
Intestinal Epithelial Barrier Maturation by Enteric Glial Cells Is GDNF-Dependent

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
2.1. EGCs Express and Secrete GDNF in Vivo and In Vitro at Significant Levels
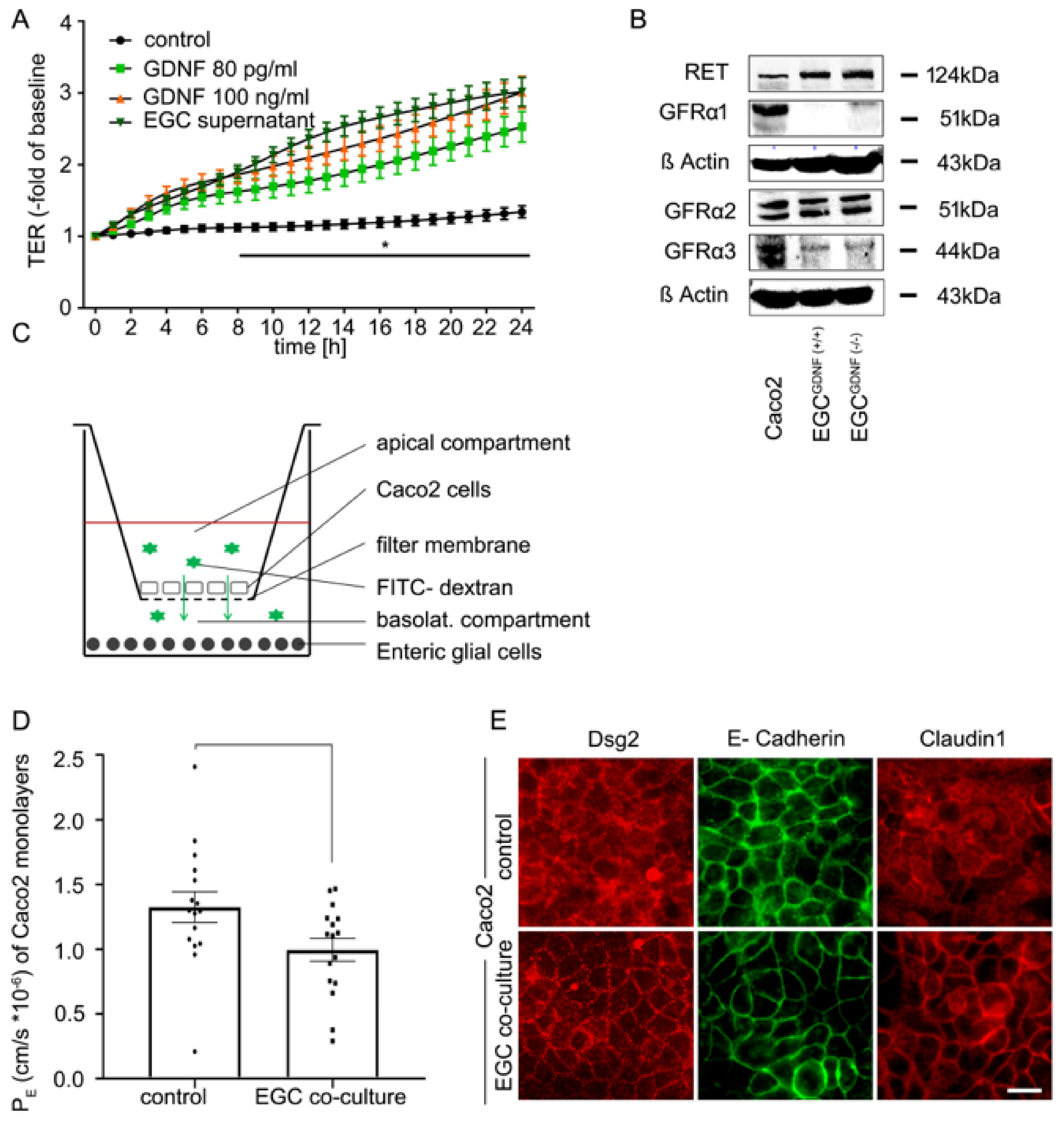
2.2. Co-Culture of EGCs and Intestinal Epithelial Cells Leads to Barrier Maturation
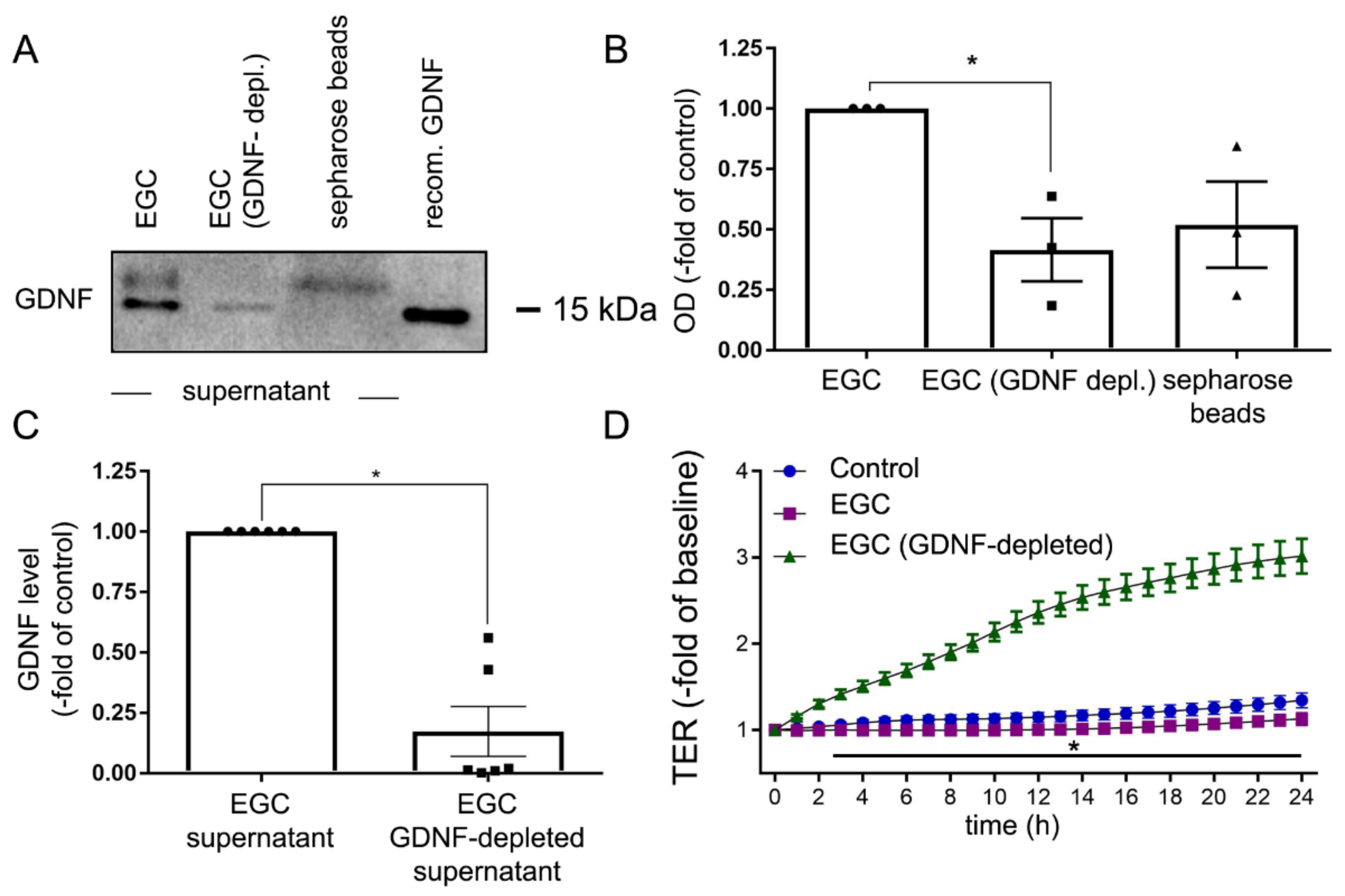
2.3. Depletion of GDNF from EGC Supernatants Blunts Intestinal Barrier Maturation
2.4. Knockdown of GDNF in EGCs Attenuates Intestinal Epithelial Barrier Maturation
2.5. Effects of EGCs on the Distribution of Junctional Proteins at the Cell Borders Are GDNF-Dependent
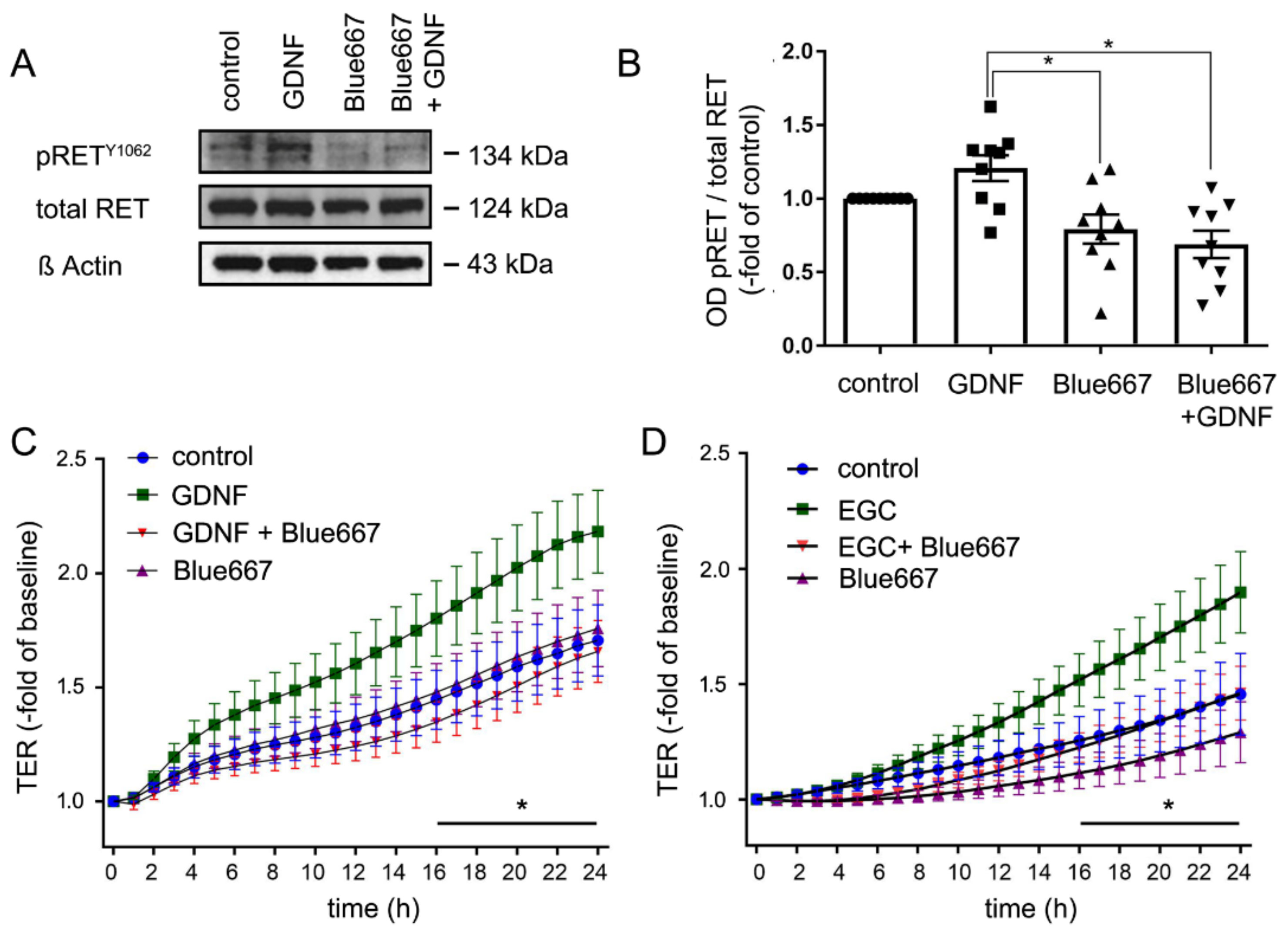
2.6. EGC-Mediated Epithelial Barrier Stabilization Is Dependent on the GDNF Receptor RET
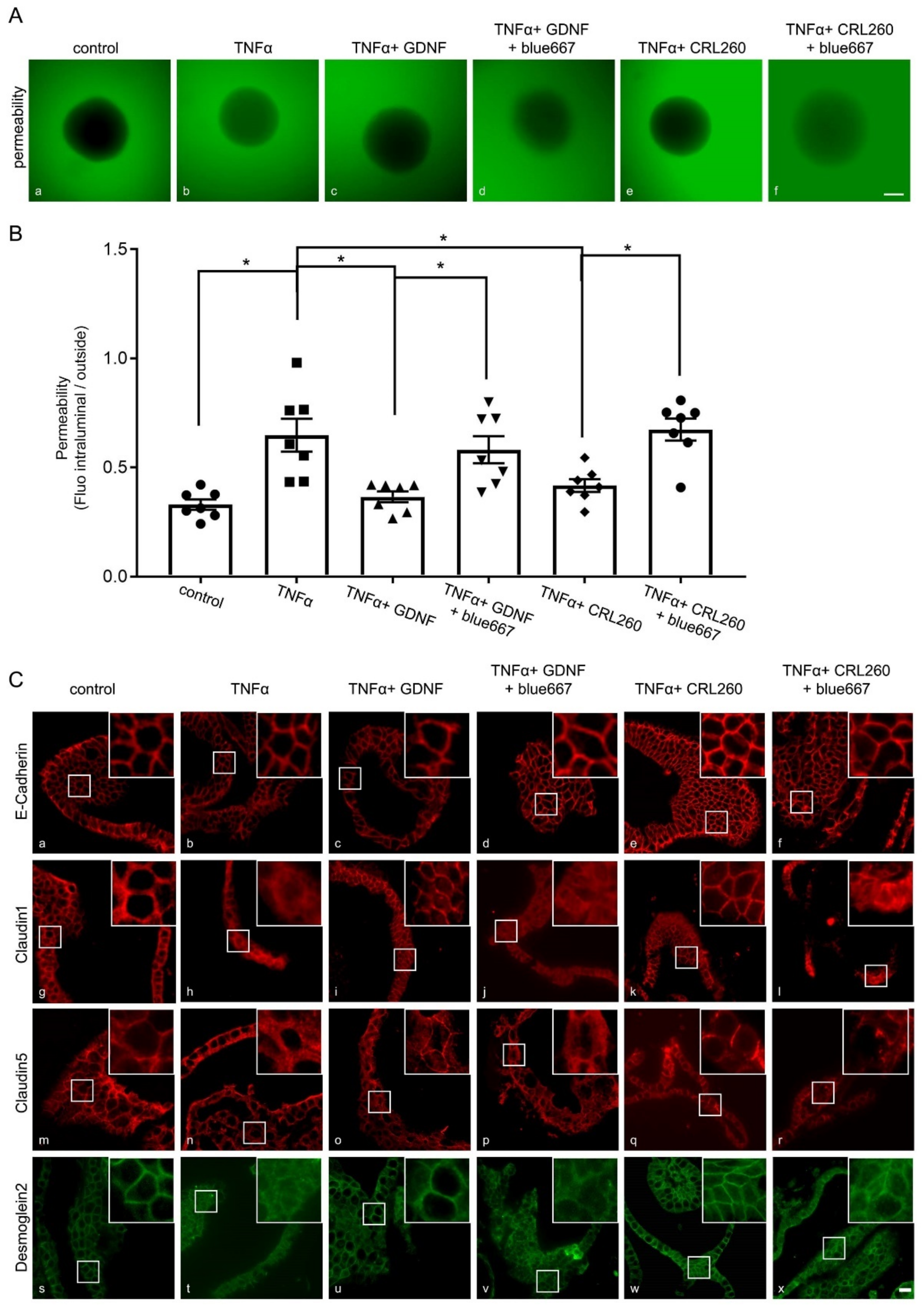
2.7. The Effects of EGC Co-Culture on the Inflammation-Induced Breakdown of Intestinal Epithelial Barrier Function Are GDNF-Dependent
2.8. EGC-Mediated Effects on IEB Function on Junctional Proteins in Intestinal Organoids Are GDNF-Dependent
3. Discussion
3.1. Considerations of the Co-Culture System of EGCs and Caco2 Cells
3.2. EGC-Mediated Effects on Intestinal Epithelial Barrier Function
3.3. GDNF Is a Key Mediator of Barrier Stabilization by EGCs In Vitro
3.4. GDNF Secretion from EGCs Is Stimulated by Inflammatory Mediators Leading to Epithelial Barrier Protection
4. Materials and Methods
4.1. Cell-Culture
4.2. Animals and Intestinal Organoid Generation
4.3. Organoid Generation
4.4. Generation and Cultivation of Primary EGC
4.5. Gene Expression Analysis of Tdtomato Positive EGCs
4.6. Cell Culture Supernatant
4.7. Human Tissue Samples
4.8. Enteroid Permeability Assay
4.9. Test Reagents
4.10. Western Blot
4.11. Immunocytochemistry
4.12. qRT-PCR
4.12.1. Measurement of FITC-Dextran Flux across Monolayers of Cultured Epithelial Cells
4.12.2. Depletion of GDNF form EGC Supernatants by Immunoprecipitation
4.12.3. Generation of Cripsr/Cas9-Mediated Gene Knock-Down for GDNF in EGCs
4.12.4. Measurements of Transepithelial Electrical Resistance (TER)
4.13. Quantification of Immunostaining
4.14. GDNF ELISA
4.15. Cell Viability Assay
4.16. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luissint, A.C.; Parkos, C.A.; Nusrat, A. Inflammation and the Intestinal Barrier: Leukocyte-Epithelial Cell Interactions, Cell Junction Remodeling, and Mucosal Repair. Gastroenterology 2016, 151, 616–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability—A new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, N.; Boerner, K.; Waschke, J. Targeting desmosomal adhesion and signalling for intestinal barrier stabilization in inflammatory bowel diseases-Lessons from experimental models and patients. Acta Physiol. 2021, 231, e13492. [Google Scholar] [CrossRef]
- Martini, E.; Krug, S.M.; Siegmund, B.; Neurath, M.F.; Becker, C. Mend Your Fences: The Epithelial Barrier and its Relationship with Mucosal Immunity in Inflammatory Bowel Disease. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neunlist, M.; Toumi, F.; Oreschkova, T.; Denis, M.; Leborgne, J.; Laboisse, C.L.; Galmiche, J.P.; Jarry, A. Human ENS regulates the intestinal epithelial barrier permeability and a tight junction-associated protein ZO-1 via VIPergic pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G1028–G1036. [Google Scholar] [CrossRef] [Green Version]
- Rao, M.; Rastelli, D.; Dong, L.; Chiu, S.; Setlik, W.; Gershon, M.D.; Corfas, G. Enteric Glia Regulate Gastrointestinal Motility but Are Not Required for Maintenance of the Epithelium in Mice. Gastroenterology 2017. [Google Scholar] [CrossRef]
- Grubisic, V.; Gulbransen, B.D. Enteric glial activity regulates secretomotor function in the mouse colon but does not acutely affect gut permeability. J. Physiol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Cornet, A.; Savidge, T.C.; Cabarrocas, J.; Deng, W.L.; Colombel, J.F.; Lassmann, H.; Desreumaux, P.; Liblau, R.S. Enterocolitis induced by autoimmune targeting of enteric glial cells: A possible mechanism in Crohn’s disease? Proc. Natl. Acad. Sci. USA 2001, 98, 13306–13311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, T.G.; Savidge, T.C.; Freeman, T.C.; Cox, H.J.; Campbell, E.A.; Mucke, L.; Johnson, M.H.; Sofroniew, M.V. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell 1998, 93, 189–201. [Google Scholar] [CrossRef] [Green Version]
- von Boyen, G.B.; Schulte, N.; Pfluger, C.; Spaniol, U.; Hartmann, C.; Steinkamp, M. Distribution of enteric glia and GDNF during gut inflammation. BMC Gastroenterol. 2011, 11, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pochard, C.; Coquenlorge, S.; Jaulin, J.; Cenac, N.; Vergnolle, N.; Meurette, G.; Freyssinet, M.; Neunlist, M.; Rolli-Derkinderen, M. Defects in 15-HETE Production and Control of Epithelial Permeability by Human Enteric Glial Cells from Patients With Crohn’s Disease. Gastroenterology 2016, 150, 168–180. [Google Scholar] [CrossRef]
- Neunlist, M.; Van Landeghem, L.; Mahe, M.M.; Derkinderen, P.; des Varannes, S.B.; Rolli-Derkinderen, M. The digestive neuronal-glial-epithelial unit: A new actor in gut health and disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 90–100. [Google Scholar] [CrossRef]
- Savidge, T.C.; Newman, P.; Pothoulakis, C.; Ruhl, A.; Neunlist, M.; Bourreille, A.; Hurst, R.; Sofroniew, M.V. Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology 2007, 132, 1344–1358. [Google Scholar] [CrossRef]
- Bach-Ngohou, K.; Mahe, M.M.; Aubert, P.; Abdo, H.; Boni, S.; Bourreille, A.; Denis, M.G.; Lardeux, B.; Neunlist, M.; Masson, D. Enteric glia modulate epithelial cell proliferation and differentiation through 15-deoxy-12,14-prostaglandin J2. J. Physiol. 2010, 588, 2533–2544. [Google Scholar] [CrossRef]
- Meir, M.; Burkard, N.; Ungewiss, H.; Diefenbacher, M.; Flemming, S.; Kannapin, F.; Germer, C.T.; Schweinlin, M.; Metzger, M.; Waschke, J.; et al. Neurotrophic factor GDNF regulates intestinal barrier function in inflammatory bowel disease. J. Clin. Investig. 2019, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruhl, A.; Trotter, J.; Stremmel, W. Isolation of enteric glia and establishment of transformed enteroglial cell lines from the myenteric plexus of adult rat. Neurogastroenterol. Motil. 2001, 13, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Meir, M.; Flemming, S.; Burkard, N.; Bergauer, L.; Metzger, M.; Germer, C.T.; Schlegel, N. Glial cell line-derived neurotrophic factor (GDNF) promotes barrier maturation and wound healing in intestinal epithelial cells in vitro. Am. J. Physiol. Gastrointest. Liver Physiol. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, N.; Meir, M.; Spindler, V.; Germer, C.T.; Waschke, J. Differential role of Rho GTPases in intestinal epithelial barrier regulation in vitro. J. Cell Physiol. 2011, 226, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Gainor, J.F.; Rahal, R.; Brubaker, J.D.; Kim, J.L.; Maynard, M.; Hu, W.; Cao, Q.; Sheets, M.P.; Wilson, D.; et al. Precision Targeted Therapy with BLU-667 for RET-Driven Cancers. Cancer Discov. 2018, 8, 836–849. [Google Scholar] [CrossRef] [Green Version]
- von Boyen, G.B.; Steinkamp, M.; Reinshagen, M.; Schafer, K.H.; Adler, G.; Kirsch, J. Nerve growth factor secretion in cultured enteric glia cells is modulated by proinflammatory cytokines. J. Neuroendocrinol. 2006, 18, 820–825. [Google Scholar] [CrossRef]
- Brun, P.; Giron, M.C.; Qesari, M.; Porzionato, A.; Caputi, V.; Zoppellaro, C.; Banzato, S.; Grillo, A.R.; Spagnol, L.; De Caro, R.; et al. Toll-like receptor 2 regulates intestinal inflammation by controlling integrity of the enteric nervous system. Gastroenterology 2013, 145, 1323–1333. [Google Scholar] [CrossRef] [Green Version]
- Bardenbacher, M.; Ruder, B.; Britzen-Laurent, N.; Schmid, B.; Waldner, M.; Naschberger, E.; Scharl, M.; Muller, W.; Gunther, C.; Becker, C.; et al. Permeability analyses and three dimensional imaging of interferon gamma-induced barrier disintegration in intestinal organoids. Stem Cell Res. 2019, 35, 101383. [Google Scholar] [CrossRef] [PubMed]
- Meir, M.; Flemming, S.; Burkard, N.; Wagner, J.; Germer, C.T.; Schlegel, N. The glial cell-line derived neurotrophic factor: A novel regulator of intestinal barrier function in health and disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G1118–G1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soret, R.; Coquenlorge, S.; Cossais, F.; Meurette, G.; Rolli-Derkinderen, M.; Neunlist, M. Characterization of human, mouse, and rat cultures of enteric glial cells and their effect on intestinal epithelial cells. Neurogastroenterol. Motil 2013, 25, e755–e764. [Google Scholar] [CrossRef] [PubMed]
- Fogh, J.; Orfeo, T.; Tiso, J.; Sharkey, F.E.; Fogh, J.M.; Daniels, W.P. Twenty-three new human tumor lines established in nude mice. Exp. Cell Biol. 1980, 48, 229–239. [Google Scholar] [CrossRef]
- Schlegel, N.; Meir, M.; Heupel, W.M.; Holthofer, B.; Leube, R.E.; Waschke, J. Desmoglein 2-mediated adhesion is required for intestinal epithelial barrier integrity. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G774–G783. [Google Scholar] [CrossRef] [Green Version]
- Langness, S.; Kojima, M.; Coimbra, R.; Eliceiri, B.P.; Costantini, T.W. Enteric glia cells are critical to limiting the intestinal inflammatory response after injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G274–G282. [Google Scholar] [CrossRef]
- MacEachern, S.J.; Patel, B.A.; Keenan, C.M.; Dicay, M.; Chapman, K.; McCafferty, D.M.; Savidge, T.C.; Beck, P.L.; MacNaughton, W.K.; Sharkey, K.A. Inhibiting Inducible Nitric Oxide Synthase in Enteric Glia Restores Electrogenic Ion Transport in Mice with Colitis. Gastroenterology 2015, 149, 445–455.e443. [Google Scholar] [CrossRef] [Green Version]
- Lomasney, K.W.; Houston, A.; Shanahan, F.; Dinan, T.G.; Cryan, J.F.; Hyland, N.P. Selective influence of host microbiota on cAMP-mediated ion transport in mouse colon. Neurogastroenterol. Motil. 2014, 26, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Van Landeghem, L.; Chevalier, J.; Mahe, M.M.; Wedel, T.; Urvil, P.; Derkinderen, P.; Savidge, T.; Neunlist, M. Enteric glia promote intestinal mucosal healing via activation of focal adhesion kinase and release of proEGF. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G976–G987. [Google Scholar] [CrossRef] [Green Version]
- Mulligan, L.M. GDNF and the RET Receptor in Cancer: New Insights and Therapeutic Potential. Front. Physiol. 2018, 9, 1873. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.M.; Bocci, G.; Di Desidero, T.; Ruffilli, I.; Elia, G.; Ragusa, F.; Fioravanti, A.; Orlandi, P.; Paparo, S.R.; Patrizio, A.; et al. Vandetanib has antineoplastic activity in anaplastic thyroid cancer, in vitro and in vivo. Oncol. Rep. 2018, 39, 2306–2314. [Google Scholar] [CrossRef]
- Spindler, V.; Meir, M.; Vigh, B.; Flemming, S.; Hutz, K.; Germer, C.T.; Waschke, J.; Schlegel, N. Loss of Desmoglein 2 Contributes to the Pathogenesis of Crohn’s Disease. Inflamm. Bowel Dis. 2015. [Google Scholar] [CrossRef]
- Rodrigues, D.M.; Li, A.Y.; Nair, D.G.; Blennerhassett, M.G. Glial cell line-derived neurotrophic factor is a key neurotrophin in the postnatal enteric nervous system. Neurogastroenterol. Motil. 2011, 23, e44–e56. [Google Scholar] [CrossRef] [PubMed]
- von Boyen, G.B.; Steinkamp, M.; Geerling, I.; Reinshagen, M.; Schafer, K.H.; Adler, G.; Kirsch, J. Proinflammatory cytokines induce neurotrophic factor expression in enteric glia: A key to the regulation of epithelial apoptosis in Crohn’s disease. Inflamm. Bowel Dis. 2006, 12, 346–354. [Google Scholar] [CrossRef]
- Rosenbaum, C.; Schick, M.A.; Wollborn, J.; Heider, A.; Scholz, C.J.; Cecil, A.; Niesler, B.; Hirrlinger, J.; Walles, H.; Metzger, M. Activation of Myenteric Glia during Acute Inflammation In Vitro and In Vivo. PLoS ONE 2016, 11, e0151335. [Google Scholar] [CrossRef]
- Meir, M.; Salm, J.; Fey, C.; Schweinlin, M.; Kollmann, C.; Kannapin, F.; Germer, C.T.; Waschke, J.; Beck, C.; Burkard, N.; et al. Enteroids generated from patients with severe inflammation in Crohn’s disease maintain alterations of junctional proteins. J. Crohn’s Colitis 2020. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.; Leven, P.; Glowka, T.; Kuzmanov, I.; Lysson, M.; Schneiker, B.; Miesen, A.; Baqi, Y.; Spanier, C.; Grants, I.; et al. A novel P2X2-dependent purinergic mechanism of enteric gliosis in intestinal inflammation. EMBO Mol. Med. 2020, e12724. [Google Scholar] [CrossRef]
- Liu, T.Y.; Yang, X.Y.; Zheng, L.T.; Wang, G.H.; Zhen, X.C. Activation of Nur77 in microglia attenuates proinflammatory mediators production and protects dopaminergic neurons from inflammation-induced cell death. J. Neurochem. 2017, 140, 589–604. [Google Scholar] [CrossRef]
- Schwandner, R.; Dziarski, R.; Wesche, H.; Rothe, M.; Kirschning, C.J. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 1999, 274, 17406–17409. [Google Scholar] [CrossRef] [Green Version]
- Gross, A.; Pack, L.A.P.; Schacht, G.M.; Kant, S.; Ungewiss, H.; Meir, M.; Schlegel, N.; Preisinger, C.; Boor, P.; Guldiken, N.; et al. Desmoglein 2, but not desmocollin 2, protects intestinal epithelia from injury. Mucosal. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungewiss, H.; Vielmuth, F.; Suzuki, S.T.; Maiser, A.; Harz, H.; Leonhardt, H.; Kugelmann, D.; Schlegel, N.; Waschke, J. Desmoglein 2 regulates the intestinal epithelial barrier via p38 mitogen-activated protein kinase. Sci. Rep. 2017, 7, 6329. [Google Scholar] [CrossRef]
- Labun, K.; Montague, T.G.; Gagnon, J.A.; Thyme, S.B.; Valen, E. CHOPCHOP v2: A web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 2016, 44, W272–W276. [Google Scholar] [CrossRef] [PubMed]
- Flemming, S.; Burkard, N.; Renschler, M.; Vielmuth, F.; Meir, M.; Schick, M.A.; Wunder, C.; Germer, C.T.; Spindler, V.; Waschke, J.; et al. Soluble VE-cadherin is involved in endothelial barrier breakdown in systemic inflammation and sepsis. Cardiovasc. Res. 2015, 107, 32–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence |
|---|---|
| mSox10_fw | GGACTACAAGTACCAACCTCGG |
| mSox10_rv | GGACTGCAGCTCTGTCTTTGG |
| mGFAP_fw | ACATCGAGATCGCCACCTAC |
| mGFAP_rv | CCTTCTGACACGGATTTGGT |
| mGDNF_fw | CAGTGACTCCAATATGCCTGA |
| mGDNF_rv | CCGCTTGTTTATCTGGTGAC |
| Primer Name | Sequence |
|---|---|
| rat-ccGDNF 1f | CACCGTTCGAGAAGCGTCTTACCGG |
| rat-ccGDNF 1r | AAACCCGGTAAGACGCTTCTCGAAC |
| rat-ccGDNF 2f | CACCGTCACCAGATAAACAAGCGG |
| rat-ccGDNF 2r | AAACCCGCTTGTTTATCTGGTGAC |
| qPCR Primer | Sequence |
| rat-GDNF forward | AAGAGAGAGGAACCGGCAAG |
| rat-GDNF reverse | CGACCTTTCCCTCTGGAAT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meir, M.; Kannapin, F.; Diefenbacher, M.; Ghoreishi, Y.; Kollmann, C.; Flemming, S.; Germer, C.-T.; Waschke, J.; Leven, P.; Schneider, R.; et al. Intestinal Epithelial Barrier Maturation by Enteric Glial Cells Is GDNF-Dependent. Int. J. Mol. Sci. 2021, 22, 1887. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041887
Meir M, Kannapin F, Diefenbacher M, Ghoreishi Y, Kollmann C, Flemming S, Germer C-T, Waschke J, Leven P, Schneider R, et al. Intestinal Epithelial Barrier Maturation by Enteric Glial Cells Is GDNF-Dependent. International Journal of Molecular Sciences. 2021; 22(4):1887. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041887
Chicago/Turabian StyleMeir, Michael, Felix Kannapin, Markus Diefenbacher, Yalda Ghoreishi, Catherine Kollmann, Sven Flemming, Christoph-Thomas Germer, Jens Waschke, Patrick Leven, Reiner Schneider, and et al. 2021. "Intestinal Epithelial Barrier Maturation by Enteric Glial Cells Is GDNF-Dependent" International Journal of Molecular Sciences 22, no. 4: 1887. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041887