
Probing Protein Folding with Sequence-Reversed α-Helical Bundles

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
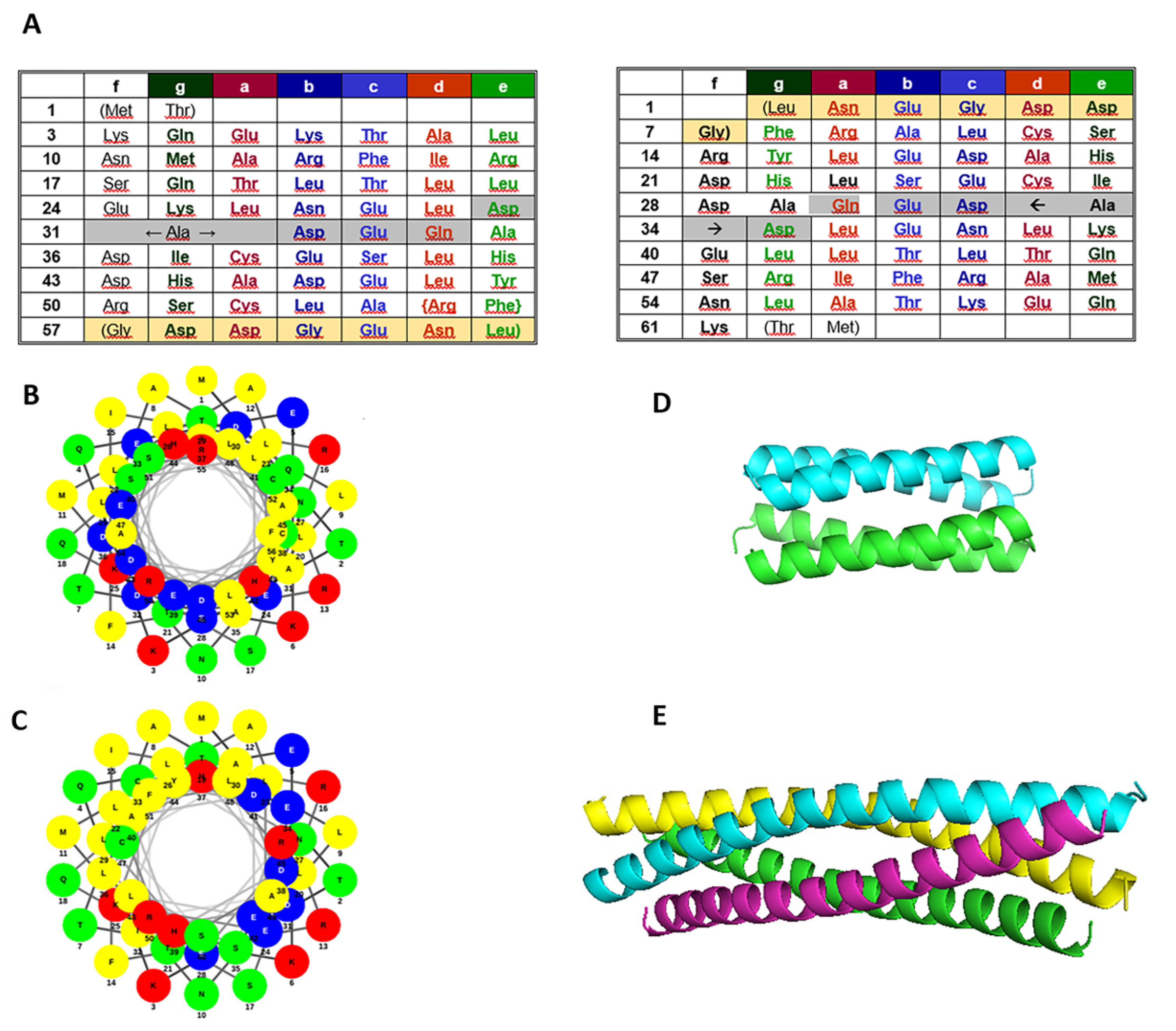
2.1. Sequence Analysis
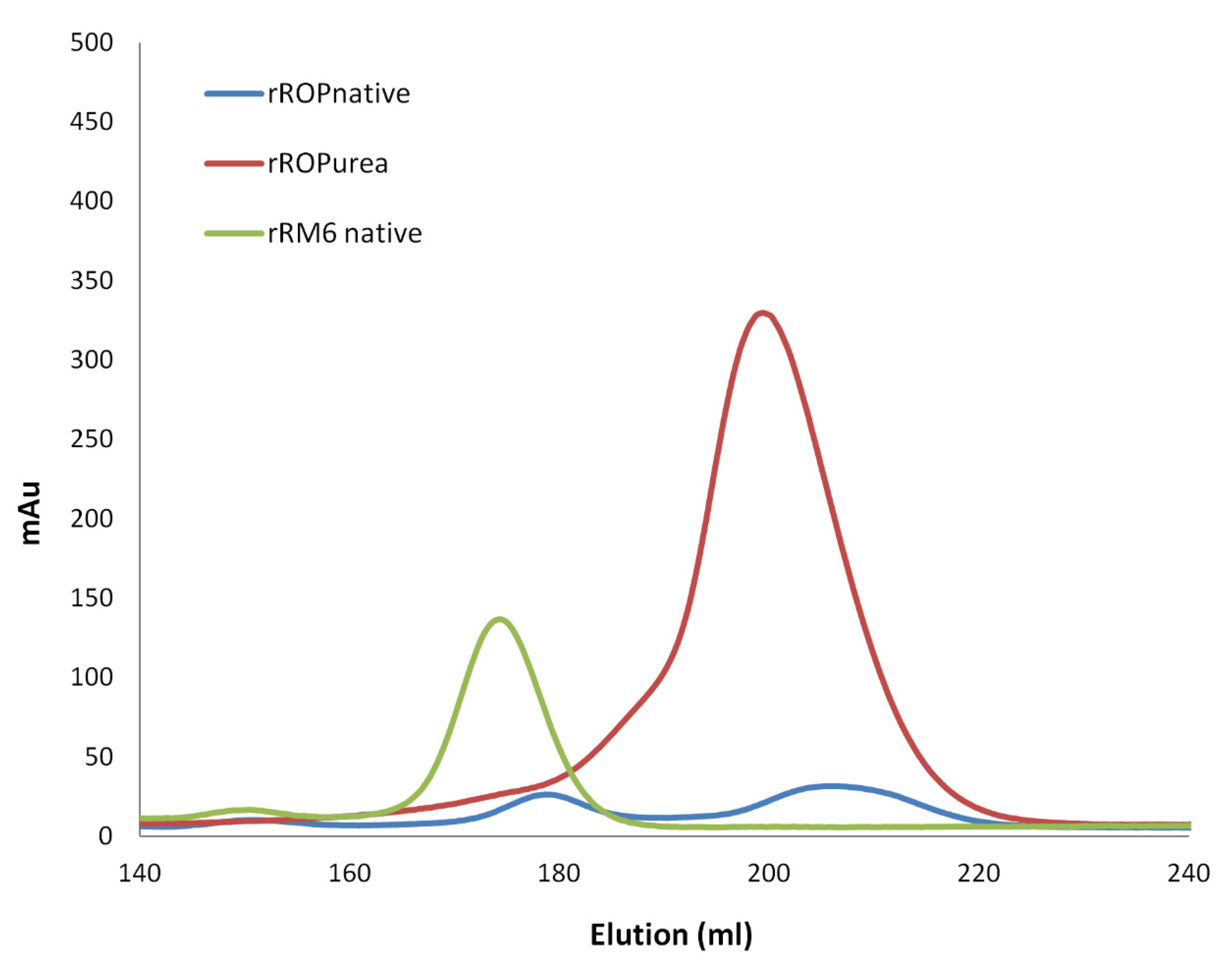
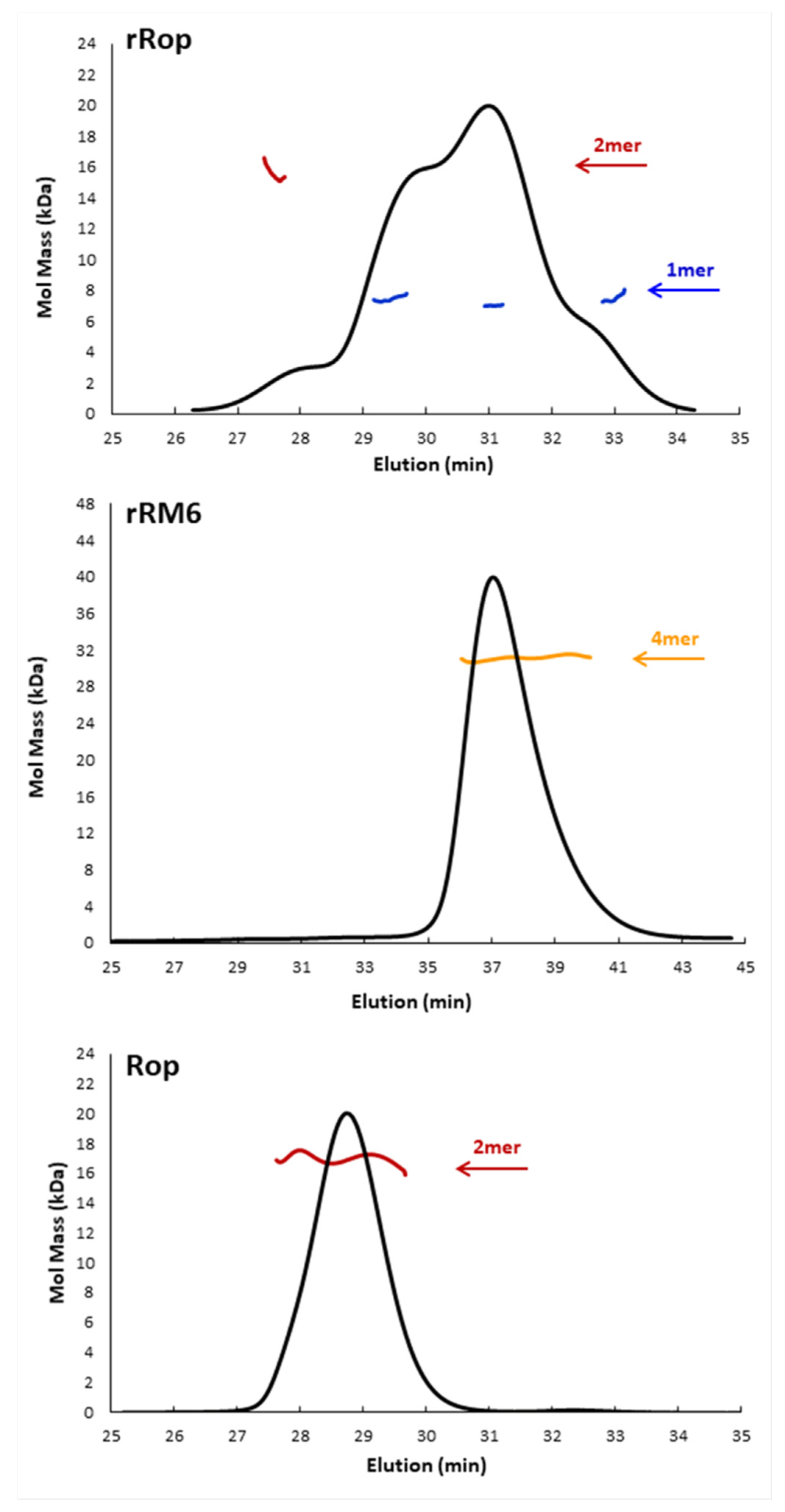
2.2. Sequence Reversal Affects Differently the Oligomerization Propensities of the Retro-Molecules
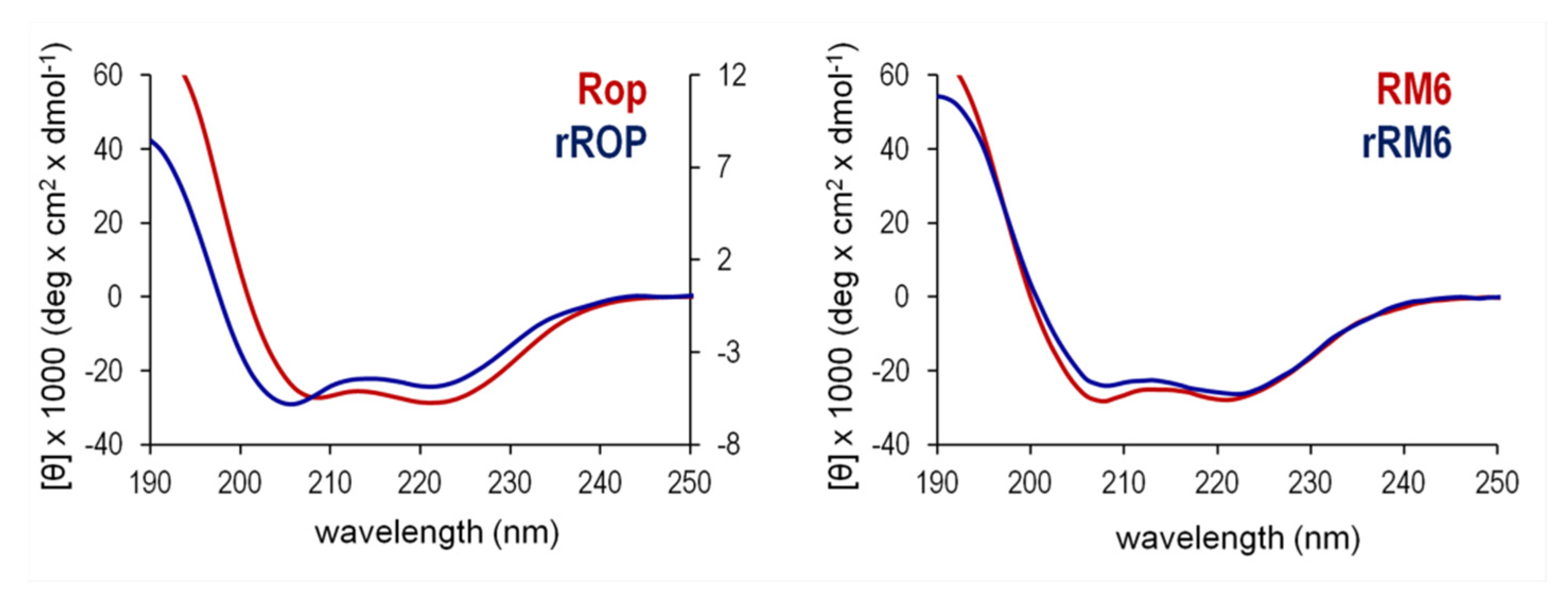
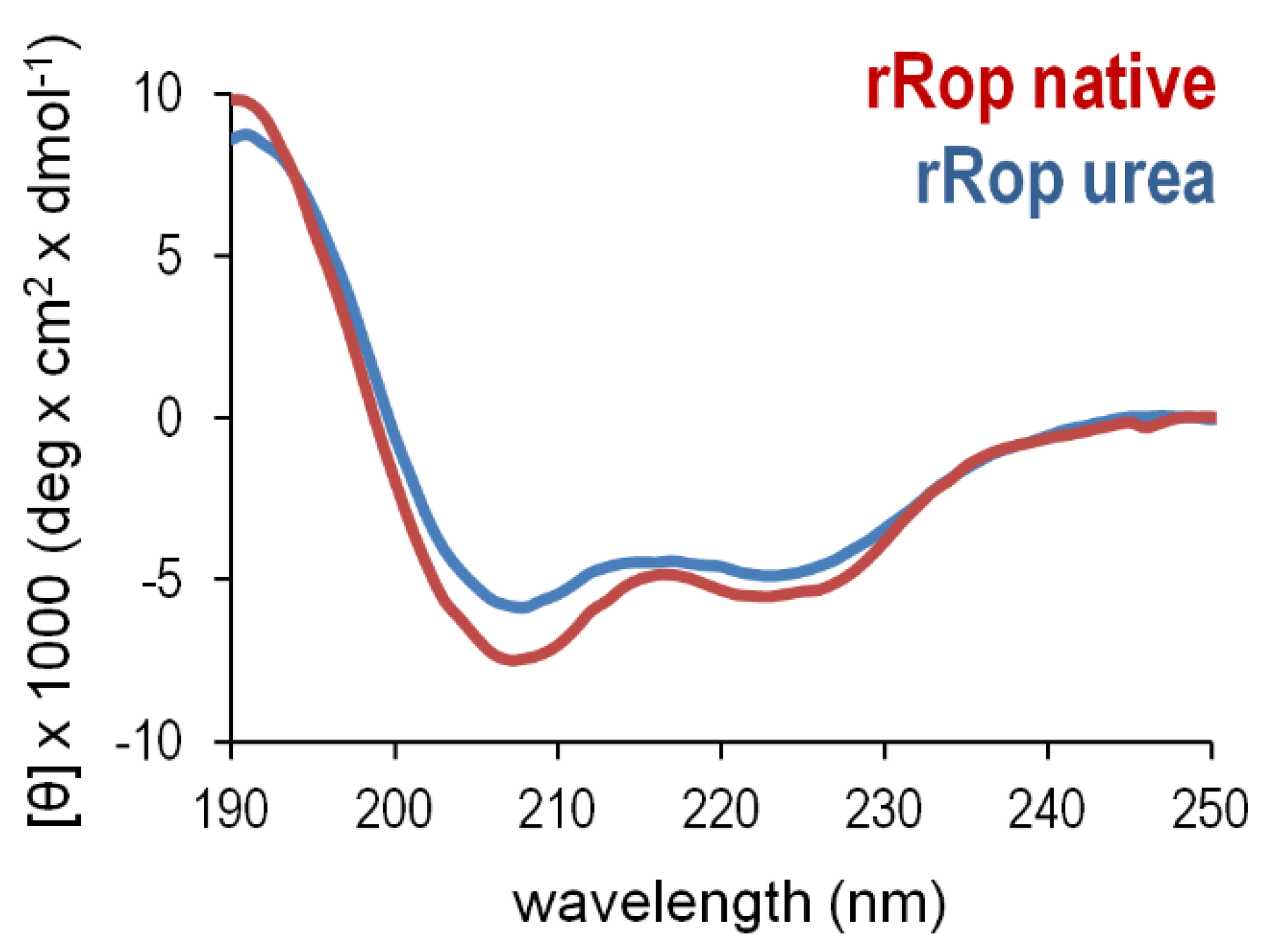
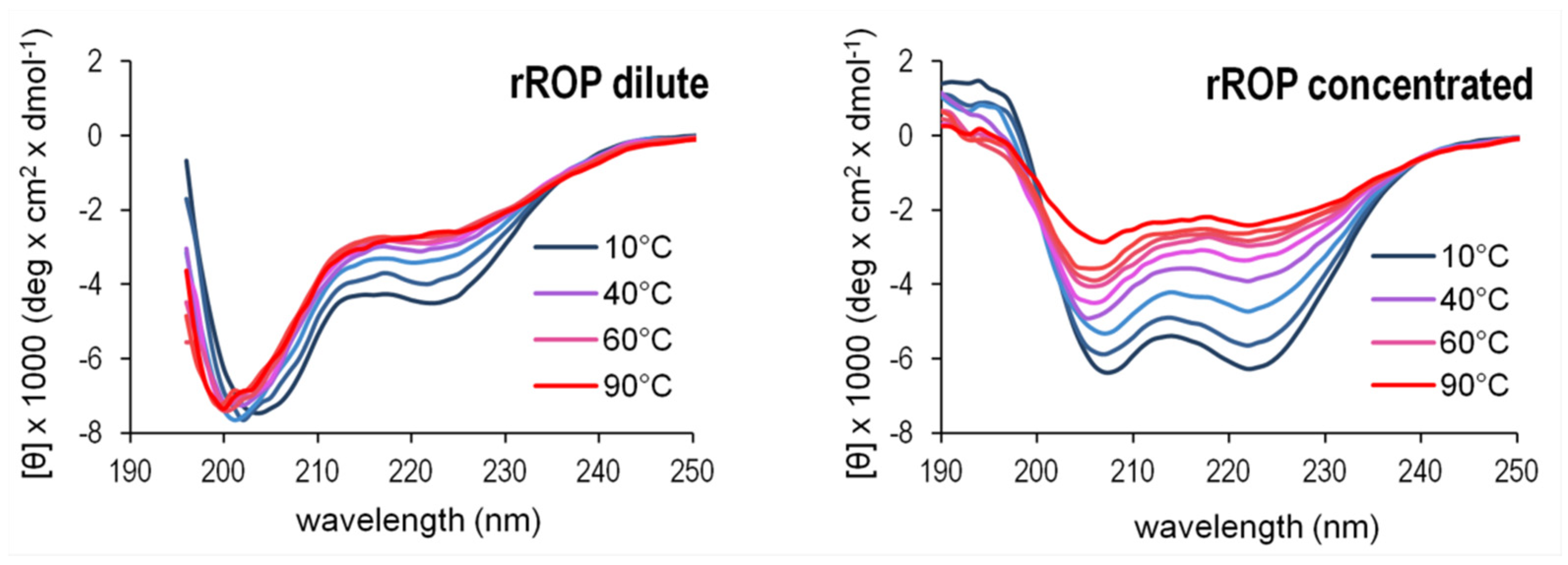
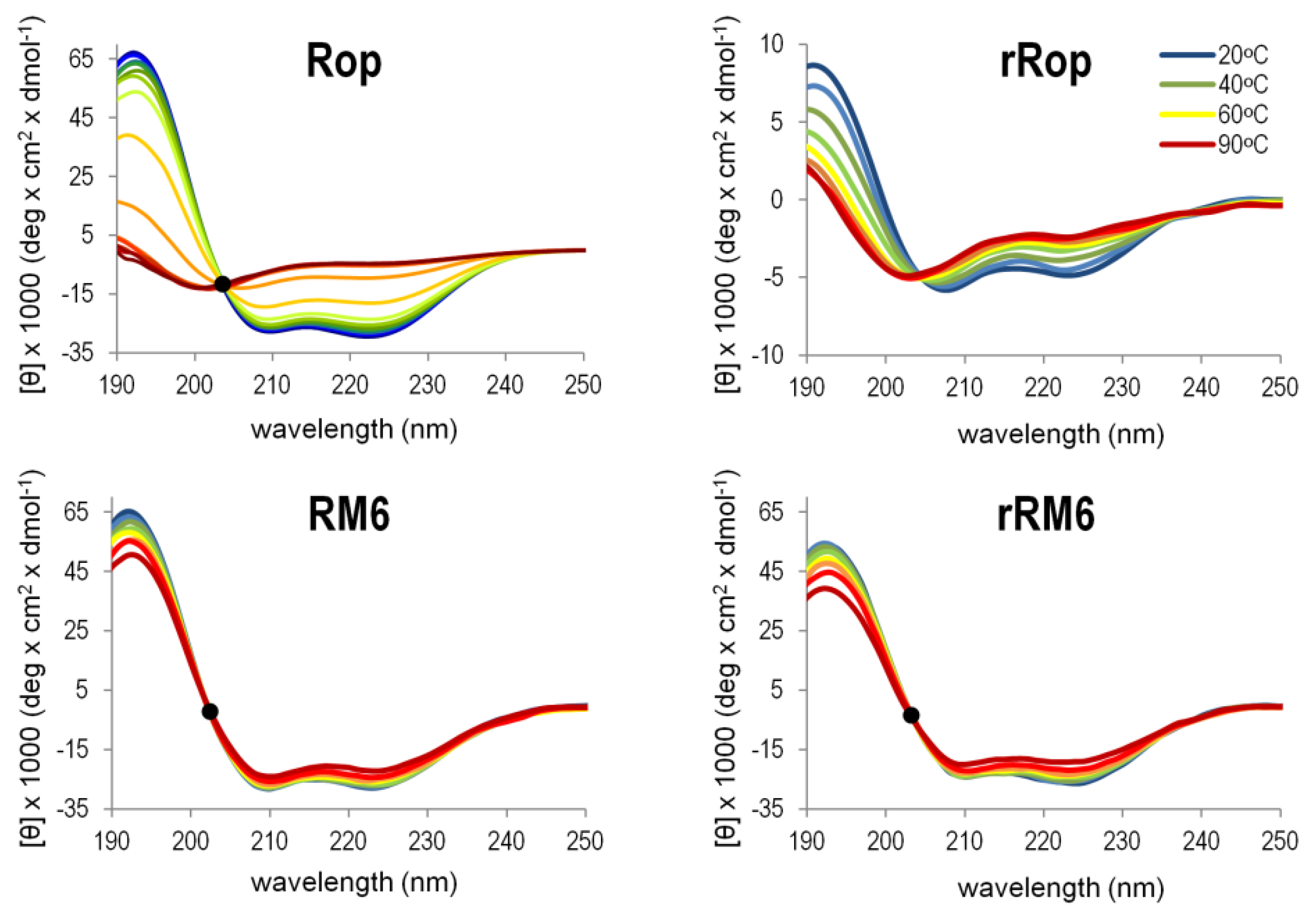
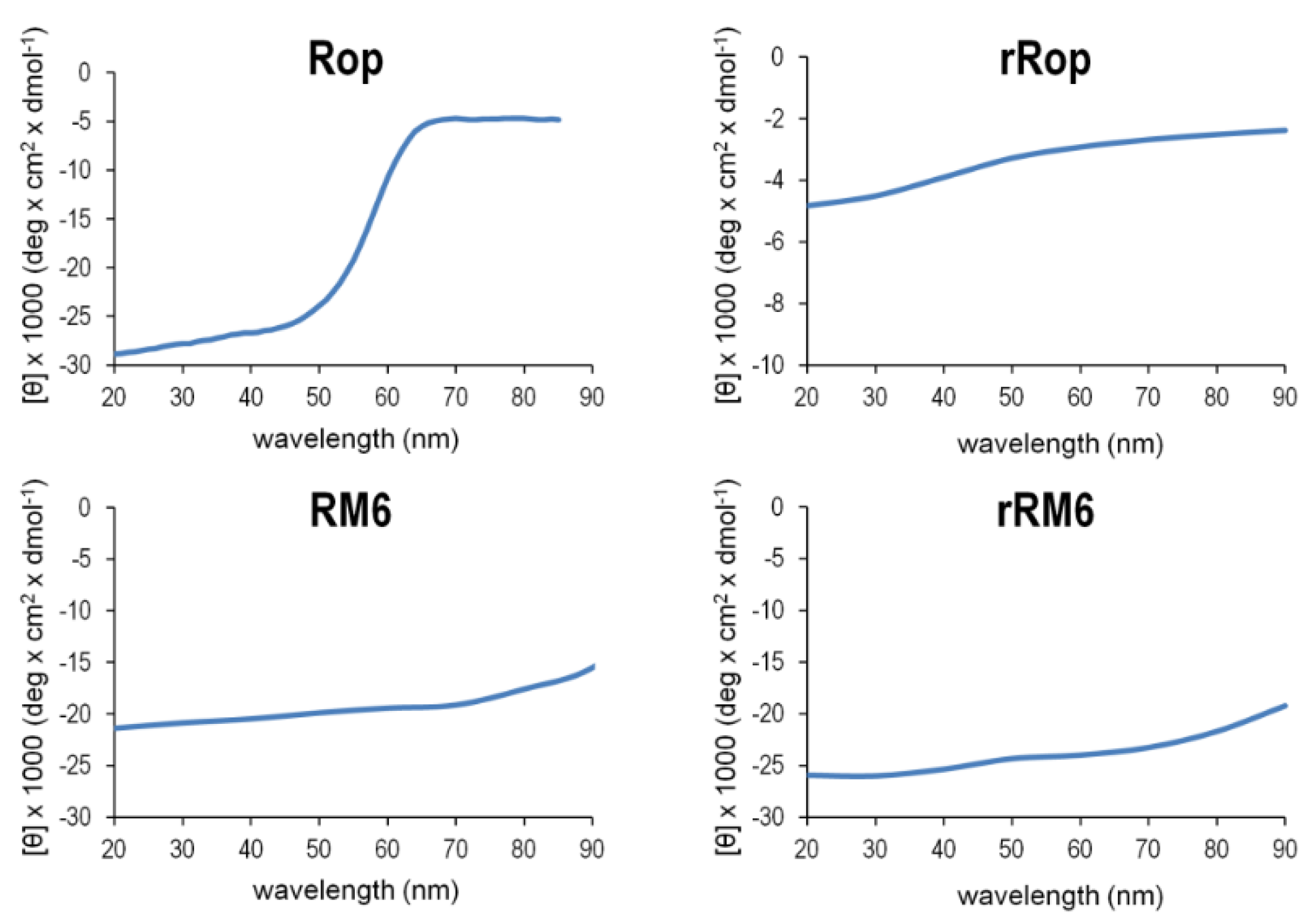
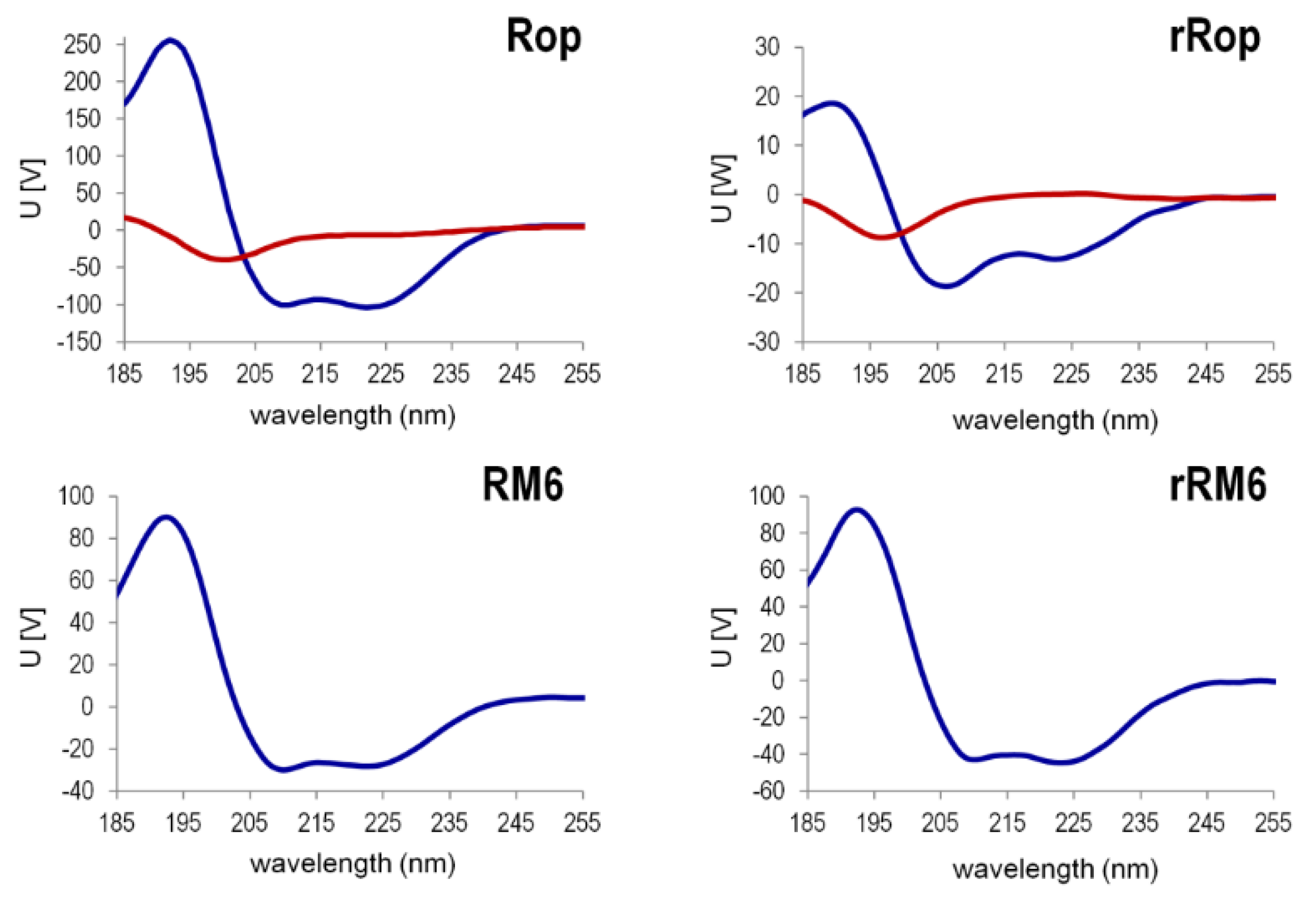
2.3. Sequence Reversal Affects Differently the α-Helical Content, α-Helix Association, and the Stability of the Two Retro-Proteins, While Maintaining Their Overall α-Helical Character
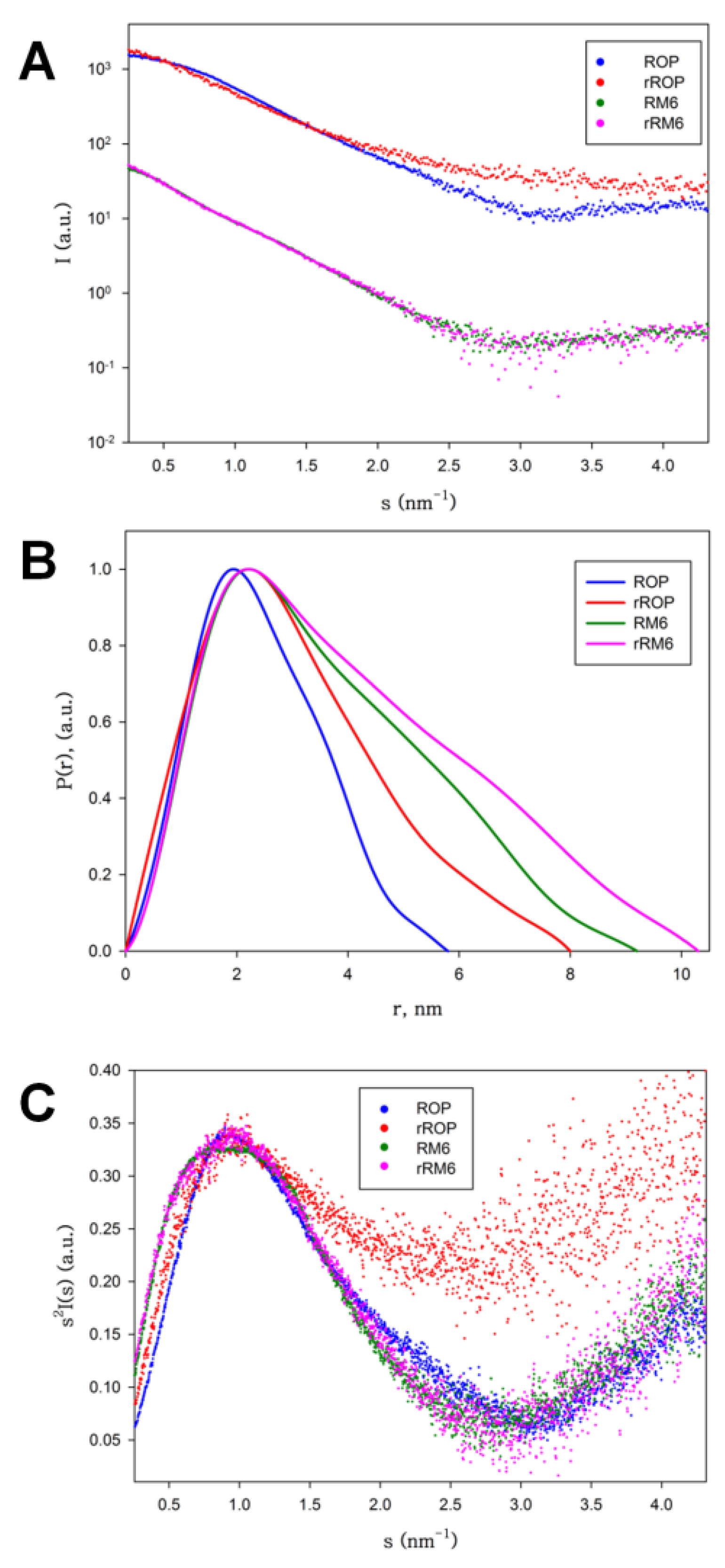
2.4. SAXS Experiments Reveal the Shapes and Folding States of the Retro Proteins
3. Discussion
3.1. The Retro Protein Sequences
3.2. Chromatographic Behavior and Oligomerization Propensities
3.3. Secondary Structure, Stability, and Folding States
3.4. Helix Dipoles
3.5. Sequence Reversal Affects Differently the Folding State of Each Retro Protein
4. Materials and Methods
4.1. Sequence Alignment
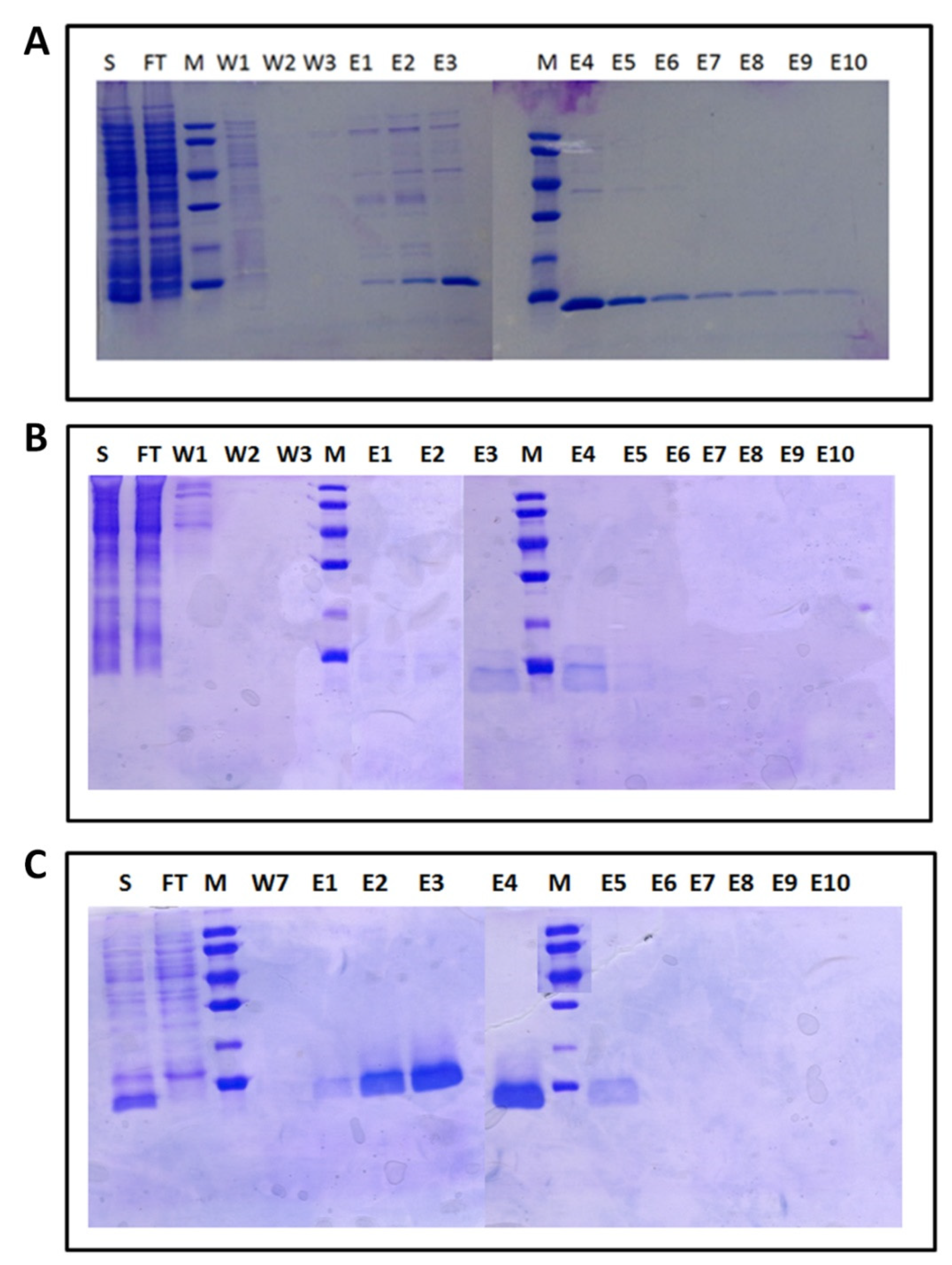
4.2. Synthesis, Expression, and Purification
4.3. SEC-MALS Analysis
4.4. Circular Dichroism Measurements
4.5. SAXS Measurements
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MALS | Multiangle Laser Light Scattering |
| MRE | Mean Residue Ellipticity |
| SAXS | Small Angle X-ray Scattering |
| SEC | Size Exclusion Chromatography |
| CD | Circular Dichroism |
| PDB | Protein Data Bank |
References
- John, M.; Leppik, R.; Busch, S.J.; Granger-Schnarr, M.; Schnarr, M. DNA binding of Jun and Fos bZip domains: Homodimers and heterodimers induce a DNA conformational change in solution. Nucleic Acids Res. 1996, 24, 4487–4494. [Google Scholar] [CrossRef] [Green Version]
- Lupas, A.N.; Bassler, J.; Dunin-Horkawicz, S. The Structure and Topology of alpha-Helical Coiled Coils. Subcell Biochem. 2017, 82, 95–129. [Google Scholar]
- Parry, D.A.; Fraser, R.D.; Squire, J.M. Fifty years of coiled-coils and alpha-helical bundles: A close relationship between sequence and structure. J. Struct. Biol. 2008, 163, 258–269. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.; Kim, P.S.; Berger, B. MultiCoil: A program for predicting two- and three-stranded coiled coils. Protein Sci. 1997, 6, 1179–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazi, A.D.; Bastaki, M.; Charova, S.N.; Gkougkoulia, E.A.; Kapellios, E.A.; Panopoulos, N.J.; Kokkinidis, M. Evidence for a coiled-coil interaction mode of disordered proteins from bacterial type III secretion systems. J. Biol. Chem. 2008, 283, 34062–34068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, A.L.; Woolfson, D.N. De novo designed peptides for biological applications. Chem. Soc. Rev. 2011, 40, 4295–4306. [Google Scholar] [CrossRef]
- Khoury, G.A.; Smadbeck, J.; Kieslich, C.A.; Floudas, C.A. Protein folding and de novo protein design for biotechnological applications. Trends Biotechnol. 2013, 32, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paliakasis, C.D.; Kokkinidis, M. Relationships between sequence and structure for the four-alpha-helix bundle tertiary motif in proteins. Protein Eng. 1992, 5, 739–748. [Google Scholar] [CrossRef]
- Crick, F.H.C. The packing of α-helices: Simple coiled-coils. Acta Cryst. 1953, 6, 689–697. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, E.K.; Klemm, J.D.; Kim, P.S.; Alber, T. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science 1991, 254, 539–544. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, E.K.; Rutkowski, R.; Kim, P.S. Evidence that the leucine zipper is a coiled coil. Science 1989, 243, 538–542. [Google Scholar] [CrossRef] [Green Version]
- Amprazi, M.; Kotsifaki, D.; Providaki, M.; Kapetaniou, E.G.; Fellas, G.; Kyriazidis, I.; Pérez, J.; Kokkinidis, M. Structural plasticity of 4-alpha-helical bundles exemplified by the puzzle-like molecular assembly of the Rop protein. Proc. Natl. Acad. Sci. USA 2014, 111, 11049–11054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacroix, E.; Viguera, A.R.; Serrano, L. Reading protein sequences backwards. Fold. Des. 1998, 3, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. What does it mean to be natively unfolded? Eur. J. Biochem. 2002, 269, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Shukla, A.; Guptasarma, P. Folding behavior of a backbone-reversed protein: Reversible polyproline type II to beta-sheet thermal transitions in retro-GroES multimers with GroES-like features. Biochim. Biophys. Acta 2008, 1784, 916–923. [Google Scholar] [CrossRef] [PubMed]
- Kutyshenko, V.P.; Prokhorov, D.A.; Molochkov, N.V.; Sharapov, M.G.; Kolesnikov, I.; Uversky, V.N. Dancing retro: Solution structure and micelle interactions of the retro-SH3-domain, retro-SHH-‘Bergerac’. J. Biomol. Struct. Dyn. 2014, 32, 257–272. [Google Scholar] [CrossRef]
- Mittl, P.R.; Deillon, C.; Sargent, D.; Liu, N.; Klauser, S.; Thoman, R.M.; Gutte, B.; Grütter, M.G. The retro-GCN4 leucine zipper sequence forms a stable three-dimensional structure. Proc. Natl. Acad. Sci. USA. 2000, 97, 2562–2566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamtekar, S.; Hecht, M.H. Protein Motifs. 7. The four-helix bundle: What determines a fold? FASEB J. 1995, 9, 1013–1022. [Google Scholar] [CrossRef]
- Ambrazi, M.; Fellas, G.; Kapetaniou, E.G.; Kotsifaki, D.; Providaki, M.; Kokkinidis, M. Purification, crystallization and preliminary X-ray diffraction analysis of a variant of the ColE1 Rop protein. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2008, 64, 432–434. [Google Scholar] [CrossRef] [Green Version]
- Banner, D.W.; Kokkinidis, M.; Tsernoglou, D. Structure of the ColE1 rop protein at 1.7 A resolution. J. Mol. Biol. 1987, 196, 657–675. [Google Scholar] [CrossRef]
- Itoh, T.; Tomizawa, J. Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proc. Natl. Acad. Sci. USA 1980, 77, 2450–2454. [Google Scholar] [CrossRef] [Green Version]
- Polisky, B. ColE1 replication control circuitry: Sense from antisense. Cell 1988, 55, 929–932. [Google Scholar] [CrossRef]
- Tomizawa, J. Control of ColE1 plasmid replication: The process of binding of RNA I to the primer transcript. Cell 1984, 38, 861–870. [Google Scholar] [CrossRef]
- Predki, P.F.; Regan, L. Redesigning the topology of a four-helix-bundle protein: Monomeric Rop. Biochemistry 1995, 34, 9834–9839. [Google Scholar] [CrossRef]
- Glykos, N.M.; Papanikolau, Y.; Vlassi, M.; Kotsifaki, D.; Cesareni, G.; Kokkinidis, M. Loopless Rop: Structure and dynamics of an engineered homotetrameric variant of the repressor of primer protein. Biochemistry 2006, 45, 10905–10919. [Google Scholar] [CrossRef]
- Kefala, A.; Kotsifaki, D.; Providaki, D.; Amprazi, M.; Kokkinidis, M. Expression, purification and crystallization of a protein resulting from the inversion of the amino-acid sequence of a helical bundle. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2017, 73, 51–53. [Google Scholar] [CrossRef] [Green Version]
- Divan, A.; Royds, J. Tools and Techniques in Biomolecular Science; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Micsonai, A.; Wien, F.; Bulyáki, É.; Kun, J.; Moussong, É.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.E.; Kay, C.M.; Hodges, R.S. Synthetic model proteins. Positional effects of interchain hydrophobic interactions on stability of two-stranded alpha-helical coiled-coils. J. Biol. Chem. 1992, 267, 2664–2670. [Google Scholar] [CrossRef]
- Manavalan, P.; Johnson, W.C.J. Variable selection method improves the prediction of protein secondary structure from circular dichroism spectra. Anal. Biochem. 1987, 167, 76–85. [Google Scholar] [CrossRef]
- Sridhar, S.; Nagamruta, M.; Guruprasad, K. Analyses of the Sequence and Structural Properties Corresponding to Pentapeptide and Large Palindromes in Proteins. PLoS ONE 2015, 10, e0139568. [Google Scholar] [CrossRef]
- Vieille, C.; Zeikus, G.J. Hyperthermophilic enzymes: Sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev. 2001, 65, 1. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Deillon, C.; Klauser, S.; Gutte, B.; Thomas, R.M. Synthesis, physicochemical characterization, and crystallization of a putative retro-coiled coil. Protein Sci. 1998, 7, 1214–1220. [Google Scholar] [CrossRef] [Green Version]
- White, S.J.; Johnson, S.D.; Sellick, M.A.; Bronowska, A.; Stockley, P.G.; Wälti, C. The influence of two-dimensional organization on peptide conformation. Angew. Chem. Int. Ed. Engl. 2015, 54, 974–978. [Google Scholar] [CrossRef] [Green Version]
- Sala, F.A.; Valadares, N.F.; Macedo, J.N.A.; Borges, J.C.; Garratt, R.C. Heterotypic Coiled-Coil Formation is Essential for the Correct Assembly of the Septin Heterofilament. Biophys J. 2016, 111, 2608–2619. [Google Scholar] [CrossRef] [Green Version]
- Hol, W.G.; Halie, L.M.; Sander, C. Dipoles of the alpha-helix and beta-sheet: Their role in protein folding. Nature 1981, 294, 532–536. [Google Scholar] [CrossRef]
- Park, C.; Goddard, W.A. Stabilization of α-Helices by Dipole−Dipole Interactions within α-Helices. J. Phys. Chem. B 2000, 104, 7784–7789. [Google Scholar] [CrossRef]
- Perczel, A.; Hollósi, M.; Tusnády, G.; Fasman, G.D. Convex constraint analysis: A natural deconvolution of circular dichroism curves of proteins. Protein Eng. 1991, 4, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Presta, L.G.; Rose, G.D. Helix signals in proteins. Science 1988, 240, 1632–1641. [Google Scholar] [CrossRef]
- Gilson, M.K.; Honig, B. Destabilization of an alpha-helix-bundle protein by helix dipoles. Proc. Natl. Acad. Sci. USA 1989, 86, 1524–1528. [Google Scholar] [CrossRef] [Green Version]
- Kutyshenko, V.P.; Mikoulinskaia, G.V.; Molochkov, N.V.; Prokhorov, D.A.; Taran, S.; Uversky, V.N. Structure and dynamics of the retro-form of the bacteriophage T5 endolysin. Biochim. Biophys. Acta 2016, 1864, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Ruvo, M.; Fassina, G. Folding behaviour of retro-rubredoxin. Protein Pept. Lett. 1996, 3, 241–246. [Google Scholar]
- Shukla, A.; Raje, M.; Guptasarma, P. A backbone-reversed all-beta polypeptide (retro-CspA) folds and assembles into amyloid nanofibres. Protein Eng. 2003, 16, 875–879. [Google Scholar] [CrossRef] [Green Version]
- Holtzer, M.E.; Holtzer, A. Alpha-helix to random coil transitions: Interpretation of the CD in the region of linear temperature dependence. Biopolymers 1992, 32, 1589–1591. [Google Scholar] [CrossRef]
- Kuwajima, K. The Molten Globule, and Two-State vs. Non-Two-State Folding of Globular Proteins. Biomolecules 2020, 10, 407. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Weber, J.K.; Zhou, R. Folding and Stabilization of Native-Sequence-Reversed Proteins. Sci. Rep. 2016, 6, 25138. [Google Scholar] [CrossRef] [Green Version]
- Glykos, N.M.; Kokkinidis, M. Structural polymorphism of a marginally stable 4-α-helical bundle. Images of a trapped molten globule? Proteins 2004, 56, 420–425. [Google Scholar] [CrossRef]
- Kamtekar, S.; Schiffer, J.M.; Xiong, H.; Babik, J.M.; Hecht, M.H. Protein design by binary patterning of polar and nonpolar amino acids. Science 1993, 262, 1680–1685. [Google Scholar] [CrossRef]
- Konno, T. Conformational diversity of acid-denatured cytochrome c studied by a matrix analysis of far-UV CD spectra. Protein Sci. 1998, 7, 975–982. [Google Scholar] [CrossRef] [Green Version]
- Blanchet, C.E.; Spilotros, A.; Schwemmer, F.; Graewert, M.A.; Klikhney, A.; Jeffries, C.M.; Franke, D.; Mark, D.; Zengerle, R.; Cipriani, F.; et al. Versatile sample environments and automation for biological solution X-ray scattering experiments at the P12 beamline (PETRA III, DESY). J. Appl. Crystallogr. 2015, 48, 431–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, D.; Petroukhov, M.V.; Konarev, P.V.; Panjkovich, A.; Tuukkanen, A.; Mertens, H.D.T.; Kikhney, A.G.; Hajizadeh, N.R.; Franklin, J.M.; Jeffries, C.M.; et al. ATSAS 2.8: A comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017, 50, 1212–1225. [Google Scholar] [CrossRef] [Green Version]
- Konarev, P.V.; Volkov, V.V.; Sokolova, A.V.; Koch, M.H.J.; Svergun, D.I. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Guinier, A. La diffraction des rayons X aux très petits angles: Application à l’étude de phénomènes ultramicroscopiques. Ann. Phys. 2017, 11, 161–237. [Google Scholar] [CrossRef]
- Svergun, D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Durand, D.; Vivès, C.; Cannella, D.; Pérez, J.; Pebay-Peyroula, E.; Vachette, P.; Fieschi, F. NADPH oxidase activator p67(phox) behaves in solution as a multidomain protein with semi-flexible linkers. J. Struct. Biol. 2010, 169, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Silverman, B.D. Hydrophobic moments of protein structures: Spatially profiling the distribution. Proc. Natl. Acad. Sci. USA 2001, 98, 4996–5001. [Google Scholar] [CrossRef] [Green Version]
- Guptasarma, P. Reversal of peptide backbone direction may result in the mirroring of protein structure. FEBS Lett. 1992, 310, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Olszewski, K.A.; Kolinski, A.; Skolnick, J. Does a backwardly read protein sequence have a unique native state? Protein Eng. 1996, 9, 5–14. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kefala, A.; Amprazi, M.; Mylonas, E.; Kotsifaki, D.; Providaki, M.; Pozidis, C.; Fotiadou, M.; Kokkinidis, M. Probing Protein Folding with Sequence-Reversed α-Helical Bundles. Int. J. Mol. Sci. 2021, 22, 1955. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041955
Kefala A, Amprazi M, Mylonas E, Kotsifaki D, Providaki M, Pozidis C, Fotiadou M, Kokkinidis M. Probing Protein Folding with Sequence-Reversed α-Helical Bundles. International Journal of Molecular Sciences. 2021; 22(4):1955. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041955
Chicago/Turabian StyleKefala, Aikaterini, Maria Amprazi, Efstratios Mylonas, Dina Kotsifaki, Mary Providaki, Charalambos Pozidis, Melina Fotiadou, and Michael Kokkinidis. 2021. "Probing Protein Folding with Sequence-Reversed α-Helical Bundles" International Journal of Molecular Sciences 22, no. 4: 1955. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041955