
Homocysteine in Neurology: A Possible Contributing Factor to Small Vessel Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
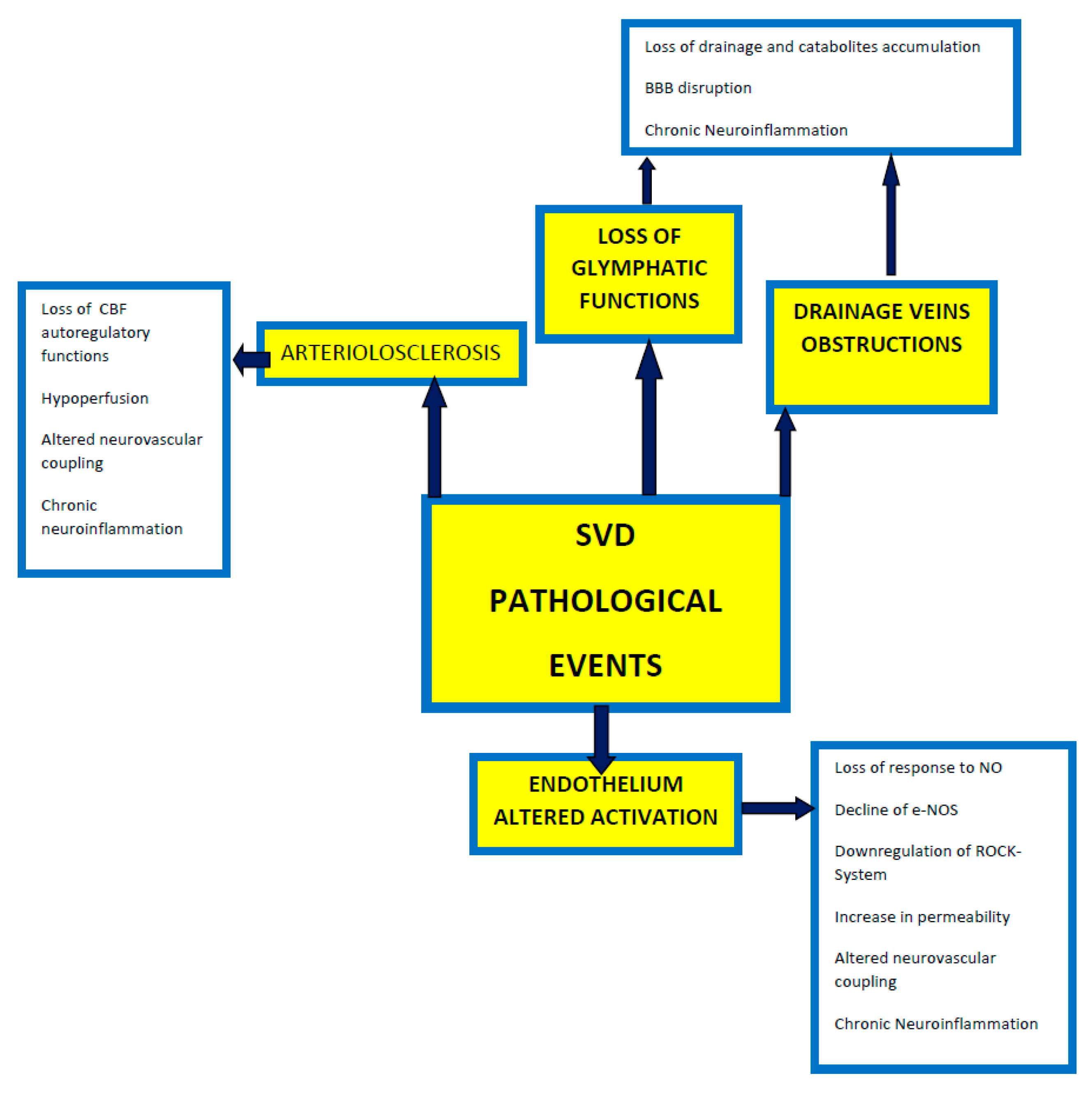
2. Cerebral Small Vessel Disease
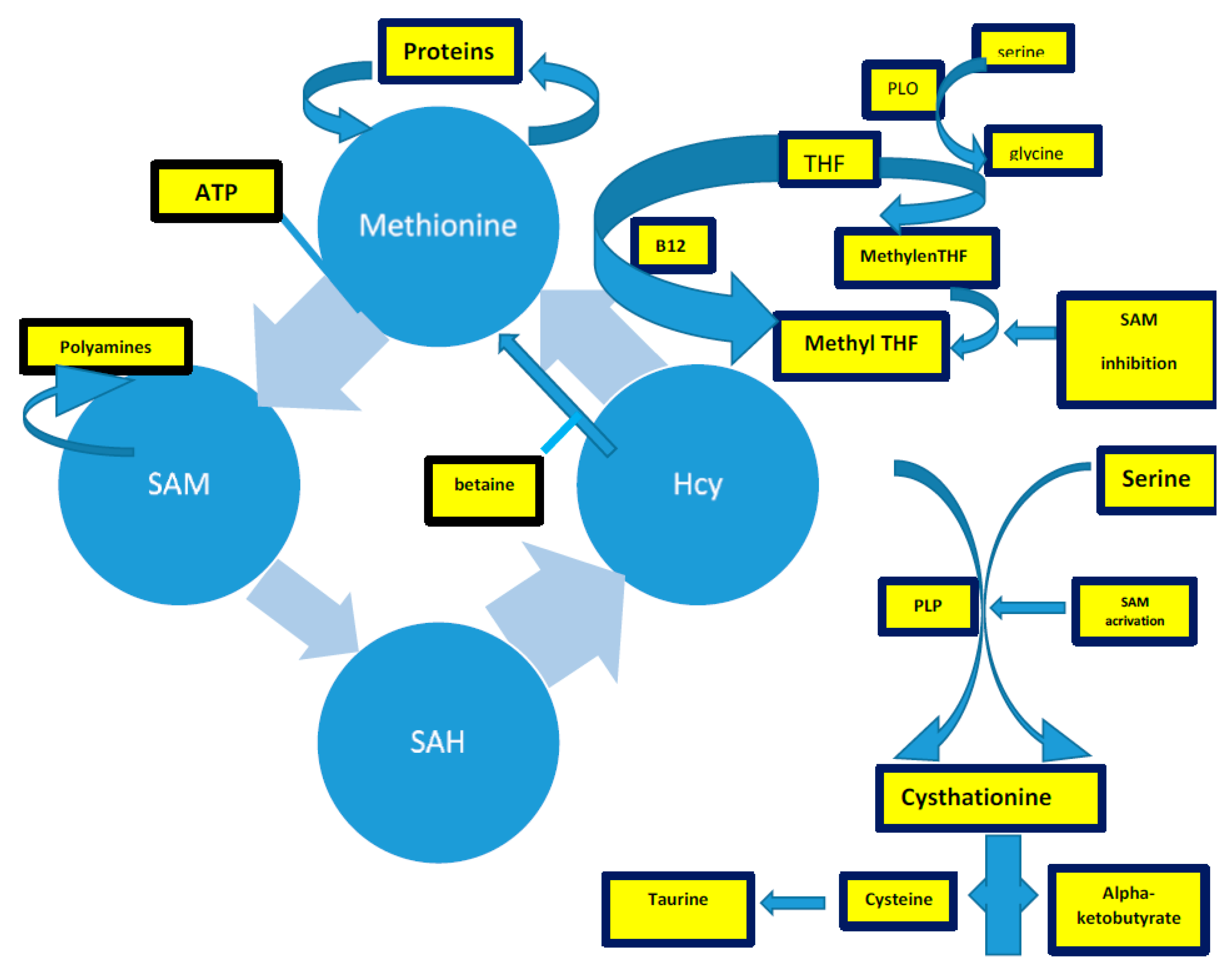
3. Homocysteine and Brain
4. Homocysteine and Neurodegeneration
5. Homocysteine and Neuroinflammation
6. Homocysteine and Oxidative Stress
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5-methylTHF | 5:10-methylenetetrahydrofolate to 5-methyltetrahydrofolate |
| ATP | Adenosine triphosphate |
| SAHH | AdoHcy hydrolase |
| AGEs | Advanced glycation end products |
| AD | Alzheimer’s disease |
| APP | Amyloid precursor |
| ADMA | Asymmetric dimethylarginine |
| BHMT | Betaine-homocysteine methyltransferase |
| BBB | Blood–brain barrier |
| CAA | Cerebral amyloid angiopathy |
| CBF | Cerebral blood flow |
| SVD | Cerebral small vessel disease |
| CSF | Cerebrospinal fluid |
| CRP | C-reactive protein |
| cGMP | Cyclic guanosine monophosphate |
| cGMP | Cyclic guanosine-30,5-monophosphate |
| CBS | Cystathionine β-synthase |
| CSE | Cystathionine γ-lyase |
| DDAH | Dimethylarginine dimethylaminohydrolase |
| DUSPs | Dual-specificity phosphatases |
| HERP | Endoplasmic protein |
| ER | Endoplasmic reticulum |
| eNOS | Endothelial NO synthase |
| EDHF | Endothelium-derived hyperpolarizing factors |
| ERK | Extracellular signal-regulated kinase |
| ERM | Ezrin, radixin, and moesin |
| GABA | Gamma-amino butyric |
| PQC | Global cellular protein quality control |
| Hcy | Homocysteine |
| HERP | Hcy-induced Endoplasmic Reticulum Protein |
| HHcy | hyperhomocysteinemia |
| P-tau | Hyperphosphorylated tau protein |
| HIF 1 | Hypoxia-inducible factor 1 |
| ICAM-1 | Intercellular adhesion molecule-1 |
| IL-6 | Interleukin-6 |
| LOX | Lectin-like low-density lipoprotein |
| MMP | Matrix-metalloprotease |
| MST | 3-mercaptopyruvate sulfurtransferase |
| MAT | Methionine adenosyltransferase |
| MTR | Methionine synthase |
| MetRS | Methionyl-tRNA synthase |
| MT | Methyltransferase |
| PPM1 and PPM2A | Methyltransferase systems |
| MAP | Mitogen-activated protein kinases |
| MLCP | Myosin light-chain phosphatase |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate Hydrogen |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| NMDA | N-methyl D-Aspartate |
| SID | N-[(1,1-dimethyl ethoxy)carbonyl]-L-tryptophan-2-[[[2-[(2- ethyl phenyl)amino]-2-oxoethyl]thio]carbonyl]hydrazide |
| NFK-Beta | Nuclear factor kappa light-chain enhancer of activated B cells |
| PVS | Perivascular spaces |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PHOX | Phagocytic NADPH oxidase |
| PC | Phosphatidylcholines |
| PEMT | Phosphatidylethanolamine N-methyltransferase |
| PS1 | Presenilin 1 |
| PRMTs | Protein arginine N-methyltransferases |
| PKM-2 | Pyruvate kinase muscle isoenzyme 2 |
| ROS | Reactive oxygen species |
| ROCK | Rho-associated protein kinase |
| SAM | S-adenosyl methionine |
| AdoHcy | S-adenosylhomocysteine |
| SAH | S-adenosyl-L-homocysteine |
| AdoMet | S-adenosylmethionine |
| STATs | Signal transducer and activator of transcription proteins |
| STAT3 | Signal transducer and activator of transcription 3 |
| SIRT1-HSF1 axis | Sirtuin/heat shock factor 1/heat shock protein axis |
| sTM | Soluble thrombomodulin |
| sVAD | Subcortical vascular dementia |
| SOD | Superoxide dismutase |
| DNMT | Toll like receptor/NF-KB-DNA methyltransferase |
| TLR | Toll-like receptor |
| UPS | Ubiquitin-proteasome system |
| UPR | Unfolded protein response |
| VEGF | Vascular endothelial growth factor |
| Cbl | Vitamin B12 |
References
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gotzsche, P.C.; Ioannidis, J.P.A.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: Explanation and elaboration. J. Clin. Epidemiol. 2009, 62, e1–e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantoni, L.; Gorelick, P. Cerebral Small Vessel Disease, 1st ed.; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar]
- Xu, W.H. Large artery: An important target for cerebral small vessel diseases. Ann. Transl. Med. 2014, 2, 78. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.; Caruso, P. The Controversial Role of Homocysteine in Neurology: From Labs to Clinical Practice. Int. J. Mol. Sci. 2019, 20, 231. [Google Scholar] [CrossRef] [Green Version]
- Vinciguerra, L.; Lanza, G.; Puglisi, V.; Fisicaro, F.; Pennisi, M.; Bella, R.; Cantone, M. Update on the Neurobiology of Vascular Cognitive Impairment: From Lab to Clinic. Int. J. Mol. Sci. 2020, 21, 2977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantoni, L. Cerebral small vessel disease: From pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010, 9, 689–701. [Google Scholar] [CrossRef]
- Zhang, E.T.; Inman, C.B.; Weller, R.O. Interrelationships of the pia mater and the perivascular (Wirchov-Robin) spaces in the human cerebrum. J. Anatom. 1990, 170, 111–123. [Google Scholar]
- Iadecola, C. The neurovascular Unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 2017, 27, 17–42. [Google Scholar] [CrossRef] [Green Version]
- Caruso, P.; Signori, R.; Moretti, R. Small vessel disease to subcortical dementia: A dynamic model, which interfaces aging, cholinergic dysregulation and the neurovascular unit. Vasc. Health Risk Manag. 2019, 15, 259–281. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Pizzo, M.E.; Preston, J.E.; Janigro, D.; Thorne, R.G. The role of brain barriers in fluid movement in the CNS: Is there a ‘glymphatic’ system? Acta Neuropathol. 2018, 135, 387–407. [Google Scholar] [CrossRef] [Green Version]
- Huijts, M.; Duits, A.; Staals, J.; Kroon, A.A.; Leeuw, P.W.D.; Oostenbrugge, R.J.V. Basal ganglia enlarged perivascular spaces are linked to cognitive function in patients with cerebral small vessel disease. Curr. Neurovasc. Res. 2014, 11, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Balado, J.; Riba-Llena, I.; Garde, E.; Valor, M.; Gutiérrez, B.; Pujadas, F.; Delgado, P. Prevalence of hippocampal enlarged perivascular spaces in a sample of patients with hypertension and their relation with vascular risk factors and cognitive function. J. Neurol. Neurosurg. Psychiatry 2018, 89, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.H.; Lassen, N.A.; Weiller, C.; Sperling, B.; Nakagawara, J. Ischemic stroke and incomplete infarction. Stroke 1996, 27, 761–765. [Google Scholar] [CrossRef]
- Dalkara, T.; Alarcon-Martinez, L. Cerebral micro-vascular signaling in health and disease. Brain Res. 2015, 1623, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, P.; Gold, G.; Kowaru, E.; von Gunten, A.; Imhof, A.; Bouras, C.; Hof, P.R. Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: The Geneva experience. Acta Neuropathol. 2007, 113, 1–12. [Google Scholar] [CrossRef]
- Launer, L.J.; Hughes, T.M.; White, L.R. Microinfarcts, brain atrophy, and cognitive function: The Honolulu Asia Aging Study Autopsy Study. Ann. Neurol. 2011, 70, 774–780. [Google Scholar] [CrossRef] [Green Version]
- Munoz, D.G.; Hastak, S.M.; Harper, B.; Lee, D.; Hachinski, V.C. Pathologic correlates of increased signals of the centrum ovale on magnetic resonance imaging. Arch. Neurol. 1993, 50, 492–497. [Google Scholar] [CrossRef]
- Mirski, M.A. Pharmacology of Blood Pressure Management during Cerebral Ischemia; American Academy of Neurology (AAN): Miami, FL, USA, 2005; 5PC-004; pp. 456–469. [Google Scholar]
- Wallin, A.; Blennow, K.; Gottfries, C.G. Neurochemical abnormalities in vascular dementia. Dementia 1989, 1, 120–130. [Google Scholar]
- Jani, B.I.; Rajkumar, C. Ageing and vascular ageing. Postgrad. Med. J. 2006, 82, 357–362. [Google Scholar] [CrossRef]
- De la Torre, J.C. Vascular basis of Alzheimer’s pathogenesis. Ann. N. Y. Acad. Sci. 2002, 977, 196–215. [Google Scholar] [CrossRef]
- Mathias, C.J.; Kimber, J.R. Postural hypotension: Causes, clinical features, investigation, and management. Annu. Rev. Med. 1999, 50, 317–336. [Google Scholar] [CrossRef]
- Roriz-Filho, J.S.; Bernardes Silva Filho, S.R.; Rosset, I.; Roriz-Cruz, M. Postural blood pressure dysregulation and dementia: Evidence for a vicious circle and implications for neurocardiovascular rehabilitation. In Cardiac Rehabilitation; Halliday, J.T., Ed.; Novascience Publisher Inc: New York, NY, USA, 2009; pp. 1–37. ISBN 987-1-60741-918-1. [Google Scholar]
- Kumar, V.; Cotran, R.S.; Robbins, S.L. Basic Pathology, 8th ed.; Saunders: Philadelphia, PA, USA, 2007. [Google Scholar]
- Lodder, J.; Bamford, J.M.; Sandercock, P.A.; Jones, L.N.; Warlow, C.P. Are hypertension or cardiac embolism likely causes of lacunar infarction? Stroke 1990, 21, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Gamble, C. The pathogenesis of hyaline arteriosclerosis. Am. J. Pathol. 1986, 122, 410–420. [Google Scholar]
- Moritz, A.; Oldt, M. Arteriolar sclerosis in hypertensive and non-hypertensive individuals. Am. J. Pathol. 1937, 13, 679. [Google Scholar]
- Pavelka, M.; Roth, J. Hyaline Arteriolosclerosis. In Functional Ultrastructure; Springer: Vienna, Austria, 2010; pp. 256–257. [Google Scholar]
- Najjar, S.S.; Scuteri, A.; Lakatta, E.G. Arterial aging: Is it an immutable cardiovascular risk factor? Hypertension 2005, 46, 454–462. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, M.F.; Safar, M.E. Relationship between aortic stiffening and microvascular disease in brain and kidney: Cause and logic of therapy. Hypertension 2005, 46, 200–204. [Google Scholar] [CrossRef]
- Cervós-Navarro, J.; Matakas, F.; Roggendorf, W.; Christmann, U. The morphology of spastic intracerebral arterioles. Neuropathol Appl. Neurobiol. 1978, 4, 369–379. [Google Scholar] [CrossRef]
- Wakita, H.; Tomimoto, H.; Akiguchi, I.; Kimura, J. Glial activation and white matter changes in the rat brain induced by chronic cerebral hypoperfusion: An immunoistochemical study. Acta Neuropathol. 1994, 87, 484–492. [Google Scholar] [CrossRef]
- Farkas, E.; Donka, G.; de Vous, R.A.I.; Mihaly, A.; Bari, F.; Luiten, P.G.M. Experimental cerebral hypoeprfusion induces white matter injury and microglial activation in the rat brain. Acta Neuropathol. 2004, 108, 57–64. [Google Scholar] [CrossRef]
- Zhang, Z.G.; Bower, L.; Zhang, R.L.; Chen, S.; Windham, J.P.; Chopp, M. Three dimensional measurement of cerebral microvascular plasma perfusion, glial fibrillary acid protein and microtubule associated P-2 immunoreactivity after embolic stroke in rats: A double fluorescent labeled laser scanning confocal microscopic study. Brain Res. 1999, 844, 55–66. [Google Scholar] [CrossRef]
- Jung, S.; Zarow, C.; Mack, W.J.; Zheng, L.; Vinters, H.V.; Ellis, W.G.; Lyness, S.A.; Chui, H.C. Preservation of neurons of the nucleus basalis in subcortical ischemic vascular disease. Arch. Neurol. 2012, 69, 879–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swartz, R.H.; Sahlas, D.J.; Black, S.E. Strategic involvement of cholinergic pathways and executive dysfunction: Does location of white matter signal hyperintensities matter? J. Stroke Cerebrovasc. Dis. 2003, 12, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Yang, G.; Ebner, T.J.; Chen, G. Local and propagated vascular responses evoked by focal synaptic activity in cerebellar cortex. J. Neurophysiol. 1997, 78, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Salloway, S. Subcortical Vascular Dementia: Binswanger’s and CADASIL; American Academy of Neurology (AAN): Honolulu, HI, USA, 2003; 8AC.006-2; pp. 1–29. [Google Scholar]
- Pantoni, L.; Garcia, J.H.; Gutierrez, J.A. Cerebral white matter is highly vulnerable to ischemia. Stroke 1996, 27, 1641–1647. [Google Scholar] [CrossRef]
- Schmidt, R.; Schmidt, H.; Haybaeck, J.; Loitfelder, M.; Weis, S.; Cavalieri, M.; Seiler, S.; Enzinger, C.; Ropele, S.; Erkinjuntti, T.; et al. Heterogeneity in age-related white matter changes. Acta Neuropathol. 2011, 122, 171–185. [Google Scholar] [CrossRef]
- Hommet, C.; Mondon, K.; Constans, T.; Beaufils, E.; Desmidt, T.; Camus, V.; Cottier, J.P. Review of cerebral microangiopathy and Alzheimer’s disease: Relation between white matter hyperintensities and microbleeds. Dement. Geriatr. Cogn. Disord. 2011, 32, 367–378. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Muller, M.L.T.M.; Kuwabara, H.; Ocnstantien, G.M.; Studentski, S.A. Age-associated leukoaraiosis and cortical cholinergic deafferentation. Neurology 2009, 72, 1411–1416. [Google Scholar] [CrossRef] [Green Version]
- Román, G.C. Brain hypoperfusion: A critical factor in vascular dementia. Neurol. Res. 2004, 26, 454–458. [Google Scholar] [CrossRef]
- Zhan, S.S.; Beyreuther, K.; Schmitt, H.P. Synaptophysin immunoreactivity of the cortical neuropil in vascular dementia of Binswanger type compared with the dementia of Alzheimer type and non-demented controls. Dementia 1994, 5, 79–87. [Google Scholar] [CrossRef]
- Ahtiluoto, S.; Polvikoski, T.; Peltonen, M.; Solomon, A.; Tuomilehto, J.; Winblad, B.; Sulkava, R.; Kivipelto, M. Diabetes, Alzheimer disease, and vascular dementia: A population-based neuropathologic study. Neurology 2010, 75, 1195–1202. [Google Scholar] [CrossRef]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Conejero-Goldberg, C.; Davies, P.; Ulloa, L. Alpha7 nicotinic acetylcholine receptor: A link between inflammation and neurodegeneration. Neurosci. Biobehav. Rev. 2008, 32, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Pavlov, V.A.; Tracey, K.J. Controlling inflammation: The cholinergic anti-inflammatory pathway. Biochem. Soc. Trans. 2006, 34, 1037–1040. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Maestre, G.E.; Arizaga, R.; Friedland, R.P.; Galasko, D.; Hall, K.; Luchsinger, J.A.; Ogunniyi, A.; Perry, E.K.; Potocnik, F.; et al. World Federation of Neurology Dementia Research Group. Alzheimer’s disease and vascular dementia in developing countries: Prevalence, management, and risk factors. Lancet Neurol. 2008, 7, 812–826. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Moon, W.J.; Han, S.H. Differential cholinergic pathway involvement in Alzheimer’s disease and subcortical ischemic vascular dementia. J. Alzheimers Dis. 2013, 35, 129–136. [Google Scholar] [CrossRef]
- Kim, S.H.; Kang, H.S.; Kim, H.J.; Moon, Y.; Ryu, H.J.; Kim, M.Y.; Han, S.H. The effect of ischemic cholinergic damage on cognition in patients with subcortical vascular cognitive impairment. J. Geriatr. Psychiatry Neurol. 2012, 25, 122–127. [Google Scholar] [CrossRef]
- Liu, Q.; Zhu, Z.; Teipel, S.J.; Yang, J.; Xing, Y.; Tang, Y.; Jia, J. White Matter Damage in the Cholinergic System Contributes to Cognitive Impairment in Subcortical Vascular Cognitive Impairment, No Dementia. Front. Aging Neurosci. 2017, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- Roman, G.C.; Kalaria, R.N. Vascular determinants of cholinergic deficits in AD and vascular dementia. Neurobiol. Aging 2006, 27, 1769–1785. [Google Scholar] [CrossRef] [PubMed]
- Low, A.; Mak, E.; Rowe, J.B.; Markus, H.S.; O’Brien, J.T. Inflammation and cerebral small vessel disease: A systematic review. Ageing Res. Rev. 2019, 53, 100916. [Google Scholar] [CrossRef]
- Tomimoto, H.; Akiguchi, I.; Wakita, H.; Svenaga, T.; Nakamura, S.; Kimura, J. Regressive changes of astroglia in white matter lesions in cerebrovascular disease and AD patients. Acta Neuropathol. 1997, 94, 146–152. [Google Scholar] [CrossRef]
- O’Brien, J.T.; Thomas, A. Vascular dementia. Lancet 2015, 386, 1698–1706. [Google Scholar] [CrossRef] [Green Version]
- Filous, A.S.; Silver, J. Targeting astrocytes in CNS injury and disease: A translational research approach. Prog. Neurobiol. 2016, 144, 173–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.; Akinyemi, R.O.; Hase, Y.; Firbank, M.J.; Ndung’u, M.N.; Foster, V.; Craggs, L.J.; Washida, K.; Okamoto, Y.; Thomas, A.J.; et al. Frontal white matter hyperintensities, clasmatodendrosis and gliovascular abnormalities in ageing and post-stroke dementia. Brain 2016, 139, 242–258. [Google Scholar] [CrossRef]
- Tong, X.K.; Hamel, E. Regional cholinergic denervation of cortical microvessels and nitric oxid synthase-containing neurons in AD. Neuroscience 1999, 92, 163–175. [Google Scholar] [CrossRef]
- Cauli, B.; Tong, X.K.; Rancillac, A.; Serluca, N.; Lambolez, B.; Rossier, J.; Hamel, E. Cortical GABA interneurons in neurovascular coupling: Relays for the subcortical vasoactive pathways. J. Neurosci. 2004, 24, 8940–8949. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Smith, C.; Dichgans, M. Mechanism of sporadic cerebral small vessel disease: Insight from neuroimaging. Lancet Neurol. 2013, 12, 483–497. [Google Scholar] [CrossRef] [Green Version]
- Englund, E.A.; Person, B. Correlations between histopathologic white matter changes and proton MR relaxation times in dementia. Alzheimer Dis. Assoc. Disord. 1987, 1, 156–170. [Google Scholar] [CrossRef]
- Román, G.C. Senile dementia of the Binswanger type: A vascular form of dementia in the elderly. JAMA 1987, 258, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Vinters, H.V.; Ellis, W.G.; Zarow, C.; Zaias, B.W.; Jagust, W.J.; Mack, W.J.; Chui, H.C. Neuropathological substrate of ischemic vascular dementia. J. Neuropathol. Exp. Neurol. 2000, 59, 931–945. [Google Scholar] [CrossRef]
- Moody, D.M.; Brown, W.R.; Challa, V.R.; Anderson, R.L. Periventricular venous collagenosis: Association with leukoaraiosis. Radiology 1995, 194, 469–476. [Google Scholar] [CrossRef]
- Craggs, L.J.; Hagel, C.; Kuhlenbaeumer, G.; Borjesson-Hanson, A.; Andersen, O.; Viitanen, M.; Kalimo, H.; McLean, C.A.; Slade, J.Y.; Hall, R.A.; et al. Quantitative vascular pathology and phenotyping familial and sporadic cerebral small vessel diseases. Brain Pathol. 2013, 23, 547–557. [Google Scholar] [CrossRef]
- Hainsworth, A.H.; Oommen, A.T.; Bridges, L.R. Endothelial Cells and Human Cerebral Small Vessel Disease. Brain Pathol. 2015, 25, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Frischer, J.M.; Pipp, I.; Stavrou, I.; Trattnig, S.; Hainfellner, J.A.; Knosp, E. Cerebral cavernous malformations: Congruency of histopathological features with the current clinical definition. J. Neurol. Neurosurg. Psychiatry 2008, 79, 783–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giwa, M.O.; Williams, J.; Elderfield, K.; Jiwa, N.S.; Bridges, L.R.; Kalaria, R.N.; Markus, H.S.; Esiri, M.M.; Hainsworth, A.H. Neuropathologic evidence of endothelial changes in cerebral small vessel disease. Neurology 2012, 78, 167–174. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–728. [Google Scholar] [CrossRef]
- Cai, W.; Zhang, K.; Li, P.; Zhu, L.; Xu, J.; Yang, B.; Hu, X.; Lu, Z.; Chen, J. Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: An aging effect. Ageing Res. Rev. 2017, 34, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Prisby, R.D.; Ramsey, M.W.; Behnke, B.J.; Dominguez, J.M.; Donato, A.J.; Allen, M.R.; Delp, M.D. Aging reduces skeletal blood flow endothelium dependent vasodilation, and NO bioavailability in Rats. J. Bone Miner. Res. 2007, 22, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, W.T.; Vaa, B.; Hesse, C.; Eisenach, J.H.; Joyner, M.J. Aging is associated with reduced prostacyclin-mediated dilation in the human forearm. Hypertenison 2009, 53, 973–978. [Google Scholar] [CrossRef] [Green Version]
- Long, D.A.; Newaz, M.A.; Prabahakar, S.S.; Price, K.L.; Truong, L.; Feng, L.; Mu Oyekan, A.O.; Johnson, R.J. Loss of nitric oxide and endothelial-derived hyperpolarizing factor-mediated responses in ageing. Kidney Int. 2005, 68, 2154–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deplanque, D.; Lavallee, P.C.; Labreuche, J.; Gongora-Rivera, F.; Jaramillo, A.; Brenner, D.; Abboud, H.; Klein, I.F.; Touboul, P.J.; Vicaut, E.; et al. Cerebral and extracerebral vasoreactivity in symptomatic lacunar stroke patients: A case-control study. Int. J. Stroke 2013, 8, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Gunarathne, A.; Patel, J.V.; Kausar, S.; Gammon, B.; Hughes, E.A.; Lip, G.Y. Glycemic status underlies increased arterial stiffness and impaired endothelial function in migrant South Asian stroke survivors compared to European Caucasians: Pathophysiological insights from the West Birmingham Stroke Project. Stroke 2009, 40, 2298–2306. [Google Scholar] [CrossRef] [Green Version]
- Markus, H.S.; Lythgoe, D.J.; Ostegaard, L.; O’Sullivan, M.; Williams, S.C. Reduced cerebral blood flow in white matter in ischaemic leukoaraiosis demonstrated using quantitative exogenous contrast based perfusion MRI. J. Neurol. Neurosurg. Psychiatry 2000, 69, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, M.; Lythgoe, D.J.; Pereira, A.C.; Summers, P.E.; Jarosz, J.M.; Williams, S.C.; Markus, H.S. Patterns of cerebral blood flow reduction in patients with ischemic leukoaraiosis. Neurology 2002, 59, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Markus, H.S.; Allan, C.L.; Ebmeier, K.P. Cerebral hemodynamics in cerebral small vessel disease. In Cerebral Small Vessel Disease; Pantoni, L., Gorelick, P.B., Eds.; Cambridge University Press: Cambridge, UK, 2014; pp. 180–191. [Google Scholar]
- Van der Loo, B.; Labugger, R.; Skepper, J.N.; BAchschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callendere, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T. Enhanced peroxynitrite formation is associated with vascular ageing. J. Exp. Med. 2000, 192, 1731–1744. [Google Scholar] [CrossRef] [Green Version]
- Puca, A.A.; Carrizzo, A.; Ferrario, A.; Villa, F.; Vecchione, C. Endothelial nitric oxide synthase, vascular integrity and human exceptional longevity. Immun. Ageing 2012, 9, 26. [Google Scholar] [CrossRef] [Green Version]
- Flentje, A.; Kalsi, R.; Monahan, T.S. Small GTPases and Their Role in Vascular Disease. Int. J. Mol. Sci. 2019, 20, 917. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, S.; Ridley, A.J.; Lutz, S. The Function of Rho-Associated Kinases ROCK1 and ROCK2 in the Pathogenesis of Cardiovascular Disease. Front. Pharmacol. 2015, 6, 276. [Google Scholar] [CrossRef]
- Pestonjamasp, K.; Amieva, M.R.; Strassel, C.P.; Nauseef, W.M.; Furthmayr, H.; Luna, E.J. Moesin, ezrin, and p205 are actin-binding proteins associated with neutrophil plasma membranes. Mol. Biol. Cell. 1995, 6, 247–259. [Google Scholar] [CrossRef]
- Van Nieuw Amerongen, G.P.; Koolwijk, P.; Versteilen, A.; van Hinsbergh, V.W. Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arter. Thromb. Vasc. Biol. 2003, 23, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, Y.; Uwatoku, T.; Oi, K.; Abe, K.; Hattori, T.; Morishige, K.; Eto, Y.; Fukumoto, Y.; Nakamura, K.I.; Shibata, Y.; et al. Long-term inhibition of Rho-kinase suppresses neointimal formation after stent implantation in porcine coronary arteries: Involvement of multiple mechanisms. Arter. Thromb. Vasc. Biol. 2004, 24, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Szulcek, R.; Beckers, C.M.; Hodzic, J.; de Wit, J.; Chen, Z.; Grob, T.; Musters, R.J.; Minshall, R.D.; van Hinsbergh, V.W.; van Nieuw Amerongen, G.P. Localized RhoA GTPase activity regulates dynamics of endothelial monolayer integrity. Cardiovasc. Res. 2013, 99, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Van Nieuw Amerongen, G.P.; Beckers, C.M.; Achekar, I.D.; Zeeman, S.; Musters, R.J.; van Hinsbergh, V.W. Involvement of Rho kinase in endothelial barrier maintenance. Arter. Thromb. Vasc. Biol. 2007, 27, 2332–2339. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, H.; Chen, B.; Li, Q.; Huang, X.; Wang, L.; Guo, X.; Huang, Q. RhoA/ROCK-dependent moesin phosphorylation regulates AGE-induced endothelial cellular response. Cardiovasc. Diabetol. 2012, 11, 7. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Breslin, J.W.; Zhu, J.; Yuan, S.Y.; Wu, M.H. Rho and ROCK signaling in VEGF-induced microvascular endothelial hyperpermeability. Microcirculation 2006, 13, 237–247. [Google Scholar] [CrossRef]
- Gradinaru, D.; Borsa, C.; Ionescu, C.; Prada, G.I. Oxidized LDL and NO synthesis-biomarkers of endothelial dysfunction and ageing. Mech. Ageing Dev. 2015, 151, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Cicek, F.A.; Kandilci, H.B.; Turan, B. Role of ROCK upregulation in endothelial and smooth muscle vascular functions in diabetic rat aorta. Cardiovasc. Diabetol. 2013, 12, 51. [Google Scholar] [CrossRef] [Green Version]
- Noma, K.; Oyama, N.; Liao, J.K. Physiological role of ROCKs in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2006, 290, C661–C668. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Gormley, K.; O’Sullivan, M.; Knight, J.; Sham, P.; Vallance, P.; Bamford, J. Markus H Endothelial Nitric Oxide Gene Haplotypes and Risk of Cerebral Small-Vessel Disease. Stroke 2004, 35, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Knottnerus, I.L.; Cate, H.; Lodder, J.; Kessels, F.; van Oostenbrugge, R.J. Endothelial dysfunction in lacunar stroke: A systematic review. Cerebrovasc. Dis. 2009, 27, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Esiri, M.M.; Wilcock, G.K.; Morris, J.H. Neuropathological ssessment of the lesions of significance in vascular dementia. J. Neurol. Neurosurg. Psychiatry 1997, 63, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, C.T.; Iadecola, C. The role of the neuronal signaling in controlling cerebral blood flow. Brain Lang. 2007, 102, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallin, J.I.; Snyderman, R. Inflammation: Basic Principles and Clinical Correlates, 3rd ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 1999. [Google Scholar]
- de Leeuw, F.E.; de Kleine, M.; Frijns, C.J.; Fijnheer, R.; van Gijn, J.; Kappelle, L.J. Endothelial cell activation is associated with cerebral white matter lesions in patients with cerebrovascular disease. Ann. N. Y. Acad. Sci. 2002, 977, 306–314. [Google Scholar] [CrossRef]
- Rouhl, R.P.; van Oostenbrugge, R.J.; Theunissen, R.O.; Knottnerus, I.L.; Staals, J.; Henskens, L.H. Autoantibodies against oxidized low-density lipoprotein in cerebral small vessel disease. Stroke 2010, 41, 2687–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, M.; Takahashi, Y.; Iseki, C.; Kawanami, T.; Daimon, M.; Kato, T. Plasma fibrinogen, global cognitive function, and cerebral small vessel disease: Results of a cross-sectional study in community-dwelling Japanese elderly. Intern. Med. 2011, 50, 999–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knottnerus, I.L.; Govers-Riemslag, J.W.; Hamulyak, K.; Rouhl, R.P.; Staals, J.; Spronk, H.M. Endothelial activation in lacunar stroke subtypes. Stroke 2010, 41, 1617–1622. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, S.F.; Doubal, F.N.; Shuler, K.; Wardlaw, J.M. A systematic review of dynamic cerebral and peripheral endothelial function in lacunar stroke versus controls. Stroke 2010, 41, e434–e442. [Google Scholar] [CrossRef] [Green Version]
- Markus, H.S.; Hunt, B.; Palmer, K.; Enzinger, C.; Schmidt, H.; Schmidt, R. Markers of endothelial and hemostatic activation and progression of cerebral white matter hyperintensities: Longitudinal results of the Austrian Stroke Prevention Study. Stroke 2005, 36, 1410–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, M.S.; Simpson, J.E.; Matthews, F.; Brayne, C.; Lewis, C.E.; Barber, R.; Kalaria, R.N.; Forster, G.; Esteves, F.; Wharton, S.B.; et al. White matter lesions in an unselected cohort of the elderly: Molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 2006, 37, 1391–1398. [Google Scholar] [CrossRef] [Green Version]
- Tallini, Y.N.; Brekke, J.F.; Shui, B.; Doran, R.; Hwang, S.M.; NAkai, J.; Salama, G.; Segal, S.S.; Kotlikoff, M.I. Propagated endothelial Ca++ waves and arteriolar dilatation in vivo: Measurements in Cx40 BAC GCaMP2 transgenic mice. Circ. Res. 2007, 101, 1300–1309. [Google Scholar] [CrossRef] [Green Version]
- Segal, S.S. Integration and modulation of intracellular signaling underlying blood flow control. J. Vasc. Res. 2015, 52, 136–157. [Google Scholar] [CrossRef] [Green Version]
- Hen, B.P.; Kozberg, M.G.; Bouchard, M.B.; Shaik, M.A.; Hillman, E.M.C. A critical role for the vascular endothelium in functional neurovascular coupling in the brain. J. Am. Heart Assoc. 2014, 3, e000787. [Google Scholar] [CrossRef] [Green Version]
- Longden, T.A.; Hill-Eubanks, D.C.; Nelosn, M.T. Ion channel networks in the control of cerebral blood flow. J. Cer. Blood Flow. Metab. 2016, 36, 492–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagher, P.; Segal, S.S. Regulation of blood flow in the microcirculation: Role of the conducted vasodilation. Acta Physiol. 2011, 202, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Uhurovoa, H.; Kilic, K.; Tian, P.; Thunemann, M.; Desjardins, M.; Saisan, P.A.; Sakadžić, S.; Ness, T.V.; Mateo, C.; Cheng, Q.; et al. Cell-type specificity of neurovascular coupling in cerebral cortex. ELife 2016, 5, 155. [Google Scholar] [CrossRef]
- Longden, T.A.; Dabertrand, F.; Koide, M.; Gonzales, A.L.; Tykochi, N.T.; Brayden, J.E.; Hill-Eubanks, D.; Nelosn, M.T. Capillary K+ sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat. Neurosci. 2017, 20, 717–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Veen, P.H.; Muller, M.; Vinken, K.L.; Hendrikse, J.; Mali, W.P.; van der Graaf, Y.; Geerlings, M.I.; SMART Study Group. Longitudinal relationship between cerebral small vessel disease and cerebral blood flow. The second manifestations of arterial disease-magnetic resonance study. Stroke 2015, 46, 1233–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouw, A.A.; van Der Flier, W.M.; Fazekas, F.; van Straaten, E.C.; Pantoni, L.; Poggesi, A.; Inzitari, D.; Erkinjuntti, T.; Wahlund, L.O.; Waldemar, G.; et al. Progression of white matter hyperintensities and incidence of new lacunes over a 3-year period: The leukoaraiosis and disability study. Stroke 2008, 39, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Seiler, S.; Loitfelder, M. Longitudinal change of small vessel disease related brain abnormalities. J. Cerebr. Blood Flow Metab. 2016, 36, 26–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Maniega, S.; Chappell, F.M.; Valdes-Henrandez, M.C.; Armitage, P.A.; Makin, S.D.; Heye, A.K.; Thrippleton, M.J.; Sakka, E.; Shuler, K.; Dennis, M.S.; et al. Integrity of normal appearing white matter: Influence of age, visible lesion burden and hypertension in patients with small-vessel disease. J. Cerebr. Blood Flow Metab. 2016, 37, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Smallwood, A.; Oulhaj, A.; Joachim, C.; Christie, S.; Sloan, C.; Smith, A.D.; Esiri, M. Cerebral subcortical small vessel disease and its relation to cognition in elderly subjects: A pathological study in the Oxford Project to Investigate Memory and Ageing (OPTIMA) cohort. Neuropathol. Appl. Neurobiol. 2012, 38, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.H.; Reed, B.R.; Mungas, D.; Weiner, M.W.; Chui, H. Executive dysfunction in subcortical ischaemic vascular disease. J. Neurol. Neurosurg. Psychiatr. 2002, 72, 217–220. [Google Scholar] [CrossRef] [Green Version]
- Burton, E.; Ballard, C.; Stephens, S.; Kenny, R.A.; Kalaria, R.; Barber, R.; O’Brien, J. Hyperintensities and fronto-subcortical atrophy on MRI are substrates of mild cognitive deficits after stroke. Dement. Geriatr. Cogn. Disord. 2003, 16, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Golsari, A.; Fiehler, J.; Rosenkranz, M.; Gerloff, C.; Thomalla, G. Dynamics of regional distribution of ischemic lesions in middle cerebral artery trunk occlusion relates to collateral circulation. J. Cereb. Blood Flow Metab. 2010, 31, 36–40. [Google Scholar] [CrossRef] [Green Version]
- Dijkhuizen, R.M.; Knollema, S.; van der Worp, H.B.; Ter Horst, G.J.; De Wildt, D.J.; Berkelbach van der Sprenkel, J.W.; Tulleken, K.A.; Nicolay, K. Dynamics of cerebral tissue injury and perfusion after temporary hypoxia-ischemia in the rat: Evidence for region-specific sensitivity and delayed damage. Stroke 1998, 29, 695–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.H.; Liu, K.F.; Ye, Z.R.; Gutierrez, J.A. Incomplete infarct and delayed neuronal death after transient middle cerebral artery occlusion in rats. Stroke 1997, 28, 2303–2309. [Google Scholar] [CrossRef]
- Konaka, K.; Miyashita, K.; Naritomi, H. Changes in diffusion-weighted magnetic resonance imaging findings in the acute and subacute phases of anoxic encephalopathy. J. Stroke Cerebrovasc. Dis. 2007, 16, 82–83. [Google Scholar] [CrossRef]
- Ravens, J.R. Vascular changes in the human senile brain. Adv. Neurol. 1978, 20, 487–501. [Google Scholar]
- Klassen, A.C.; Sung, J.H.; Stadlan, E.M. Histological changes in cerebral arteries with increasing age. J. Neuropathol. Exp. Neurol. 1968, 27, 607–623. [Google Scholar] [CrossRef]
- Cummings, J.L. Frontal-subcortical circuits and human behavior. Arch. Neurol. 1993, 50, 873–880. [Google Scholar] [CrossRef]
- Mega, M.S.; Cummings, J.L. Frontal-subcortical circuits and neuropsychiatric disorders. J. Neuropsychiatry Clin. Neurosci. 1994, 6, 358–370. [Google Scholar]
- Smith, A.D.; Refsum, H. Homocysteine, B vitamins, and cognitive impairment. Annu. Rev. Nutr. 2016, 36, 211–239. [Google Scholar] [CrossRef] [PubMed]
- Blom, H.J.; Smulders, Y. Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J. Inherit. Metab. Dis. 2011, 34, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Loscalzo, J.; Handy, D.E. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. 2013 Grover Conference Series. Pulm. Circ. 2014, 482, 169–174. [Google Scholar]
- Miles, L.; Allen, E.; Mills, K.; Clarke, R.; Uauy, R.; Dangour, A.D. Vitamin B12 status and neurologic function in older people: A cross-sectional analysis of baseline trial data from the Older People and Enhanced Neurological Function (OPEN) study. Am. J. Clin. Nutr. 2016, 104, 790–796. [Google Scholar] [CrossRef] [Green Version]
- Obeid, R.; Herrmann, W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006, 580, 2994–3005. [Google Scholar] [CrossRef] [Green Version]
- Price, B.R.; Wilcock, D.M.; Weekman, E.M. Hyeprhomocysteinemia as a risk factor for vascular contributions to cognitive impairment and dementia. Front. Aging Neurosci. 2018, 10, 305. [Google Scholar] [CrossRef] [Green Version]
- Mudd, S.H.; Cantoni, G.L. Activation of methionine for transmethylation. III. The methionine-activating enzyme of Bakers’ yeast. J. Biol. Chem. 1958, 231, 481–492. [Google Scholar] [CrossRef]
- Mato, J.M.; Alvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280. [Google Scholar] [CrossRef] [Green Version]
- Taha, S.; Azzi, A.; Ozer, N.K. Homocysteine induces DNA synthesis and proliferation of vascular smooth muscle cells by a hydrogen peroxide-independent mechanism. Antioxid. Redox Signal. 1999, 1, 365–369. [Google Scholar] [CrossRef]
- Robinson, J.L.; McBreairty, L.E.; Randell, E.W.; Harding, S.V.; Bartlett, R.K.; Brunton, J.A.; Bertolo, R.F. Betaine or folate can equally furnish remethylation to methionine and increase transmethylation in methionine-restricted neonates. J. Nutr. Biochem. 2018, 59, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Kotb, M.; Mudd, S.H.; Mato, J.M. Consensus nomenclature for the mammalian methionine adenosyltransferase genes and gene products. Trends Genet. 1997, 13, 51–52. [Google Scholar] [CrossRef]
- Smulders, Y.M.; Blom, H.J. The homocysteine controversy. J. Inherit. Metab. Dis. 2011, 34, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Selhub, J. Homocysteine metabolism. Annu. Rev. Nutr. 1999, 19, 217–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parnetti, L.; Bottiglieri, T.; Lowenthal, D. Role of homocysteine in age-related vascular and non-vascular diseases. Aging Clin. Exp. Res. 1997, 9, 241–257. [Google Scholar] [CrossRef]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enk, C.; Hougaard, K.; Hippe, E. Reversible dementia and neuropathy associated with folate deficiency 16 years after partial gastrectomy. Scand. J. Haematol. 1980, 25, 63–66. [Google Scholar] [CrossRef]
- Bottiglieri, T. Ademetionine (S-adenosylmethionine) neuropharmacology: Implications for drug therapies in psychiatric and neurological disorders. Expert Opin. Investig. Drugs 1997, 6, 417–426. [Google Scholar] [CrossRef]
- Weir, D.G.; Keating, S.; Molloy, A. Methylation deficiency causes vitamin B12-associated neuropathy in the pig. J. Neurochem. 1988, 51, 1949–1952. [Google Scholar] [CrossRef] [PubMed]
- Surtees, R.; Leonard, J.; Austin, S. Association of demyelination with deficiency of cerebrospinal-fluid S-adenosylmethionine in inborn errors of methyl-transfer pathway. Lancet 1991, 338, 1550–1554. [Google Scholar] [CrossRef]
- Pennypacker, L.C.; Allen, R.H.; Kelly, J.P. High prevalence of cobalamin deficiency in elderly outpatients. J. Am. Geriatr. Soc. 1992, 40, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- McKeever, M.P.; Weir, D.G.; Molloy, A.; Scott, J.M. Betaine-homocysteine methyltransferase: Organ distribution in man, pig and rat and subcellular distribution in the rat. Clin. Sci. 1991, 81, 551–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclerc, D.; Wilson, A.; Dumas, R. Cloning and mapping of a cDNA for methionine synthase reductase, a flavoprotein defective in patients with homocystinuria. Proc. Natl. Acad. Sci. USA 1998, 95, 3059–3064. [Google Scholar] [CrossRef] [Green Version]
- Sunden, S.L.; Renduchintala, M.S.; Park, E.I.; Miklasz, S.D.; Garrow, T.A. Betaine-homocysteine methyltransferase expression in porcine and human tissues and chromosomal localization of the human gene. Arch. Biochem. Biophys. 1997, 345, 171–174. [Google Scholar] [CrossRef]
- Quéré, I.; Paul, V.; Rouillac, C. Spatial and temporal expression of the cystathionine beta-synthase gene during early human development. Biochem. Biophys. Res. Commun. 1999, 254, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Pietrzik, K.; Bronstrup, A. Vitamins B12, B6 and folate as determinants of homocysteine concentration in the healthy population. Eur. J. Pediatr. 1998, 157 (Suppl. S2), S135–S138. [Google Scholar] [CrossRef]
- Huang, Y.C.; Chang, S.J.; Chiu, Y.T.; Chang, H.H.; Cheng, C.H. The status of plasma homocysteine and related B-vitamins in healthy young vegetarians and nonvegetarians. Eur. J. Nutr. 2003, 42, 84–90. [Google Scholar] [CrossRef]
- Kulkarni, K.; Richard, B.C. Lifestyle, homocysteine and the metabolic syndrome. Metab. Syndr. Relat. Disord. 2003, 1, 141–147. [Google Scholar] [CrossRef]
- Ansari, R.; Mahta, A.; Mallack, E.; Luo, J.J. Hyperhomocysteinemia and neurologic disorders: A review. J. Clin. Neurol. 2014, 10, 281–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stea, T.H.; MAnsoor, M.A.; Wandel, M.; Uglem, S.; Frolich, W. Changes in predictors and status of homocysteine in young male adults after dietary intervention with vegetables, fruits and bread. Eur. J. Nutr. 2008, 47, 201–209. [Google Scholar] [CrossRef]
- Pushpakumar, S.; Kundu, S.; Sen, U. Endothelial dysfunction: The link between homocysteine and hydrogen sulfide. Curr. Med. Chem. 2014, 21, 3662–3672. [Google Scholar] [CrossRef]
- Moretti, R.; Dal Ben, M.; Gazzin, S.; Tiribelli, C. Homcysteine in neurology: From endothelium to neurodegeneration. Curr. Nutr. Food Sci. 2017, 13, 163–175. [Google Scholar] [CrossRef]
- Surtees, R.; Bowron, A.; Leonard, J. Cerebrospinal fluid and plasma total homocysteine and related metabolites in children with cystathionine beta-synthase deficiency: The effect of treatment. Pediatr. Res. 1997, 42, 577–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afman, L.A.; Blom, H.J.; Drittij, M.J.; Brouns, M.R.; van Straaten, H.W. Inhibition of transmethylation disturbs neurulation in chick embryos. Brain Res. Dev. Brain Res. 2005, 158, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Kamath, A.F.; Chauhan, A.K.; Kisucka, J. Elevated levels of homocysteine compromise blood-brain barrier integrity in mice. Blood 2006, 107, 591–593. [Google Scholar] [CrossRef] [Green Version]
- Troen, A.M. The central nervous system in animal models of hyperhomocysteinemia. Prog. NeuroPsychopharmacol. Biol. Psychiatry 2005, 29, 1140–1151. [Google Scholar] [CrossRef]
- Algaidi, S.A.; Christie, L.A.; Jenkinson, A.M. Long-term homocysteine exposure induces alterations in spatial learning, hippocampal signalling and synaptic plasticity. Exp. Neurol. 2006, 197, 8–21. [Google Scholar] [CrossRef]
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 6. [Google Scholar] [CrossRef] [Green Version]
- Sultan, M.O.; Farooque, U.; Javed, R.; Khan, M.I.; Karimi, S.; Abdul Sattar, R.; Cheema, O. Correlation of Homocysteine Level and Age in Patients with Ischemic Stroke. Cureus 2020, 12, e7785. [Google Scholar] [CrossRef] [Green Version]
- Moretti, R.; Peinkhofer, C. B Vitamins and Fatty Acids: What Do They Share with Small Vessel Disease-Related Dementia? Int. J. Mol. Sci. 2019, 20, 5797. [Google Scholar] [CrossRef] [Green Version]
- Moretti, R. Homocysteine: New Aspects of an Ancient Enigma. Cardiology 2019, 144, 36–39. [Google Scholar] [CrossRef]
- Piao, X.; Wu, G.; Yang, P.; Shen, J.; De, A.; Wu, J.; Qu, Q. Association between Homocysteine and Cerebral Small Vessel Disease: A Meta-Analysis. J. Stroke Cerebrovasc. Dis. 2018, 27, 2423–2430. [Google Scholar] [CrossRef] [PubMed]
- Rutten-Jacobs, L.C.A.; Traylor, M.; Adib-Samii, P.; Thijs, V.; Sudlow, C.; Rothwell, P.M.; Boncoraglio, G.; Dichgans, M.; Meschia, J.; Maguire, J.; et al. Association of MTHFR C677T Genotype With Ischemic Stroke Is Confined to Cerebral Small Vessel Disease Subtype. Stroke 2016, 47, 646–651. [Google Scholar] [CrossRef] [Green Version]
- Irizarry, M.C.; Gurol, M.E.; Raju, S. Association of homocysteine with plasma amyloid beta protein in aging and neurodegenerative disease. Neurology 2005, 65, 1402–1408. [Google Scholar] [CrossRef]
- Hasegawa, T.; Ukai, W.; Jo, D.-G. Homocysteic acid induces intraneuronal accumulation of neurotoxic Abeta42, implications for the pathogenesis of Alzheimer’s disease. J. Neurosci. Res. 2005, 80, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.S. Homocysteine and Alzheimer’s disease. Lancet Neurol. 2003, 2, 425–428. [Google Scholar] [CrossRef]
- Kruman, I.I.; Kumaravel, T.S.; Lohani, A. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J. Neurosci. 2002, 22, 1752–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sai, X.; Kawamura, Y.; Kokame, K. Endoplasmic reticulum stress-inducible protein, Herp, enhances presenilin-mediated generation of amyloid beta-protein. J. Biol. Chem. 2002, 277, 12915–12920. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Presenilin, Notch, and the genesis and treatment of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 11039–11041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpa, S.; Fuso, A.; D’Anselmi, F.; Cavallaro, R.A. Presenilin 1 gene silencing by S-adenosylmethionine: A treatment for Alzheimer disease? FEBS Lett. 2003, 541, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Baernstein, H.D. A modification of the method for determining methionine in proteins. J. Biol. Chem. 1934, 106, 451–456. [Google Scholar] [CrossRef]
- Jakubowski, H.; Fersht, A. Alternative pathways of rejection of noncognate amino acids by aminoacyl-tRNA synthetases. Nucleic Acids Res. 1981, 9, 3105–3117. [Google Scholar] [CrossRef] [Green Version]
- Jakubowski, H. Proofreading in vivo: Editing of homocysteine by methionyl-tRNA synthetase in Escherichia coli. Proc. Natl. Acad. Sci. USA 1990, 87, 4504–4508. [Google Scholar] [CrossRef] [Green Version]
- Jakubowski, H. Metabolism of homocysteine thiolactone in human cell cultures: Possible mechanism for pathological consequences of elevated homocysteine levels. J. Biol. Chem. 1997, 272, 1935–1942. [Google Scholar] [CrossRef]
- Sharma, G.S.; Kumar, T.; Dar, T.A.; Singh, L.R. Protein-N-Homocysteinylation: Form cellular toxicity to neurodegeneration. Biochim. Et Biophys. Acta 2015, 1850, 2239–2245. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine Thiolactone: Metabolic Origin and Protein Homocysteinylation in Humans. J. Nutr. 2000, 130, 377S–381S. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine is a protein amino acid in humans. Implications for homocysteine-linked disease. J. Biol. Chem. 2002, 277, 30425–30428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakubowski, H. Protein homocysteinylation: Possible mechanism underlying pathological consequences of elevated homocysteine levels. FASEB J. 1999, 13, 2277–2283. [Google Scholar] [CrossRef]
- Sikora, M.; Marczak, Ł.; Kubalska, J.; Graban, A.; Jakubowski, H. Identification of N-homocysteinylation sites in plasma proteins. Amino Acids 2014, 46, 235–244. [Google Scholar] [CrossRef]
- Jacovina, A.T.; Deora, A.B.; Ling, Q.; Broekman, M.J.; Almeida, D.; Greenberg, C.B.; Marcus, A.J.; Smith, J.D.; Hajjar, K.A. Homocysteine inhibits neoangiogenesis in mice through blockade of annexin A2-dependent fibrinolysis. J. Clin. Investig. 2009, 119, 3384–3394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, A.; Sengupta, S.; McComb, M.E.; Théberge, R.; Wilson, W.G.; Costello, C.E.; Jacobsen, D.W. In vitro and in vivo interactions of homocysteine with human plasma transthyretin. J. Biol. Chem. 2003, 278, 49707–49713. [Google Scholar] [CrossRef] [Green Version]
- Jakubowski, H. Homocysteine Modification in Protein Structure/Function and Human Disease. Physiol. Rev. 2019, 99, 555–604. [Google Scholar] [CrossRef]
- Hortin, G.L.; Seam, N.; Hoehn, G.T. Bound homocysteine, cysteine, and cysteinylglycine distribution between albumin and globulins. Clin. Chem. 2006, 52, 2258–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakubowski, H. Homocysteine in Protein Structure/Function and Human Disease—Chemical Biology of Homocysteine-Containing Proteins; Springer: Vienna, Austria, 2013. [Google Scholar] [CrossRef]
- Lai, W.K.; Kan, M.Y. Homocysteine-Induced Endothelial Dysfunction. Ann. Nutr. Metab. 2015, 67, 1–12. [Google Scholar] [CrossRef]
- Perla-Kajan, J.; Twardowski, T.; Jakubowski, H. Mechanisms of homocysteine toxicity in humans. Amino Acids 2007, 32, 561–572. [Google Scholar] [CrossRef]
- Frey, D.; Braun, O.; Briand, C.; Vasak, M.; Grutter, M.G. Structure of the mammalian NOS regulator dimethylarginine dimethylaminohydrolase: A basis for the design of specific inhibitors. Structure 2006, 14, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Leulliot, N.; Quevillon-Cheruel, S.; Sorel, I. Structure of protein phosphatase methyltransferase 1 (PPM1), a leucine carboxyl methyltransferase involved in the regulation of protein phosphatase 2A activity. J. Biol. Chem. 2004, 279, 8351–8358. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.; Lu, Q.; Orecchio, L.; Kosik, K.S. Selective phosphorylation of adult tau isoforms in mature hippocampal neurons exposed to fibrillar A beta. Mol. Cell Neurosci. 1997, 9, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Gong, C.X.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J. Biol. Chem. 1995, 270, 4854–4860. [Google Scholar] [CrossRef] [Green Version]
- Vogelsberg-Ragaglia, V.; Schuck, T.; Trojanowski, J.Q.; Lee, V.M. PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp. Neurol. 2001, 168, 402–412. [Google Scholar] [CrossRef]
- Sontag, E.; Hladik, C.; Montgomery, L. Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J. Neuropathol. Exp. Neurol. 2004, 63, 1080–1091. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.-Q.; Feng, C.; Alkon, D.L. Impairment of phosphatase 2A contributes to the prolonged MAP kinase phosphorylation in Alzheimer’s disease fibroblasts. Neurobiol. Dis. 2003, 14, 458–469. [Google Scholar] [CrossRef]
- Vafai, S.B.; Stock, J.B. Protein phosphatase 2A methylation: A link between elevated plasma homocysteine and Alzheimer’s Disease. FEBS Lett. 2002, 518, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Tolstykh, T.; Lee, J.; Vafai, S.; Stock, J.B. Carboxyl methylation regulates phosphoprotein phosphatase 2A by controlling the association of regulatory B subunits. EMBO J. 2000, 19, 5682–5691. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.I.; Ashline, D.; Dhitavat, S. Folate deprivation induces neurodegeneration: Roles of oxidative stress and increased homocysteine. Neurobiol. Dis. 2003, 14, 32–42. [Google Scholar] [CrossRef]
- Wuerthele, S.E.; Yasuda, R.P.; Freed, W.J.; Hoffer, B.J. The effect of local application of homocysteine on neuronal activity in the central nervous system of the rat. Life Sci. 1982, 31, 2683–2691. [Google Scholar] [CrossRef]
- Lipton, S.A.; Kim, W.K.; Choi, Y.B. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Provini, L.; Cherubini, E. L-homocysteic acid mediates synaptic excitation at NMDA receptors in the hippocampus. Neurosci. Lett. 1991, 124, 157–161. [Google Scholar] [CrossRef]
- Klancnik, J.M.; Cuénod, M.; Gähwiler, B.H.; Jiang, Z.P.; Do, K.Q. Release of endogenous amino acids, including homocysteic acid and cysteine sulphinic acid, from rat hippocampal slices evoked by electrical stimulation of Schaffer collateral-commissural fibres. Neuroscience 1992, 49, 557–570. [Google Scholar] [CrossRef]
- Kim, J.P.; Koh, J.Y.; Choi, D.W. L-homocysteate is a potent neurotoxin on cultured cortical neurons. Brain Res. 1987, 437, 103–110. [Google Scholar] [CrossRef]
- Ziemiffska, E.; Stafiej, A.; Lazarewicz, J.W. Role of group I metabotropic glutamate receptors and NMDA receptors in homocysteine-evoked acute neurodegeneration of cultured cerebellar granule neurones. Neurochem. Int. 2003, 43, 481–492. [Google Scholar] [CrossRef]
- Shi, Q.; Savage, J.E.; Hufeisen, S.J. L-homocysteine sulfinic acid and other acidic homocysteine derivatives are potent and selective metabotropic glutamate receptor agonists. J. Pharmacol. Exp. 2003, 305, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, K.; Pagès, C.; Ledru, A. Regulation of extracellular signal-regulated kinase by homocysteine in hippocampus. Neuroscience 2005, 133, 925–935. [Google Scholar] [CrossRef]
- De Lau, L.M.; Koudstaal, P.J.; van Meurs, J.B.; Uitterlinden, A.G.; Hofman, A.; Breteler, M.M. Methylenterahydrofolate reductase C677T genotype and PD. Annu. Neurol. 2005, 57, 927–930. [Google Scholar] [CrossRef]
- Zhao, P.; Yang, J.F.; Liu, W.; Wang, Y.; Sun, Y.N.; Li, Q. Effects of entacapone on plasma homocysteine in Parkinson’s Disease patients on levodopoa. Zhongha Yi Xue Za Zhi 2013, 93, 512–515. [Google Scholar]
- Mok, S.S.; Turner, B.J.; Beyreuther, K. Toxicity of substrate-bound amyloid peptides on vascular smooth muscle cells is enhanced by homocysteine. Eur. J. Biochem. FEBS 2002, 269, 3014–3022. [Google Scholar] [CrossRef]
- Pang, X.; Liu, J.; Zhao, J.; Mao, J.; Zhang, X.; Feng, L. Homocysteine induces the expression of C-reactive protein via NMDAr-ROS-MAPK-NF-KB signal pathway in rat vascular smooth muscle cells. Atherosclerosis 2014, 236, 73–81. [Google Scholar] [CrossRef]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 887–900. [Google Scholar] [CrossRef]
- Jakubowski, H. The pathophysiological hypothesis of homocysteine thiolactone-mediated vascular disease. J. Physiol. Pharmacol. 2008, 59 (Suppl. S9), 155–167. [Google Scholar]
- Mercie, P.; Garnier, O.; Lascoste, L.; Renard, M.; Closse, C.; Durrieu, F.; Marit, G.; Boisseau, R.M.; Belloc, F. Homocysteine-thiolactone induces caspase-independent vascular endothelial cell death with apoptotic features. Apoptosis 2000, 5, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Dayal, S.; Wilson, K.M.; Leo, L.; Arning, E.; Bottiglieri, T.; Lentz, S.R. Enhanced susceptibility to arterial thrombosis in a murine model of hyperhomocysteinemia. Blood 2006, 108, 2237–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Undas, A.; Brozek, J.; Szczeklik, A. Homocysteine and thrombosis: From basic science to clinical evidence. Thromb. Haemost. 2005, 94, 907–915. [Google Scholar]
- Sauls, D.L.; Lockhart, E.; Warren, M.E.; Lenkowski, A.; Wilhelm, S.E.; Hoffman, M. Modification of fibrinogen by homocysteine thiolactone increases resistance to fibrinolysis: A potential mechanism of the thrombotic tendency in hyperhomocysteinemia. Biochemistry 2006, 45, 2480–2487. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Inoue, A.; Ijiri, Y.; Naemura, A.; Yamamoto, J. Short- and long-term treatment with folic acid suppresses thrombus formation in atherogenic mice in vivo. Pathophysiology 2014, 21, 169–175. [Google Scholar] [CrossRef]
- Perna, A.F.; Ingrosso, D.; De Santo, N.G. Homocysteine and oxidative stress. Amino Acids 2003, 25, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Petras, M.; Tatarakova, Z.; Kovalska, M.; Mokra, D.; Dobrota, D.; Lehotsky, J.; Drgova, A. Hyperhomocysteinemia as a risk factor for the neuronal system disorders. J. Physiol. Pharmacol. 2014, 65, 1–23. [Google Scholar]
- Wyse, A.T.S.; Zugno, A.I.; Streck, E.L. Inhibition of Na(+), K(+)-ATPase activity in hippocampus of rats subjected to acute administration of homocysteine is prevented by vitamins E and C treatment. Neurochem. Res. 2002, 27, 1685–1689. [Google Scholar] [CrossRef]
- Bleie, O.; Semb, A.G.; Grundt, H. Homcysteine-lowering therapy does not affect inflammatory markers of atherosclerosis in patients with stable coronary disease. J. Int. Med. 2007, 262, 244–253. [Google Scholar] [CrossRef]
- Ploder, M.; Kurz, K.; Splitter, A.; Neurauter, G.; Roth, E.; Fuch, D. Early increase of plasma Hcy in sepsis patients with poor outcome. Mol. Med. 2010, 16, 498–504. [Google Scholar] [CrossRef]
- Li, J.-J.; Li, Q.; Du, H.-P. Homocysteine Triggers inflammatory responses in macrophages through inhibiting CSE-H2S signaling via DNA hypermethylation of CSE promoter. Int. J. Mol. Sci. 2015, 16, 12560–12577. [Google Scholar] [CrossRef]
- Krishna, S.M.; Dear, A.; Craig, J.M.; Norman, P.E.; Golledge, J. The potential role of homocysteine mediated DNA methylation and associated epigenetic changes in abdominal aortic aneurysm formation. Atherosclerosis 2013, 228, 295–305. [Google Scholar] [CrossRef]
- Yi-Deng, J.; Tao, S.; Hui-Ping, Z. Folate and ApoE DNA methylation induced by homocysteine in human monocytes. DNA Cell Biol. 2007, 26, 737–744. [Google Scholar] [CrossRef]
- Chang, P.-Y.; Lu, S.-C.; Lee, C.-M. Homocysteine inhibits arterial endothelial cell growth through transcriptional downregulation of fibroblast growth factor-2 involving G protein and DNA methylation. Circ. Res. 2008, 102, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Kamat, P.K.; Kalani, A.; Givvimani, S.; Sathnur, P.B.; Tyagi, S.C.; Tyagi, N. Hydrogen sulfide attenuates neurodegeneration and neurovascular dysfunction induced by intracerebral-administered homocysteine in mice. Neuroscience 2013, 252, 302–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Wu, H.; Li, W.; Gao, P. Protective effects of genistein in homocysteine-induced endothelial cell inflammatory injury. Mol. Cell Biochem. 2015, 403, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Luo, M.; Xie, N.; Wang, J.; Chen, L. Curcumin protects endothelial cells against homocysteine induced injury through inhibiting inflammation. Am. J. Transl. Res. 2016, 8, 4598–4604. [Google Scholar] [PubMed]
- Keegan, P.M.; Wilder, C.L.; Platt, M.O. Tumor necrosis factor alpha stimulates cathepsin K and V activity via juxtacrine monocyteendothelial cell signaling and JNK activation. Mol. Cell Biochem. 2012, 367, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Chen, N.L.; Wong, A.; Craik, C.S.; Brömme, D. Elastin degradation by cathepsin V requires two exosites. J. Biol. Chem. 2013, 288, 34871–34881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Cheng, X.W.; Hu, L.; Wu, H.; Guo-Ping Hao, C.N.; Jiang, H.; Zhu, E.; Huang, Z.; Inoue, A.; Sasaki, T.; et al. Cathepsin S activity controls ischemia-induced neovascularization in mice. Int. J. Cardiol. 2015, 183, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, Y.Y.; Li, Q.Y.; Cai, Z.H. Evolutionary history of cathepsin L (L-like) family genes in vertebrates. Int. J. Biol. Sci. 2015, 11, 1016–1025. [Google Scholar] [CrossRef] [Green Version]
- Pribis, J.P.; Al-Abed, Y.; Yang, H.; Gero, D.; Xu, H.; Montenegro, M.F.; Bauer, E.M.; Kim, S.; Chavan, S.S.; Cai, C.; et al. The HIV protease inhibitor saquinavir inhibits HMGB1 driven inflammation by targeting the interaction of cathepsin V with TLR4/MyD88. Mol. Med. 2015, 21, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, G.; Starzinski-Powitz, A.; Sloane, B.F.; Doll, M.; Kippenberger, S.; Bernd, A.; Kaufmann, R.; Meissner, M. PPARα agonist Wy14643 suppresses cathepsin B in human endothelial cells via transcriptional, post-transcriptional and post-translational mechanisms. Angiogenesis 2013, 16, 223–233. [Google Scholar] [CrossRef]
- Platt, M.O.; Shockey, W.A. Endothelial cells and cathepsins: Biochemical and biomechanical regulation. Biochimie 2016, 122, 314–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, Y.P.; Ma, Y.S.; Li, X.G.; Chen, R.F.; Zeng, P.Y.; Li, X.H.; Qiu, C.F.; Li, Y.P.; Zhang, Z.; Chen, A.F. l-Homocysteine-induced cathepsin V mediates the vascular endothelial inflammation in hyperhomocysteinaemia. Br. J. Pharmacol. 2018, 175, 1157–1172. [Google Scholar] [CrossRef]
- Ahmad, S.; Siddiqi, M.I. Insights from molecular modeling into the selective inhibition of cathepsin S by its inhibitor. J. Mol. Model. 2017, 23, 92. [Google Scholar] [CrossRef]
- Aavik, E.; Lumivuori, H.; Leppänen, O.; Wirth, T.; Häkkinen, S.K.; Bräsen, J.H.; Beschorner, U.; Zeller, T.; Braspenning, M.; van Criekinge, W.; et al. Global DNA methylation analysis of human atherosclerotic plaques reveals extensive genomic hypomethylation and reactivation at imprinted locus 14q32 involving induction of a miRNA cluster. Eur. Heart J. 2015, 36, 993–1000. [Google Scholar] [CrossRef]
- Boldyrev, A.; Bryshkova, E.; MAshkina, A.; Vladychenskaya, E. Why is homocysteine toxic for the nervous and immune systems? Curr. Aging Sci. 2013, 6, 29–36. [Google Scholar] [CrossRef]
- Essouma, M.; Noubiap, J.J.N. Therapeutic potential of folic acid supplementation for cardiovascular disease prevention through homocysteine lowering and blockade in rheumatoid arthritis patients. Biomark. Res. 2015, 3, 24. [Google Scholar] [CrossRef] [Green Version]
- Ying, G.; Wang, Y.; Cen, X.M.; Yang, M.; Liang, Y.; Xie, Q.B. Lipid peroxidation-mediated inflammation promotes cell apoptosis through activation of NFK-B pathway in rheumatoid arthritis synovial cells. Med. Infalmm. 2015, 2015, 1–10. [Google Scholar]
- Deng, J.; Lu, S.; Li, H. Homocysteine activates B cells via regulating PKM-2 dependent metabolic reprogramming. J. Immunol. 2017, 198, 170–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniades, C.; Tousoulis, D.; Marinou, K. Asymmetrical dimethylarginine regulates endothelial function in methionine-induced but not in chronic homocystinemia in humans: Effect of oxidative stress and proinflammatory cytokines. Am. J. Clin. Nutr. 2006, 84, 781–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwedhelm, E.; Xanthakis, V.; Maas, R. Asymmetric dimethylarginine reference intervals determined with liquid chromatography–tandem mass spectrometry: Results from the Framingham offspring cohort. Clin. Chem. 2009, 55, 1539–1545. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Sun, L.; Zhang, H. Elevated plasma homocysteine was associated with hemorrhagic and ischemic stroke, but methylenetetrahydrofolate reductase gene c677t polymorphism was a risk factor for thrombotic stroke a multicenter case-control study in China. Stroke 2003, 34, 2085–2090. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Palfrey, H.A.; Pathak, R.; Kadowitz, P.J.; Gettys, T.W.; Murthy, S.N. The metabolism and significance of homocysteine in nutrition and health. Nutr. Metab. 2017, 14, 78. [Google Scholar] [CrossRef] [Green Version]
- Li, J.G.; Chu, J.; Barrero, C.; Merali, S.; Praticò, D. Homocysteine exacerbates β-amyloid pathology, tau pathology, and cognitive deficit in a mouse model of Alzheimer disease with plaques and tangles. Ann. Neurol. 2014, 75, 851–863. [Google Scholar] [CrossRef]
- Vallance, P.; Leiper, J. Cardiovascular biology of the asymmetric dimethylarginine: Dimethylarginine dimethylaminohydrolase pathway. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1023–1030. [Google Scholar] [CrossRef]
- Lentz, S.R.; Rodionov, R.N.; Dayal, S. Hyperhomocysteinemia, endothelial dysfunction, and cardiovascular risk: The potential role of ADMA. Atheroscler. Suppl. 2003, 4, 61–65. [Google Scholar] [CrossRef]
- Dayal, S.; Lentz, S.R. ADMA and hyperhomocysteinemia. Vasc. Med. 2005, 10, S27–S33. [Google Scholar] [CrossRef]
- Li, T.; Huang, Y.; Cai, W.; Chen, X.; Men, X.; Lu, T.; Wu, A.; Lu, Z. Age-related cerebral small vessel disease and inflammaging. Cell Death Dis. 2020, 11, 932. [Google Scholar] [CrossRef]
- Hassan, A.; Hunt, B.J.; O’Sullivan, M.; Bell, R.; D’Souza, R.; Jeffery, S.; Bamford, J.M.; Markus, H.S. Homocysteine is a risk factor for cerebralr small vessel disease, acting via endothelial dysfunction. Brain 2004, 127, 212–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Zhao, L.; Song, X.; Yan, Y.; Liu, N.; Li, T.; Yan, B.; Liu, B. HSP27 inhibits homocysteien-induced endothelial apoptosis by modulation of ROS production and mithocondrial caspase-depndent apoptotic pathway. Biomed Res. Int. 2016, 2016, 4847874. [Google Scholar] [CrossRef] [PubMed]
- Hossain, G.S.; van Thienen, J.V.; Werstuck, G.H.; Zhou, J.; Sood, S.K.; Dickhout, J.G.; de Koning, A.B.; Tang, D.; Wu, D.; Falk, E.; et al. TDAG51 is induced by homocysteine, promotes detachment-mediated programmed cell death, and contributes to the development of atherosclerosis in hyperhomocysteinemia. J. Biol. Chem. 2003, 278, 30317–30327. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, M.A.; Lalla, E.; Lu, Y.; Gleason, M.R.; Wolf, B.M.; Tanji, N.; Ferran LJJr Kohl, B.; Rao, V.; Kisiel, W.; Stern, D.M.; et al. Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J. Clin. Investig. 2001, 107, 675–683. [Google Scholar] [CrossRef] [Green Version]
- McCully, K.S. Chemical pathology of homocysteine. IV. Excitotoxicity, oxidative stress, endothelial dysfunction, and inflammation. Ann. Clin. Lab. Sci. 2009, 39, 219–232. [Google Scholar] [PubMed]
- Li, T.; Chen, Y.; Li, J.; Yang, X.; Zhang, H.; Qin, X.; Hu, Y.; Mo, Z. Serum Homocysteine Concentration Is Significantly Associated with Inflammatory/Immune Factors. PLoS ONE 2015, 10, e0138099. [Google Scholar] [CrossRef] [Green Version]
- Reddy, V.S.; Trinath, J.; Reddy, G.B. Implication of homocysteine in protein quality control processes. Biochimie 2019, 165, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.; Sun, Z.; Peng, C.; Liu, L.; Xiao, X.; Li, J. Homocysteine Induces Hepatic Steatosis Involving ER Stress Response in High Methionine Diet-Fed Mice. Nutrients 2017, 9, 346. [Google Scholar] [CrossRef] [Green Version]
- Yakub, M.; Schulze, K.J.; Khatry, S.K.; Stewart, C.P.; Christian, P.; West, K.P. High plasma homocysteine increases risk of metabolic syndrome in 6 to 8 year old children in rural Nepal. Nutrients 2014, 6, 1649–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Xu, F.; Liang, H.; Cao, H.; Cai, M.; Xu, W.; Weng, J. SIRT1/HSF1/HSP pathway is essential for exenatide-alleviated, lipid-induced hepatic endoplasmic reticulum stress. Hepatology 2017, 66, 809–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Dong, Z.; Cheng, M.; Zhao, Y.; Wang, M.; Sai, N.; Wang, X.; Liu, H.; Huang, G.; Zhang, X. Homocysteine exaggerates microglia activation and neuroinflammation through microglia localized STAT3 overactivation following ischemic stroke. J. Neuroinflammation 2017, 14, 187. [Google Scholar] [CrossRef] [Green Version]
- Raible, D.J.; Frey, L.C.; Brooks-Kayal, A.R. Effects of JAK2-STAT3 signaling after cerebral insults. JAKSTAT 2014, 3, e29510. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Wu, G.; Fan, C.; Xu, J.; Jiang, S.; Yan, X.; Di, S.; Ma, Z.; Hu, W.; Yang, Y. The emerging role of signal transducer and activator of transcription 3 in cerebral ischemic and hemorrhagic stroke. Prog. Neurobiol. 2016, 137, 1–16. [Google Scholar] [CrossRef]
- Zhu, H.; Zou, L.; Tian, J.; Du, G.; Gao, Y. SMND-309, a novel derivative of salvianolic acid B, protects rat brains ischemia and reperfusion injury by targeting the JAK2/STAT3 pathway. Eur. J. Pharmacol. 2013, 714, 23–31. [Google Scholar] [CrossRef]
- Satriotomo, I.; Bowen, K.K.; Vemuganti, R. JAK2 and STAT3 activation contributes to neuronal damage following transient focal cerebral ischemia. J. Neurochem. 2006, 98, 1353–1368. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.H.; Park, S.W.; Kapadia, R.; Vemuganti, R. Role of transcription factors in mediating post-ischemic cerebral inflammation and brain damage. Neurochem. Int. 2007, 50, 1014–1027. [Google Scholar] [CrossRef] [Green Version]
- Probert, L.; Akassoglou, K.; Pasparakis, M.; Kontogeorgos, G.; Kollias, G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor alpha. Proc. Natl. Acad. Sci. USA 1995, 92, 11294–11298. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhang, L.; Miao, Y.; Yang, J.; Wang, X.; Wang, C.C.; Feng, J.; Wang, L. Homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. 2019, 20, 46–59. [Google Scholar] [CrossRef]
- Ji, C.; Kaplowitz, N. Hyperhomocysteinemia, endoplasmic reticulum stress, and alcoholic liver injury. World J. Gastroenterol. 2004, 10, 1699–1708. [Google Scholar] [CrossRef] [Green Version]
- Hohsfeld, L.A.; Humpel, C. Homocysteine enhances transmigration of rat monocytes through a brain capillary endothelial cell monolayer via ICAM-1. Curr. Neurovasc. Res. 2010, 7, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Gutteridge, J.M.; Halliwell, B. Antioxidants: Molecules, medicines, and myths. Biochem. Biophys. Res. Commun. 2010, 393, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Thampi, P.; Stewart, B.W.; Joseph, L.; Melnyk, S.B.; Hennings, L.J.; Nagarajan, S. Dietary homocysteine promotes atherosclerosis in apoE‑deficient mice by inducing scavenger receptors expression. Atherosclerosis 2008, 197, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, M.B.A.; Souza, J.M.; Romero, N.; Castro, L.; Thomson, L.; Radi, R. Mechanisms and Biological Consequences of Peroxynitrite-Dependent Protein Oxidation and Nitration. In Nitric Oxide. Biology and Pathobiology, 2nd edition; Ignaro, L., Ed.; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar] [CrossRef]
- Gao, H.M.; Zhou, H.; Hong, J.S. NADPH oxidases: Novel therapeutic targets for neurodegenerative diseases. Trends Pharmacol. Sci. 2012, 33, 295–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paravicini, T.M.; Touyz, R.M. NADPH Oxidases, Reactive Oxygen Species, and Hypertension. Diabetes Care 2008, 31 (Suppl. S2), S170–S180. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; He, G.W. Imbalance of Homocysteine and H2S: Significance, Mechanisms, and Therapeutic Promise in Vascular Injury. Oxid. Med. Cell Longev. 2019, 2019, 7629673. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.-C.; Hao, Y.-J.; Jiao, Y.; Wang, Y.-H.; Xu, L.-B.; Mao, C.-Y.; Yang, X.-L.; Yang, A.-N.; Tian, J.; Zhang, M.-H.; et al. Homocysteine‑induced oxidative stress through TLR4/NF‑κB/DNMT1‑mediated LOX‑1 DNA methylation in endothelial cells. Mol. Med. Rep. 2017, 16, 9181–9188. [Google Scholar] [CrossRef] [Green Version]
- Jellinger, K.A. Pathology and pathogenesis of vascular cognitive impairment-a critical update. Front. Aging Neurosci. 2013, 5, 17–46. [Google Scholar] [CrossRef] [Green Version]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchantchou, F.; Goodfellow, M.; Li, F.; Ramsue, L.; Miller, C.; Puche, A.; Fiskum, G. Hyperhomocysteinemia-Induced Oxidative Stress Exacerbates Cortical Traumatic Brain Injury Outcomes in Rats. Cell. Mol. Neurobiol. 2020, May 13, 1–17. [Google Scholar] [CrossRef]
- Hoffman, M. Hypothesis: Hyperhomocysteinemia is an indicator of oxidant stress. Med. Hypotheses 2011, 77, 1088–1093. [Google Scholar] [CrossRef]
- Sen, U.; Mishra, P.K.; Tyagi, N.; Tyagi, S.C. Homocysteine to hydrogen sulfide or hypertension. Cell Biochem. Biophys. 2010, 57, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawle, P.; Foresti, R.; Green, C.J.; Motterlini, R. Homocysteine attenuates endothelial heme-oxygenase-1 induction by nitric oxide (NO) and hypoxia. FEBS Lett. 2001, 508, 403–406. [Google Scholar] [CrossRef] [Green Version]
- Stuhlinger, M.C.; Tsao, P.S.; Her, J.H.; Kimoto, M.; Balint, R.F.; Cooke, J.P. Homocysteine impairs the nitric oxide synthase pathway: Role of asymmetric dimethylarginine. Circulation 2001, 104, 2569–2575. [Google Scholar] [CrossRef]
- Vallance, P.; Chan, N. Endothelial function and nitric oxide: Clinical relevance. Heart 2001, 85, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, N.; Sedoris, K.C.; Steed, M.; Ovechkin, A.V.; Moshal, K.S.; Tyagi, S.C. Mechanisms of homocysteine-induced oxidative stress. Am. J. Physiol-Heart Circ. Physiol. 2005, 289, H2649–H2656. [Google Scholar] [CrossRef] [Green Version]
- Fornier, I.; Ploye, F.; Cottet-Emard, J.M.; Brun, J.; Claustrat, B. Folate deficiency alters melatonin secretion in rats. J. Nutr. 2002, 132, 2781–2784. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Tan, D.X.; Pappolla, M.A. Melatonin relieves the neural oxidative burden tht contributes to dementias. Annu. N. Y. Acad. Sci. 2004, 1035, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Baydar, G.; Ozer, M.; Yasar, A.; Tuzcu, M.; Koz, S.T. Melatonin improves learning and memory performances impaired by hyperhomocysteinemia in rats. Brain Res. 2005, 1046, 187–194. [Google Scholar]
- Baydar, G.; Kutlu, S.; Nazirroglu, M.; Canpolat, S.; Sandal, S.; Ozcan, M.; Kelestimur, H. Inhibitory effects of melatonin on neural lipid peroxidation induced by intracerebroventricularly administered homocysteine. J. Pinel. Res. 2003, 34, 36–39. [Google Scholar]
- Curro, M.; Gugliandolo, A.; Gangemi, C.; Risitano, R.; Ientile, R.; Caccamo, D. Toxic effects of mildy elevated homocysteine concnetrations in neuronal-like cells. Neurochem. Res. 2014, 39, 1485–1495. [Google Scholar] [CrossRef]
- Sharma, M.; Rai, S.K.; Tiwari, M.; Chandra, R. Effect of hyperhomcysteinemia on cardiovascular risk factors and initiation of atherosclerosis in Wistar rats. Eur. J. Pharamcol. 2007, 574, 49–609. [Google Scholar] [CrossRef]
- Zou, C.-G.; Banerjee, R. Homocysteine and redox signaling. Antioxid. Redox Signal. 2005, 7, 547–559. [Google Scholar] [CrossRef]
- Banerjee, R.; Zou, C.-G. Redox regulation and reaction mechanism of human cystathionine-beta-synthase: A PLP-dependent hemesensor protein. Arch. Biochem. Biophys. 2005, 433, 144–156. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Cutler, P.; Melnyk, S. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudova, A.; Bauman, Z.; Braun, A. S-adenosylmethionine stabilizes cystathionine beta-synthase and modulates redox capacity. Proc. Natl. Acad. Sci. USA 2006, 103, 6489–6494. [Google Scholar] [CrossRef] [Green Version]
- Reis, E.A.; Zugno, A.I.; Franzon, R. Pretreatment with vitamins E and C prevent the impairment of memory caused by homocysteine administration in rats. Metab. Brain Dis. 2002, 17, 211–217. [Google Scholar] [CrossRef]
- Murr, C.; Widner, B.; Wirleeitner, B.; Fuchs, D. Neopterin as a marker for immune system activation. Curr. Drug Metab. 2001, 2, 175–187. [Google Scholar] [CrossRef]
- Aykutoglu, G.; Tartik, M.; Darendelioglu, E.; Ayna, A.; Baydas, G. Melatonin and vitamin E alleviate homocysteine-induced oxidative injury and apoptosis in endothelial cells. Mol. Biol. Rep. 2020, 47, 5285–5293. [Google Scholar] [CrossRef]
- Kumar, D.; Jugdutt, B.I. Apoptosis, and oxidants in the heart. J. Lab. Clin. Med. 2003, 142, 5–8. [Google Scholar] [CrossRef]
- Tartik, M.; Darendelioglu, E.; Aykutoglu, G.; Baydas, G. Turkish propolis supresses MCF-7 cell death induced by homocysteine. Biomed. Pharmacother. 2016, 82, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Cordaro, M.; Siracusa, R.; Fusco, R.; Cuzzocrea, S.; Di Paola, R.; Impellizzeri, D. Involvements of Hyperhomocysteinemia in Neurological Disorders. Metabolites 2021, 11, 37. [Google Scholar] [CrossRef]
- Toya, T.; Sara, J.D.; Lerman, B.; Ahmad, A.; Taher, R.; Godo, S.; Corban, M.T.; Lerman, L.O.; Lerman, A. Elevated plasma homocysteine levels are associated with impaired peripheral microvascular vasomotor response. Int. J. Cardiol. Heart Vasc. 2020, 28, 100515. [Google Scholar] [CrossRef]
- Ahmad, A.; Corban, M.T.; Toya, T.; Sara, J.D.; Lerman, B.; Park, J.Y.; Lerman, L.O.; Lerman, A. Coronary Microvascular Endothelial Dysfunction in Patients With Angina and Nonobstructive Coronary Artery Disease Is Associated With Elevated Serum Homocysteine Levels. J. Am. Heart Assoc. 2020, 9, e017746. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Li, X.; Teng, Z.; Li, X.; Jin, W.; Lv, P.Y. Homocysteine is Associated with the Development of Cerebral Small Vessel Disease: Retrospective Analyses from Neuroimaging and Cognitive Outcomes. J. Stroke Cerebrovasc. Dis. 2020, 29, 105393. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Park, G.M.; Ha, J.; Cho, Y.R.; Roh, J.H.; Park, E.J.; Yang, Y.; Won, K.B.; Ann, S.H.; Kim, Y.G.; et al. Homocysteine is not a risk factor for subclinical coronary atherosclerosis in asymptomatic individuals. PLoS ONE 2020, 15, e0231428. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moretti, R.; Giuffré, M.; Caruso, P.; Gazzin, S.; Tiribelli, C. Homocysteine in Neurology: A Possible Contributing Factor to Small Vessel Disease. Int. J. Mol. Sci. 2021, 22, 2051. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042051
Moretti R, Giuffré M, Caruso P, Gazzin S, Tiribelli C. Homocysteine in Neurology: A Possible Contributing Factor to Small Vessel Disease. International Journal of Molecular Sciences. 2021; 22(4):2051. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042051
Chicago/Turabian StyleMoretti, Rita, Mauro Giuffré, Paola Caruso, Silvia Gazzin, and Claudio Tiribelli. 2021. "Homocysteine in Neurology: A Possible Contributing Factor to Small Vessel Disease" International Journal of Molecular Sciences 22, no. 4: 2051. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042051