box binding protein 1 deficiency increases liver injury in mice fed a high-fat/sugar diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G965–G974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, G.C.; van Rooyen, D.; Gan, L.; Chitturi, S. NASH is an Inflammatory Disorder: Pathogenic, Prognostic and Therapeutic Implications. Gut Liver 2012, 6, 149–171. [Google Scholar] [CrossRef] [Green Version]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [Green Version]
- He, G.; Yu, G.Y.; Temkin, V.; Ogata, H.; Kuntzen, C.; Sakurai, T.; Sieghart, W.; Peck-Radosavljevic, M.; Leffert, H.L.; Karin, M. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010, 17, 286–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Oikawa, F.; Mizuno, S.; Ohno, H.; Yagishita, Y.; Satoh, A.; Osaki, Y.; Takei, K.; Kikuchi, T.; Han, S.I.; et al. Hyperlipidemia and hepatitis in liver-specific CREB3L3 knockout mice generated using a one-step CRISPR/Cas9 system. Sci. Rep. 2016, 6, 27857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, E.L.; Khan, Z. Liver Disease in Alpha-1 Antitrypsin Deficiency: Current Approaches and Future Directions. Curr. Pathobiol. Rep. 2017, 5, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Feldman, A.; Sokol, R.J. Alpha-1-Antitrypsin Deficiency: An Important Cause of Pediatric Liver Disease. Lung Health Profess. Magaz. 2013, 4, 8–11. [Google Scholar]
- Blanco, I.; Bueno, P.; Diego, I.; Pérez-Holanda, S.; Lara, B.; Casas-Maldonado, F.; Esquinas, C.; Miravitlles, M. Alpha-1 antitrypsin Pi*SZ genotype: Estimated prevalence and number of SZ subjects worldwide. Int. J. Chronic Obstruct. Pulm. Dis. 2017, 12, 1683–1694. [Google Scholar] [CrossRef] [Green Version]
- Tanash, H.A.; Piitulainen, E. Liver disease in adults with severe alpha-1-antitrypsin deficiency. J. Gastroenterol. 2019, 54, 541–548. [Google Scholar] [CrossRef] [Green Version]
- Strnad, P.; Buch, S.; Hamesch, K.; Fischer, J.; Rosendahl, J.; Schmelz, R.; Brueckner, S.; Brosch, M.; Heimes, C.V.; Woditsch, V.; et al. Heterozygous carriage of the alpha1-antitrypsin Pi*Z variant increases the risk to develop liver cirrhosis. Gut 2019, 68, 1099–1107. [Google Scholar] [CrossRef]
- Berg, N.O.; Eriksson, S. Liver disease in adults with alpha-1 -antitrypsin deficiency. N. Engl. J. Med. 1972, 287, 1264–1267. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S. Alpha 1-antitrypsin deficiency and liver cirrhosis in adults. An analysis of 35 Swedish autopsied cases. Acta Med. Scand. 1987, 221, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Topic, A.; Ljujic, M.; Radojkovic, D. Alpha-1-antitrypsin in pathogenesis of hepatocellular carcinoma. Hepat. Mon. 2012, 12, e7042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlmutter, D.H. α1-antitrypsin Deficiency: A Misfolded Secretory Protein Variant with Unique Effects on the Endoplasmic Reticulum. Endoplasmic Reticulum Stress Dis. 2016, 3, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidvegi, T.; Schmidt, B.Z.; Hale, P.; Perlmutter, D.H. Accumulation of mutant alpha1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and -12, NFkappaB, and BAP31 but not the unfolded protein response. J. Biol. Chem. 2005, 280, 39002–39015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordóñez, A.; Snapp, E.L.; Tan, L.; Miranda, E.; Marciniak, S.J.; Lomas, D.A. Endoplasmic reticulum polymers impair luminal protein mobility and sensitize to cellular stress in alpha1-antitrypsin deficiency. Hepatology 2013, 57, 2049–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponzetto, A.; Perez-Perez, G.I.; Figura, N. Alpha1-antitrypsin deficiency and c-JUN. Hepatology 2017, 66, 677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Garcia-Carbonell, R.; Yamachika, S.; Zhao, P.; Dhar, D.; Loomba, R.; Kaufman, R.J.; Saltiel, A.R.; Karin, M. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 2018, 175, 133–145.e15. [Google Scholar] [CrossRef] [Green Version]
- Pavlović, N.; Calitz, C.; Thanapirom, K.; Mazza, G.; Rombouts, K.; Gerwins, P.; Heindryckx, F. Inhibiting IRE1α-endonuclease activity decreases tumor burden in a mouse model for hepatocellular carcinoma. eLife 2020, 9. [Google Scholar] [CrossRef]
- Wu, S.; Du, R.; Gao, C.; Kang, J.; Wen, J.; Sun, T. The role of XBP1s in the metastasis and prognosis of hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 500, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Vandewynckel, Y.P.; Laukens, D.; Bogaerts, E.; Paridaens, A.; Van den Bussche, A.; Verhelst, X.; Van Steenkiste, C.; Descamps, B.; Vanhove, C.; Libbrecht, L.; et al. Modulation of the unfolded protein response impedes tumor cell adaptation to proteotoxic stress: A PERK for hepatocellular carcinoma therapy. Hepatol. Int. 2015, 9, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Lu, Q.; Liu, J.; Fan, L.; Wang, Y.; Wei, W.; Wang, H.; Sun, G. Melatonin Increases the Sensitivity of Hepatocellular Carcinoma to Sorafenib through the PERK-ATF4-Beclin1 Pathway. Int. J. Biol. Sci. 2019, 15, 1905–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pafili, K.; Roden, M. Nonalcoholic fatty liver disease (NAFLD) from pathogenesis to treatment concepts in humans. Mol. Metab. 2020, 101122. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, A.; Khan, Z.; Alloghbi, A.; Said Ahmed, T.S.; Ashraf, M.; Hammouda, D.M. Hepatocellular Carcinoma: Molecular Mechanisms and Targeted Therapies. Medicina 2019, 55, 526. [Google Scholar] [CrossRef] [Green Version]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
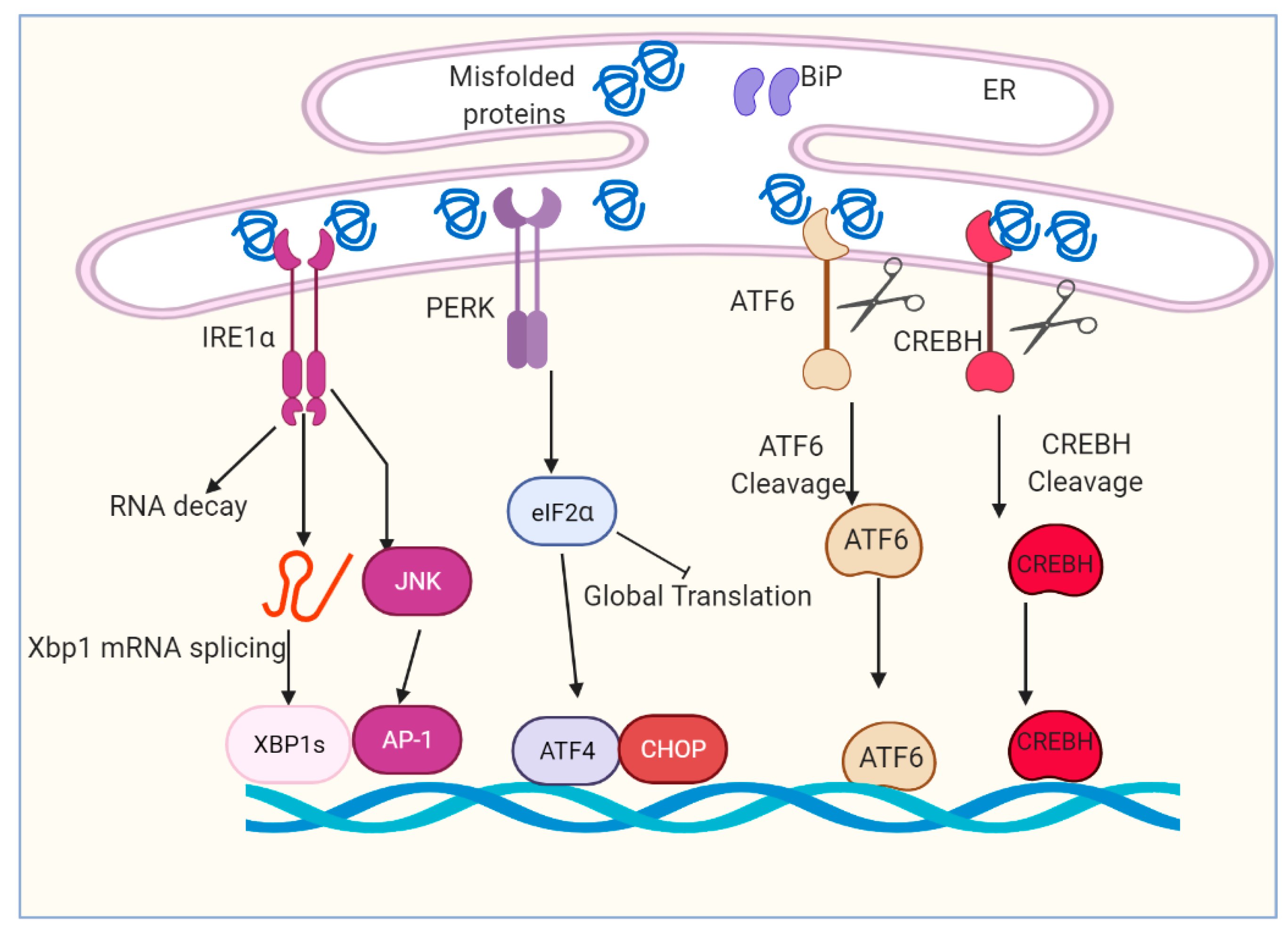
Figure 1.
Overview of ER stress and unfolded protein response (UPR) signaling pathway. The accumulation of unfolded and/or misfolded protein induces ER stress and restores ER homeostasis. The cell activates a series of signaling pathways, named the unfolded protein response (UPR). The UPR is regulated by four main proteins: IRE1α, PERK, ATF6 and CREBH. IRE1α, PERK, ATF6 and CREBH are ultimately responsible for the activation of a set of transcription factors, including spliced Xbp1, ATF4, CHOP, cleaved ATF6 and cleaved CREBH.
Figure 1.
Overview of ER stress and unfolded protein response (UPR) signaling pathway. The accumulation of unfolded and/or misfolded protein induces ER stress and restores ER homeostasis. The cell activates a series of signaling pathways, named the unfolded protein response (UPR). The UPR is regulated by four main proteins: IRE1α, PERK, ATF6 and CREBH. IRE1α, PERK, ATF6 and CREBH are ultimately responsible for the activation of a set of transcription factors, including spliced Xbp1, ATF4, CHOP, cleaved ATF6 and cleaved CREBH.

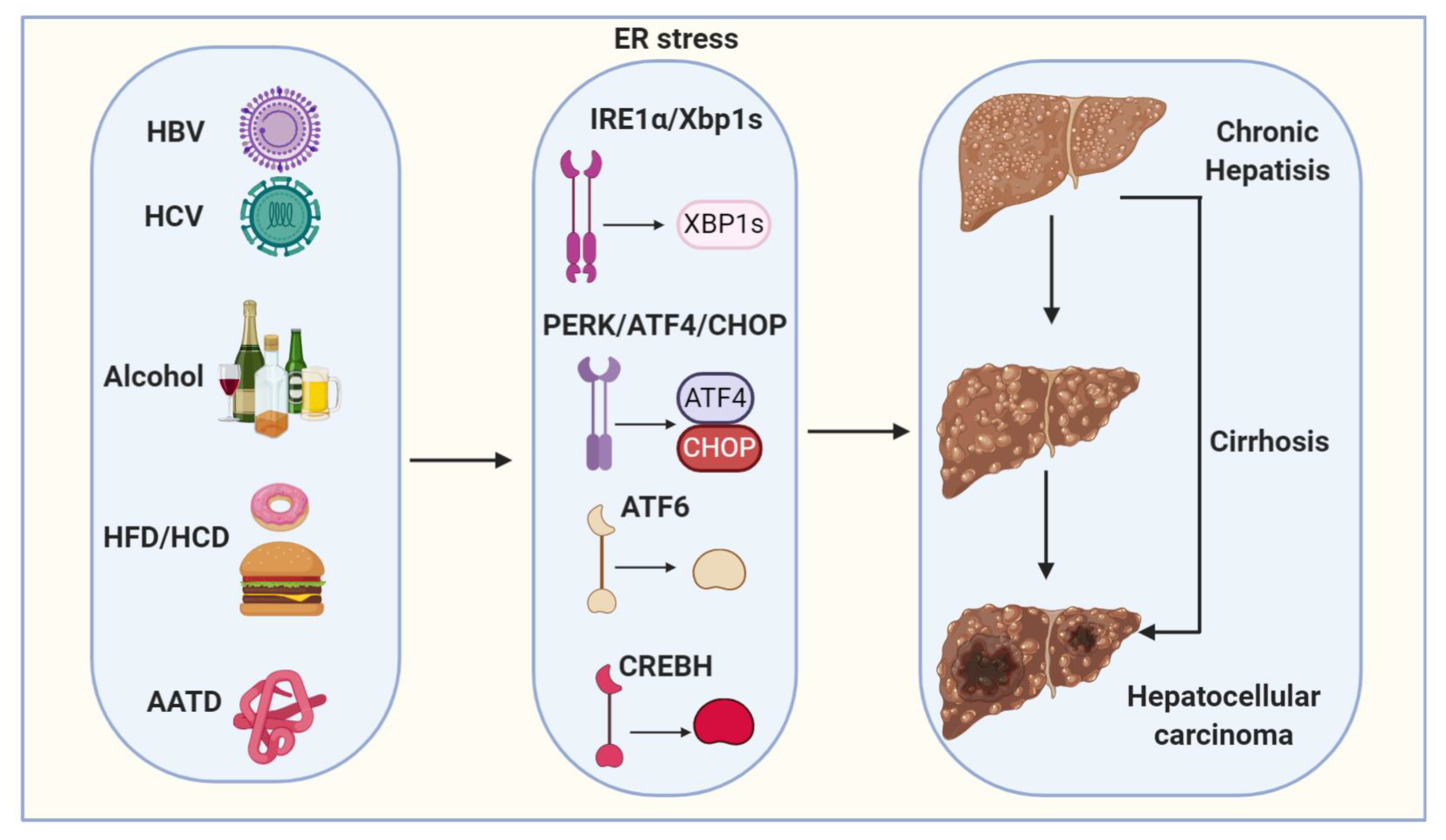
Figure 2.
Proposed model depicts ER stress mechanisms linking viral infection, alcohol, high-fat diet/high-carbohydrate diet (HFD/HCD) and Alpha1-antitrypsin deficiency (AATD) with hepatocellular carcinogenesis (HCC). Viral (HBV and HCV) infection, alcohol, HFD/HCD and AATD induce and sustain ER stress. ER stress and steatosis increase reactive oxygen species (ROS) production to cause oxidative stress and inflammation and the subsequent genomic instability, cirrhosis and HCC.
Figure 2.
Proposed model depicts ER stress mechanisms linking viral infection, alcohol, high-fat diet/high-carbohydrate diet (HFD/HCD) and Alpha1-antitrypsin deficiency (AATD) with hepatocellular carcinogenesis (HCC). Viral (HBV and HCV) infection, alcohol, HFD/HCD and AATD induce and sustain ER stress. ER stress and steatosis increase reactive oxygen species (ROS) production to cause oxidative stress and inflammation and the subsequent genomic instability, cirrhosis and HCC.

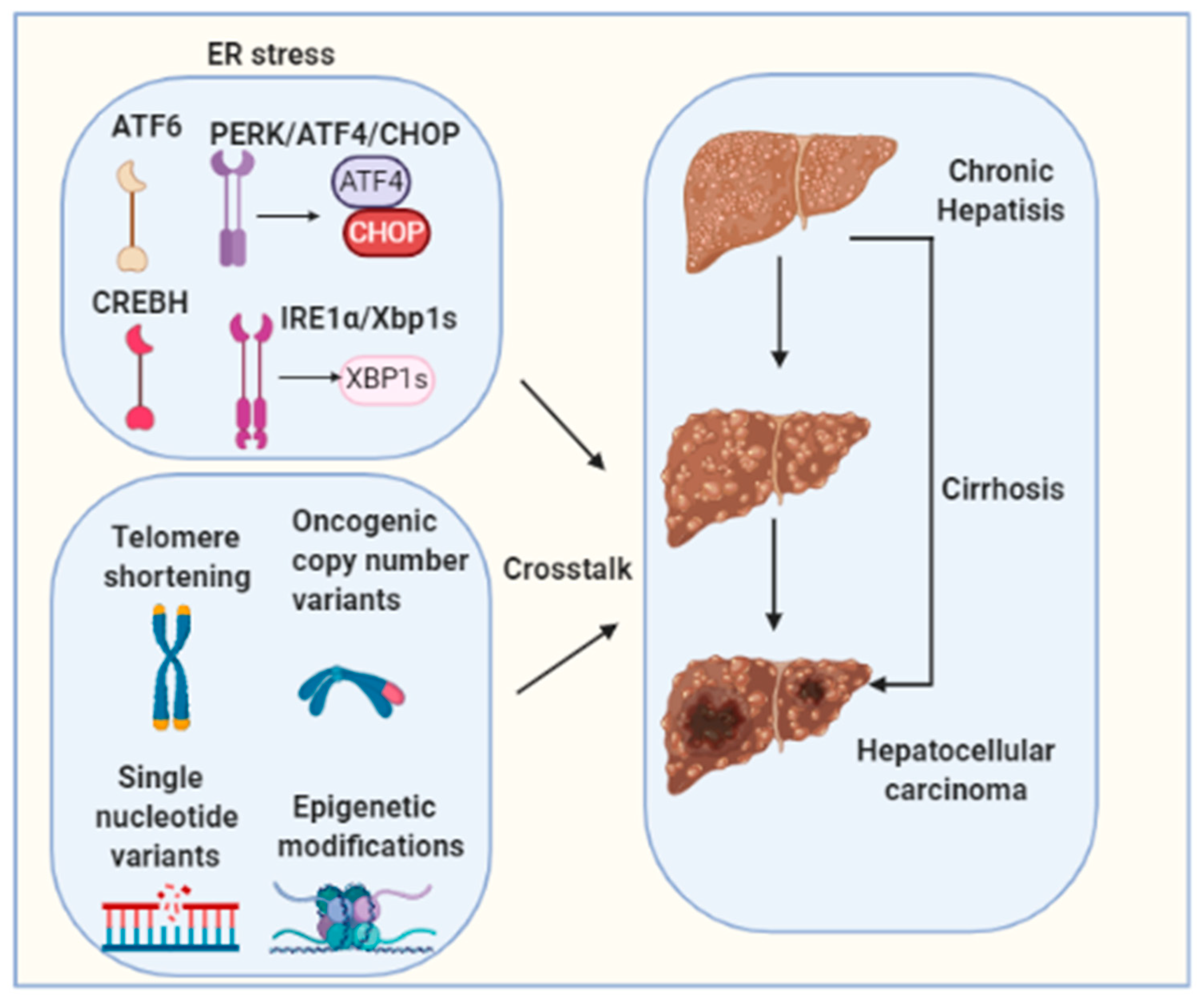
Figure 3.
Hepatocarcinogenesis is a complex multistep process involving multiple different signaling cascades’ activation. Telomere shortening, oncogenic copy number variants, single nucleotide variants and epigenetic modifications are well-established molecular mechanisms for HCC tumorigenesis. The molecular crosstalk between these factors and ER stress needs to be further investigated.
Figure 3.
Hepatocarcinogenesis is a complex multistep process involving multiple different signaling cascades’ activation. Telomere shortening, oncogenic copy number variants, single nucleotide variants and epigenetic modifications are well-established molecular mechanisms for HCC tumorigenesis. The molecular crosstalk between these factors and ER stress needs to be further investigated.


{kind=link}
{kind=link}
{kind=link}
Table 1.
ER stress in the pathogenesis of HCC.
| Risk Factor | UPR Pathway Activation | Cellular and Molecular Mechanisms of Liver Injury | Reference |
|---|---|---|---|
| Hepatitis B virus | PERK-ATF4 | Oxidative stress and apoptosis | [97] |
| ATF6 cleavage | Proliferation of hepatocellular carcinoma cells | [98,99] | |
| IRE1α-XBP1s | |||
| Hepatitis C virus | IRE1α-XBP1s | Misfolded proteins are more stable | [103] |
| IRE1α-JNK | TGFβ1 expression and proliferation | [101,105] | |
| CREBH cleavage | |||
| PERK pathway | Autophagy induction | [104] | |
| ATF6 cleavage | |||
| Alcohol | PERK-ATF4-CHOP | Apoptosis | [106] |
| NNMT expression | [107] | ||
| ATF6 cleavage | Lipogenesis and steatosis | [108] | |
| CREBH cleavage | Perturbation of bile acid homeostasis | [109] | |
| fatty liver (NAFLD/NASH) | IRE1α-XBP1s | Hepatic steatosis and insulin resistance | [110] |
| miRNA expression and liver injury | |||
| PERK-ATF4 | SCD1 and CYP2E1 expression; liver steatosis | [111,112] | |
| ATF6 cleavage | Hepatic steatosis and insulin resistance | [74] | |
| CREBH cleavage | Nonalcoholic steatohepatitis | [80] | |
| IREα—JNKs and NF-κB | Hepatic injury and fibrosis | [113] | |
| Alpha1-antitrypsin deficiency (AATD) | CHOP | Upregulates A1AT expression and apoptosis | [114] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wei, J.; Fang, D. Endoplasmic Reticulum Stress Signaling and the Pathogenesis of Hepatocarcinoma. Int. J. Mol. Sci. 2021, 22, 1799. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041799
AMA Style
Wei J, Fang D. Endoplasmic Reticulum Stress Signaling and the Pathogenesis of Hepatocarcinoma. International Journal of Molecular Sciences. 2021; 22(4):1799. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041799
Chicago/Turabian StyleWei, Juncheng, and Deyu Fang. 2021. "Endoplasmic Reticulum Stress Signaling and the Pathogenesis of Hepatocarcinoma" International Journal of Molecular Sciences 22, no. 4: 1799. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041799
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.