
Extracellular Protein Aggregates Colocalization and Neuronal Dystrophy in Comorbid Alzheimer’s and Creutzfeldt–Jakob Disease: A Micromorphological Pilot Study on 20 Brains


Abstract
:1. Introduction
2. Results
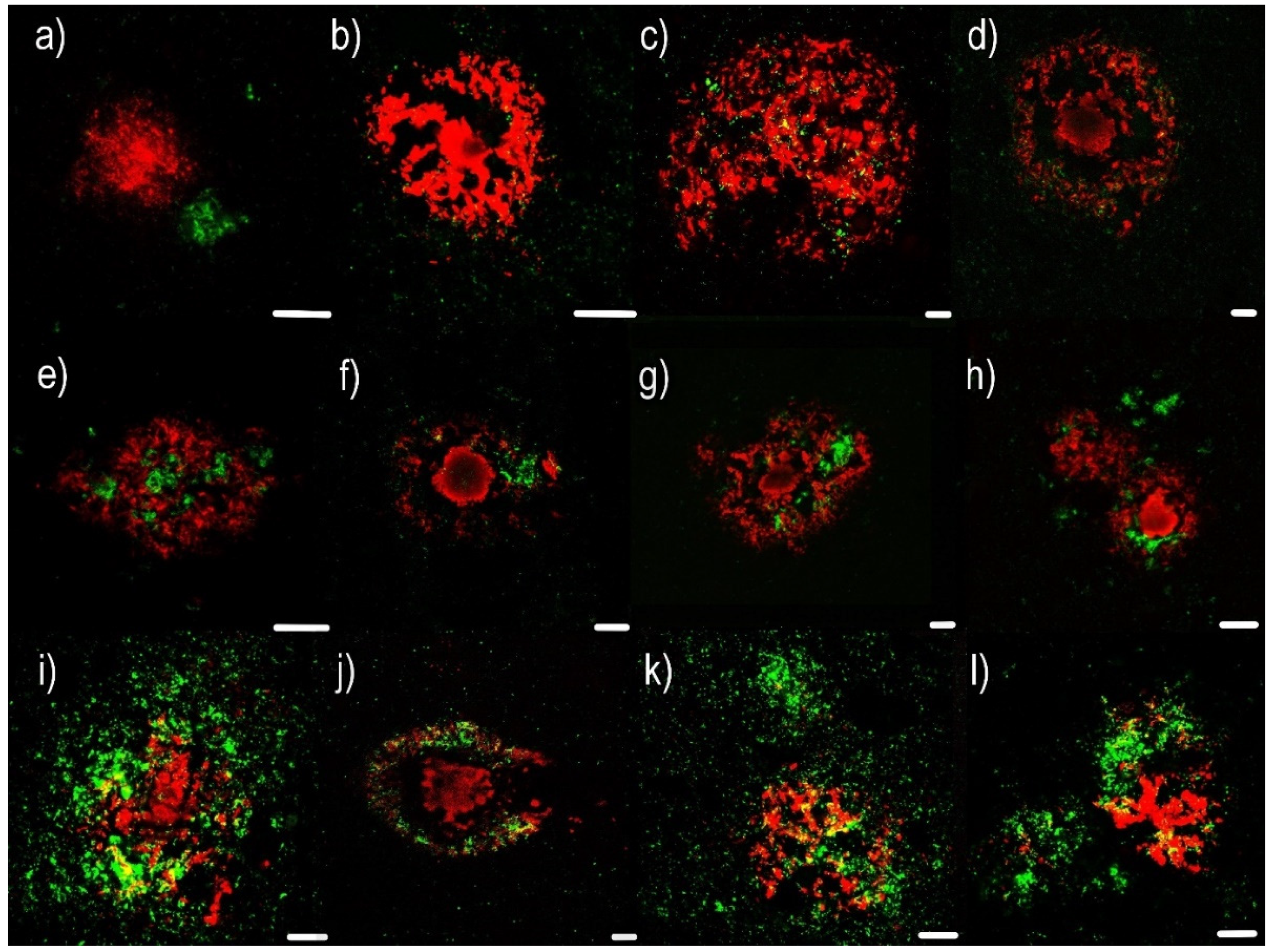
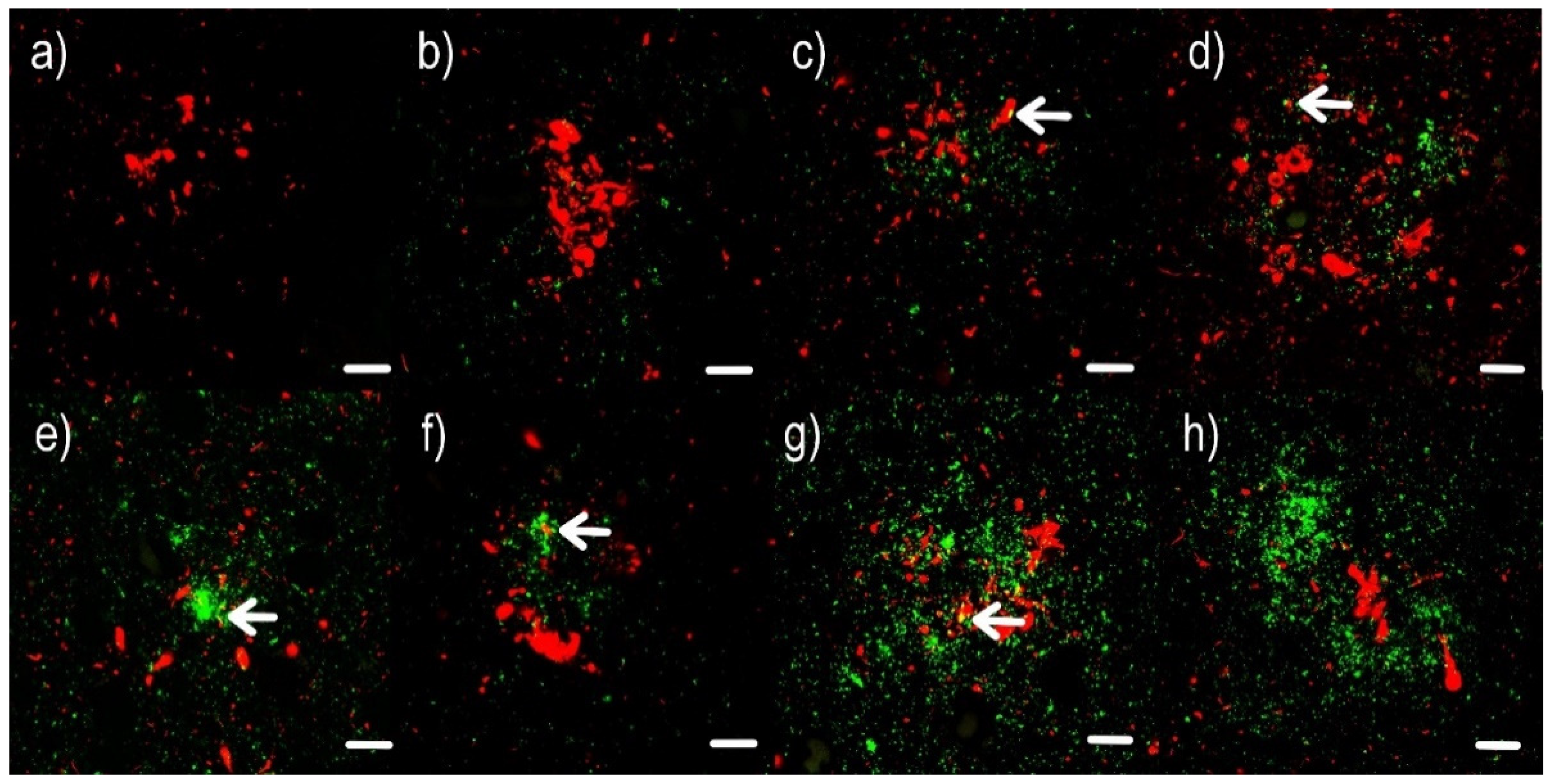
2.1. Confocal Microscopy Visualization
- (1)
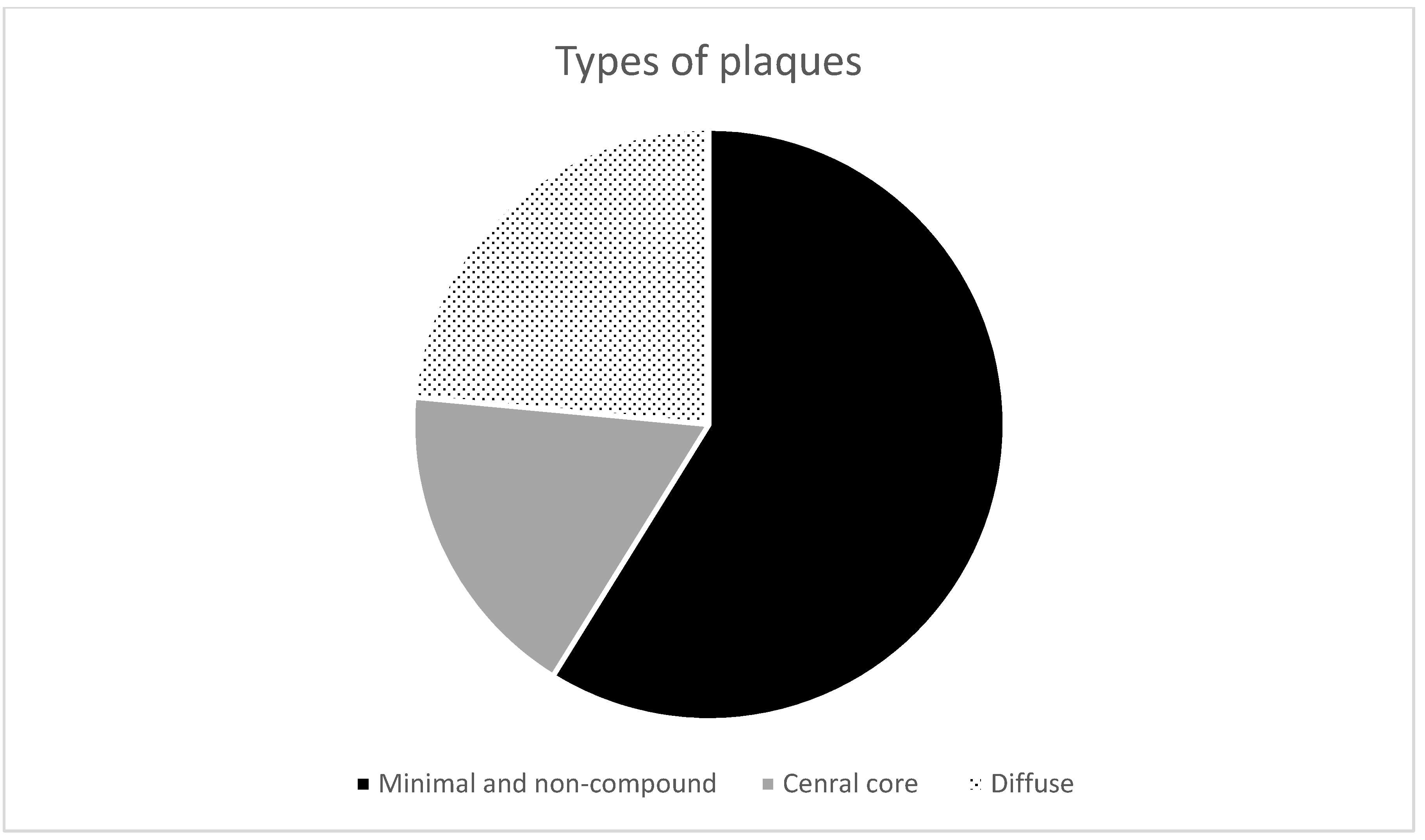
- Non-compound and minimal compound plaques (10 cases out of 17):
- Non-compound plaques (observed in 3 cases out of 17) are without co-occurrence or colocalization of Aβ and PrPSc deposits. Pure Aβ and pure PrPSc plaque exist independently of each other (Figure 1a,b).
- Minimal compound plaques (Figure 1c,d) were seen most often (7 of 17 patients in whom PrPSc aggregates were present in the neocortical and archicortical parts of the hippocampal region). The most prominent feature of minimal compound plaques was Aβ (in the form of non-cored or cored plaque); however, dotted PrPSc-immunoreactivities were also present.
- (2)
- Central core deposits—this pattern occurred in both non-cored and cored Aβ plaques (3 cases out of 17). In these cases, a rather significant PrPSc positivity was observed in central non-compact Aβ plaque structures, either with or without dense Aβ cores (Figure 1e–h).
- (3)
- Diffuse plaques (4 cases out of 17):
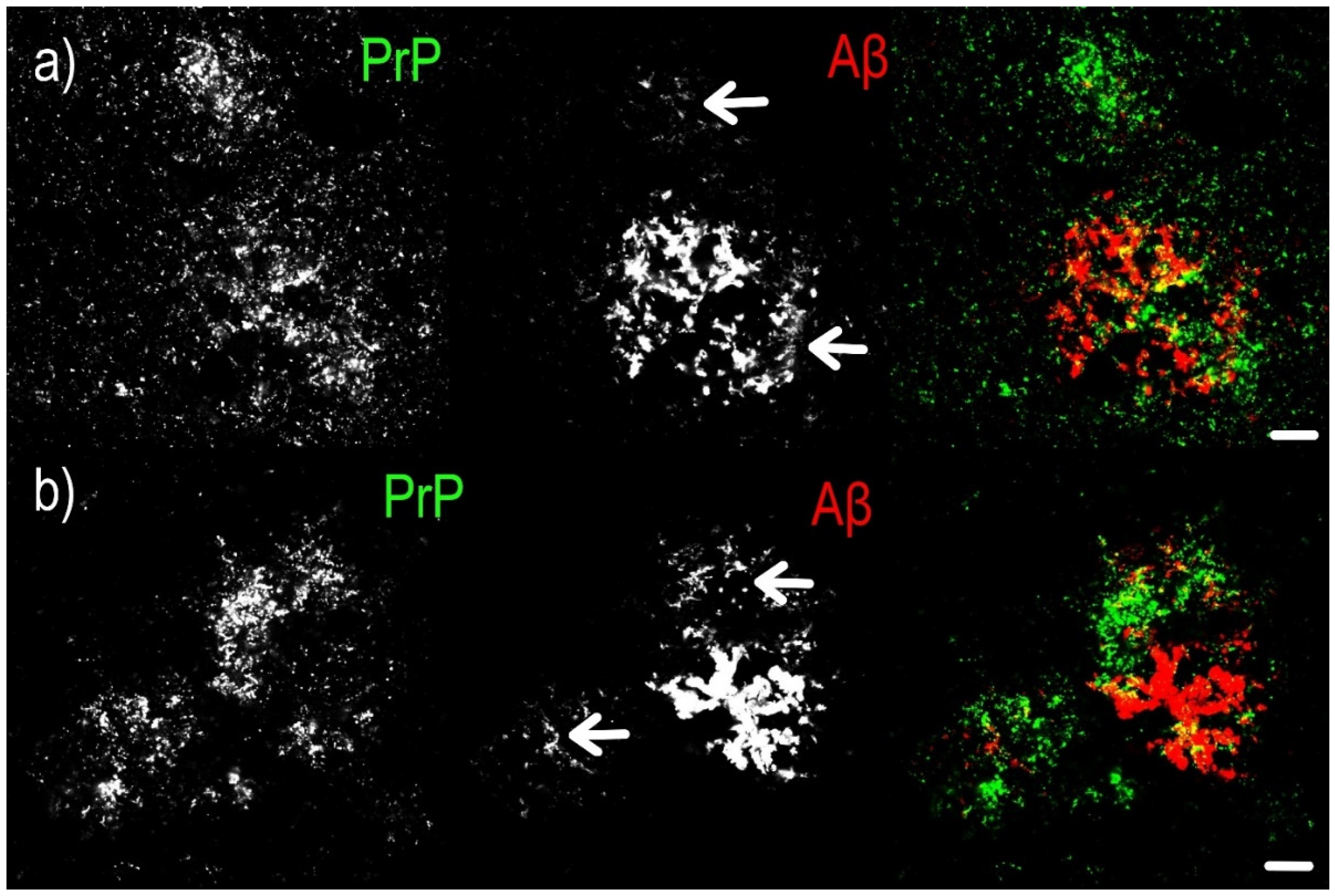
- Diffuse compound plaques (Figure 1i,j) contain PrPSc diffusely scattered in the periphery of condensed Aβ plaques and are colocalized with surrounding non-compact Aβ (seen in a single case).
- Diffuse so-called “Yin Yang” compound plaques (Figure 1k,l; Figure 2) are a particular subset of asymmetric diffuse compound plaques in which PrPSc-positive structures are polarized to the sides of asymmetric plaques in colocalization with non-compact Aβ periphery (i.e., non-compact Aβ in colocalization with PrPSc, but not surrounding the entire circumference of the compact Aβ core aggregates). This type of plaques was observed in 3 cases out of 17.
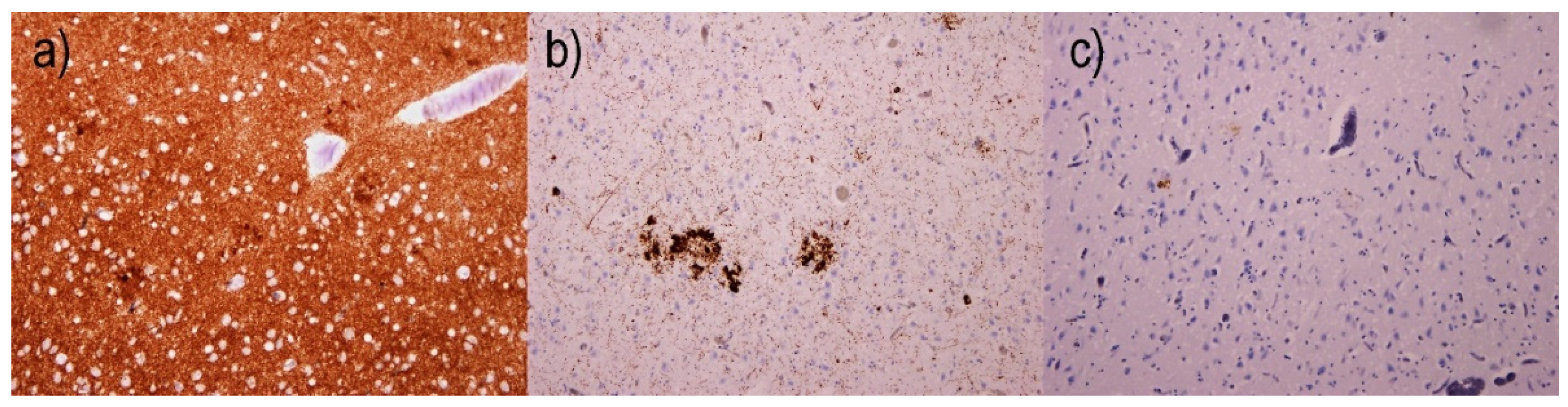
2.2. Immunohistochemical Examination
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Tissue Samples
4.3. Immunofluorescence and Immunohistochemistry
4.3.1. Primary Antibodies
4.3.2. Secondary Antibodies
4.4. Microscopy Evaluation
4.4.1. Light Microscopy
4.4.2. Confocal Microscopy
4.4.3. Classification of Aβ Plaques
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, W.-J.; Chen, W.-W.; Zhang, X. Prions mediated neurodegenerative disorders. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4028–4034. [Google Scholar]
- Thal, D.R.; Walter, J.; Saido, T.C.; Fändrich, M. Neuropathology and biochemistry of Aβ and its aggregates in Alzheimer’s disease. Acta Neuropathol. 2015, 129, 167–182. [Google Scholar] [CrossRef]
- Obeng, R. Amyloid Beta and Amyloid Beta Precursor Protein. Available online: https://www.pathologyoutlines.com/topic/stainsamyloidbetaapp.html (accessed on 12 November 2020).
- Litak, J.; Mazurek, M.; Kulesza, B.; Szmygin, P.; Litak, J.; Kamieniak, P.; Grochowski, C. Cerebral Small Vessel Disease. Int. J. Mol. Sci. 2020, 21, 9729. [Google Scholar] [CrossRef]
- Ben Halima, S.; Mishra, S.; Raja, K.M.P.; Willem, M.; Baici, A.; Simons, K.; Brüstle, O.; Koch, P.; Haass, C.; Caflisch, A.; et al. Specific Inhibition of β-Secretase Processing of the Alzheimer Disease Amyloid Precursor Protein. Cell Rep. 2016, 14, 2127–2141. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.; Udgaonkar, J.B. Molecular Mechanism of the Misfolding and Oligomerization of the Prion Protein: Current Understanding and Its Implications. Biochemistry 2015, 54, 4431–4442. [Google Scholar] [CrossRef]
- Van Der Kant, R.; Goldstein, L.S. Cellular Functions of the Amyloid Precursor Protein from Development to Dementia. Dev. Cell 2015, 32, 502–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamez, P.; Caballero, A.B. Copper in Alzheimer’s disease: Implications in amyloid aggregation and neurotoxicity. AIP Adv. 2015, 5, 092503. [Google Scholar] [CrossRef] [Green Version]
- Atwood, C.S.; Scarpa, R.C.; Huang, X.; Moir, R.D.; Jones, W.D.; Fairlie, D.P.; Tanzi, R.E.; Bush, A.I. Characterization of Copper Interactions with Alzheimer Amyloid β Peptides. J. Neurochem. 2008, 75, 1219–1233. [Google Scholar] [CrossRef]
- Watts, J.C.; Bourkas, M.E.C.; Arshad, H. The function of the cellular prion protein in health and disease. Acta Neuropathol. 2017, 135, 159–178. [Google Scholar] [CrossRef] [PubMed]
- Gunther, E.C.; Strittmatter, S.M. β-amyloid oligomers and cellular prion protein in Alzheimer’s disease. J. Mol. Med. 2010, 88, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Parkin, E.T.; Watt, N.T.; Hussain, I.; Eckman, E.A.; Eckman, C.B.; Manson, J.C.; Baybutt, H.N.; Turner, A.J.; Hooper, N.M. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2007, 104, 11062–11067. [Google Scholar] [CrossRef] [Green Version]
- Ezpeleta, J.; Baudouin, V.; Arellano-Anaya, Z.E.; Boudet-Devaud, F.; Pietri, M.; Baudry, A.; Haeberlé, A.-M.; Bailly, Y.; Kellermann, O.; Launay, J.-M.; et al. Production of seedable Amyloid-β peptides in model of prion diseases upon PrPSc-induced PDK1 overactivation. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plant, L.D.; Boyle, J.P.; Smith, I.F.; Peers, C.; Pearson, H.A. The Production of Amyloid β Peptide Is a Critical Requirement for the Viability of Central Neurons. J. Neurosci. 2003, 23, 5531–5535. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B.; Sunyach, C.; Orzechowski, H.-D.; George-Hyslop, P.S.; Checler, F. p53-Dependent Transcriptional Control of Cellular Prion by Presenilins. J. Neurosci. 2009, 29, 6752–6760. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhao, Y.; Zhang, L.; Yu, W.; Wang, Y.; Chang, W. Cellular Prion Protein as a Receptor of Toxic Amyloid-β42 Oligomers Is Important for Alzheimer’s Disease. Front. Cell. Neurosci. 2019, 13, 339. [Google Scholar] [CrossRef] [Green Version]
- Liberski, P.P. Axonal changes in experimental prion diseases recapitulate those following constriction of postganglionic branches of the superior cervical ganglion: A comparison 40 years later. Prion 2019, 13, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.A.; Hipp, S.A.; Upadhaya, A.R.; Balakrishnan, K.; Ospitalieri, S.; Koper, M.J.; Largo-Barrientos, P.; Uytterhoeven, V.; Reichwald, J.; Rabe, S.; et al. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 2019, 138, 913–941. [Google Scholar] [CrossRef]
- Ondrejcak, T.; Klyubin, I.; Corbett, G.T.; Fraser, G.; Hong, W.; Mably, A.J.; Gardener, M.; Hammersley, J.; Perkinton, M.S.; Billinton, A.; et al. Cellular Prion Protein Mediates the Disruption of Hippocampal Synaptic Plasticity by Soluble Tau In Vivo. J. Neurosci. 2018, 38, 10595–10606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankovska, N.; Olejar, T.; Matej, R. Extracellular Amyloid Deposits in Alzheimer’s and Creutzfeldt–Jakob Disease: Similar Behavior of Different Proteins? Int. J. Mol. Sci. 2020, 22, 7. [Google Scholar] [CrossRef] [PubMed]
- Jankovska, N.; Olejar, T.; Kukal, J.; Matej, R. Different Morphology of Neuritic Plaques in the Archicortex of Alzheimer’s Disease with Comorbid Synucleinopathy: A Pilot Study. Curr. Alzheimer Res. 2021, 17, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Kai, H.; Baiardi, S.; Bartoletti-Stella, A.; Carlà, B.; Zenesini, C.; Capellari, S.; Kitamoto, T.; Parchi, P. The characterization of AD/PART co-pathology in CJD suggests independent pathogenic mechanisms and no cross-seeding between misfolded Aβ and prion proteins. Acta Neuropathol. Commun. 2019, 7, 53. [Google Scholar] [CrossRef]
- Furukawa, F.; Sanjo, N.; Kobayashi, A.; Hamaguchi, T.; Yamada, M.; Kitamoto, T.; Mizusawa, H.; Yokota, T. Specific amyloid-β42 deposition in the brain of a Gerstmann-Sträussler-Scheinker disease patient with a P105L mutation on the prion protein gene. Prion 2018, 12, 315–319. [Google Scholar] [CrossRef] [Green Version]
- Miyazono, M.; Kitamoto, T.; Iwaki, T.; Tateishi, J. Colocalization of prion protein and beta protein in the same amyloid plaques in patients with Gerstmann-Sträussler Syndrome. Acta Neuropathol. 1992, 83, 333–339. [Google Scholar] [CrossRef]
- Hainfellner, J.A.; Wanschitz, J.; Jellinger, K.; Liberski, P.P.; Gullotta, F.; Budka, H. Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jakob disease. Acta Neuropathol. 1998, 96, 116–122. [Google Scholar] [CrossRef]
- Ferrer, I.; Blanco, R.; Carmona, M.; Puig, B.; Ribera, R.; Rey, M.J.; Ribalta, T. Prion protein expression in senile plaques in Alzheimer’s disease. Acta Neuropathol. 2001, 101, 49–56. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Dickson, D.W. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders, 2nd ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2011; pp. 62–68. ISBN 978140519632. [Google Scholar]
- Han, J.; Zhang, J.; Yao, H.; Wang, X.; Li, F.; Chen, L.; Gao, C.; Gao, J.; Nie, K.; Zhou, W.; et al. Study on interaction between microtubule associated protein tau and prion protein. Sci. China Ser. C Life Sci. 2006, 49, 473–479. [Google Scholar] [CrossRef]
- Dlouhy, S.R.; Hsiao, K.; Farlow, M.R.; Foroud, T.; Conneally, P.M.; Johnson, P.; Prusiner, S.B.; Hodes, M.E.; Ghetti, B. Linkage of the Indiana kindred of Gerstmann-Sträussler-Scheinker disease to the prion protein gene. Nat. Genet. 1992, 1, 64–67. [Google Scholar] [CrossRef]
- Ishizawa, K.; Mitsufuji, T.; Shioda, K.; Kobayashi, A.; Komori, T.; Nakazato, Y.; Kitamoto, T.; Araki, N.; Yamamoto, T.; Sasaki, A. An autopsy report of three kindred in a Gerstmann-Sträussler-Scheinker disease P105L family with a special reference to prion protein, tau, and beta-amyloid. Brain Behav. 2018, 8, e01117. [Google Scholar] [CrossRef]
- Race, B.; Phillips, K.; Kraus, A.; Chesebro, B. Phosphorylated human tau associates with mouse prion protein amyloid in scrapie-infected mice but does not increase progression of clinical disease. Prion 2016, 10, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2011, 123, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Parchi, P.; De Boni, L.; Saverioni, D.; Cohen, M.L.; Ferrer, I.; Gambetti, P.; Gelpi, E.; Giaccone, G.; Hauw, J.-J.; Höftberger, R.; et al. Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: An inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol. 2012, 124, 517–529. [Google Scholar] [CrossRef] [Green Version]
- Autopsy. Netherlands Brain Bank. Available online: https://www.brainbank.nl/brain-tissue/autopsy/ (accessed on 14 February 2021).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
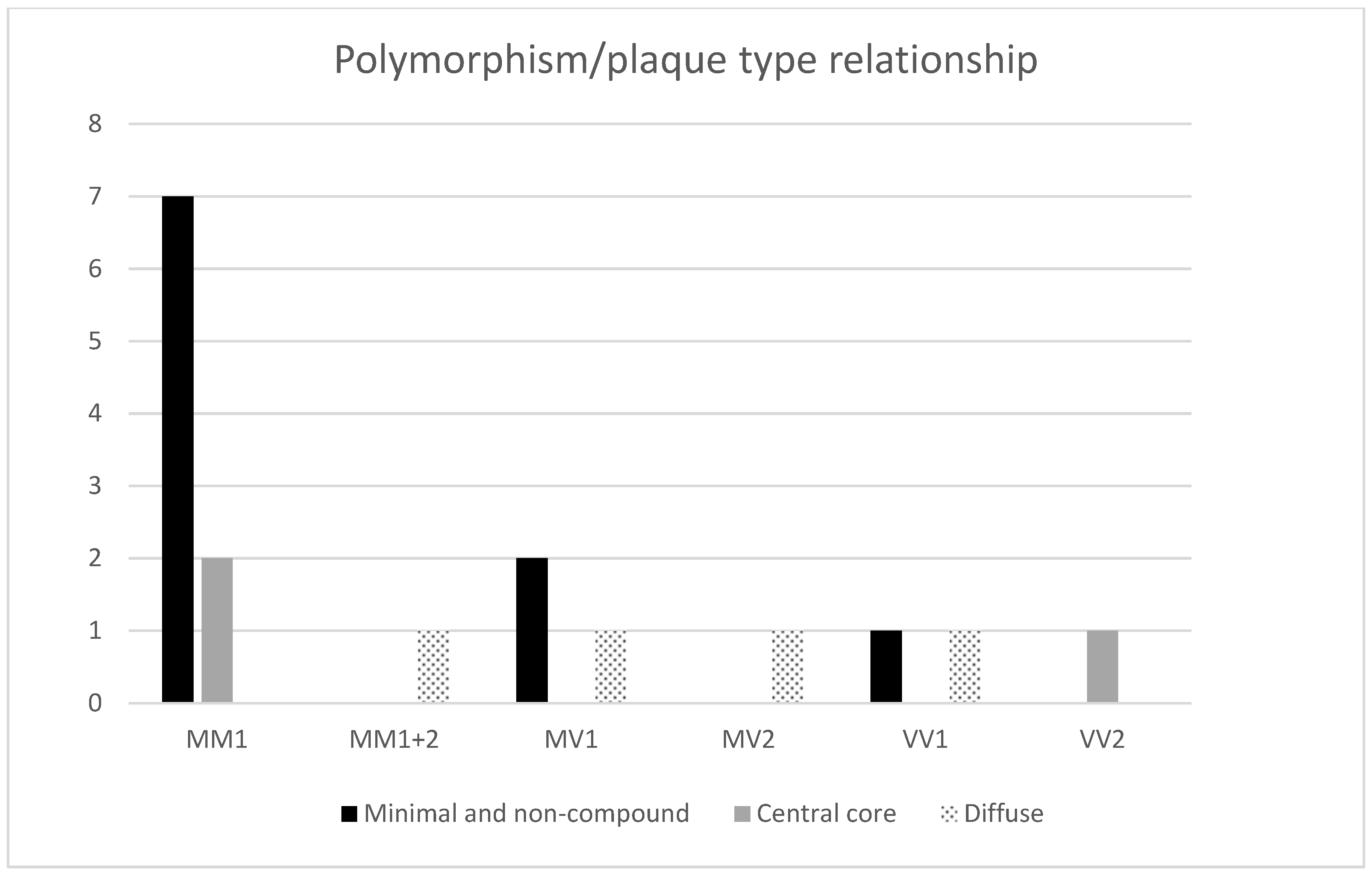
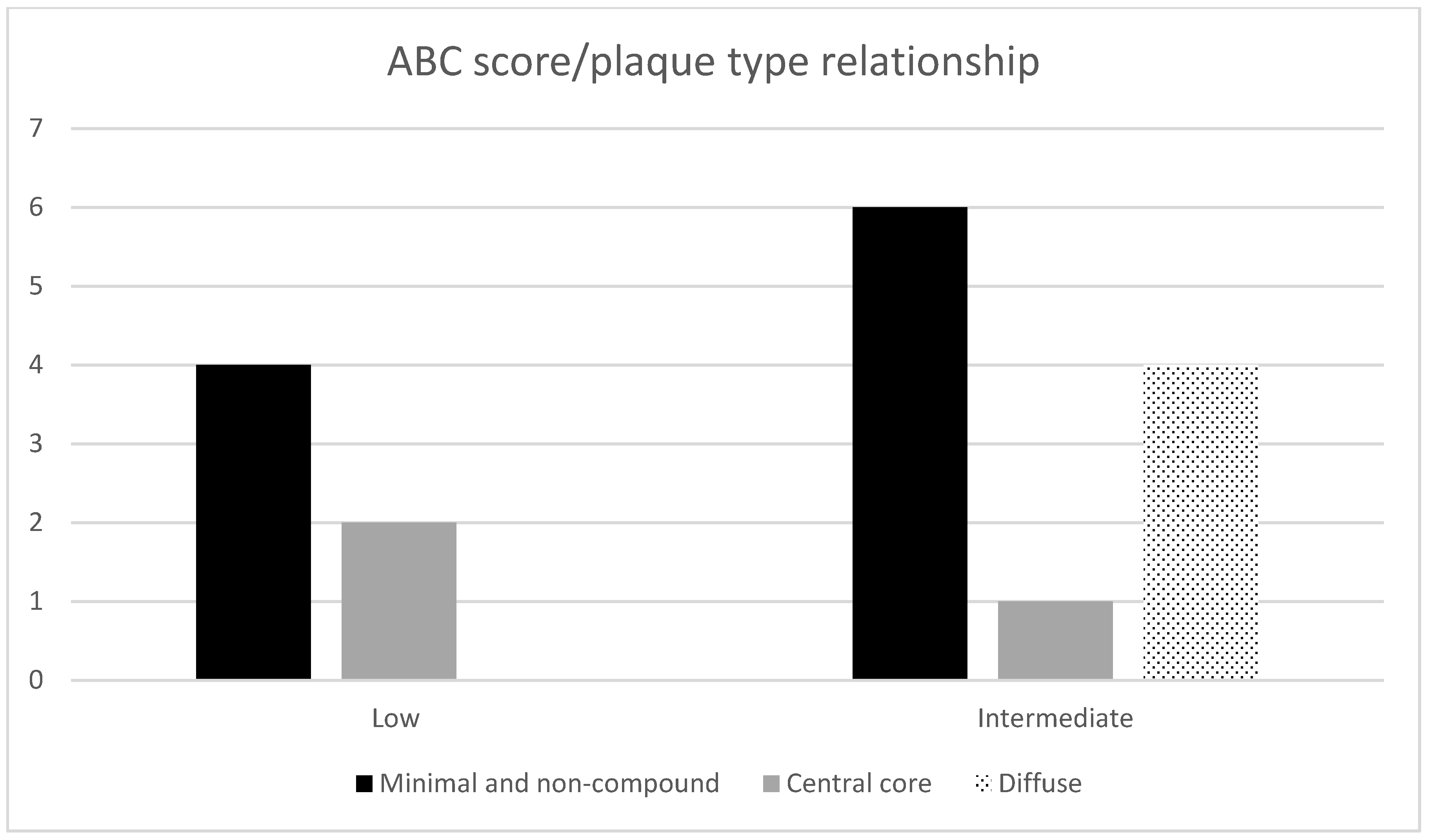
| No. | Sex | Age | Duration (Months) | Polymorph. | PrP Types Brain Tissue | Western Blot (14-3-3) CSF | AD ABC Score | Other Neuropathology | Type of Plaques |
|---|---|---|---|---|---|---|---|---|---|
| 1. | M | 75 | 2 | MM | 1 | neg. | A2B2C2 | Angiosclerotic encephalopathy, ARTAG, encephalomalacia | Non-compound |
| 2. | F | 71 | 1 | MV | 1 | low pos. | A2B1C2 | Angiosclerotic encephalopathy | Non-compound |
| 3. | F | 75 | 9 | VV | 1 | pos. | A1B1C1 | Angiosclerotic encephalopathy | Non-compound |
| 4. | F | 67 | 1 | MM | 1 | pos. | A2B1C1 | Angiosclerotic encephalopathy | Minimal compound |
| 5. | F | 69 | 2 | MM | 1 | NA | A2B2C1 | Angiosclerotic encephalopathy | Minimal compound |
| 6. | M | 62 | 6 | MV | 1 | neg. | A3B2C2 | Angiosclerotic encephalopathy | Minimal compound |
| 7. | F | 65 | 2 | MM | 1 | pos. | A2B2C2 | Angiosclerotic encephalopathy | Minimal compound |
| 8. | M | 79 | 1 | MM | 1 | pos. | A1B2C1 | Angiosclerotic encephalopathy | Minimal compound |
| 9. | M | 75 | 4 | MM | 1 | neg. | A2B2C2 | Angiosclerotic encephalopathy, ARTAG, Wernicke encephalopathy, meningioma | Minimal compound |
| 10. | F | 68 | 2 | MM | 1 | pos. | A2B2C2 | Angiosclerotic encephalopathy, AGD | Minimal compound + few compounds |
| 11. | M | 79 | 1,5 | MM | 1 | pos. | A2B2C2 | Angiosclerotic encephalopathy, ARTAG | Central core deposits |
| 12. | F | 71 | 1 | MM | 1 | pos. | A2B1C2 | Angiosclerotic encephalopathy | Central core deposits |
| 13. | F | 80 | 2 | VV | 2 | pos. | A3B1C2 | Angiosclerotic encephalopathy | Central core deposits |
| 14. | M | 64 | 2 | MV | 2 | low pos. | A2B2C2 | Angiosclerotic encephalopathy, Wernicke encephalopathy | Diffuse compound |
| 15. | F | 70 | 2 | VV | 1 | pos. | A2B2C2 | Angiosclerotic encephalopathy | Yin-Yang |
| 16. | F | 70 | 1 | MM | 1 + 2 | pos. | A2B2C3 | Angiosclerotic encephalopathy | Yin-Yang |
| 17. | F | 80 | 5 | MV | 1 | pos. | A3B2C3 | Angiosclerotic encephalopathy | Yin-Yang + few compounds |
| 18. | M | 83 | 1 | MM | 1 | low pos. | A2B2C2 | Angiosclerotic encephalopathy, ARTAG | Lacking PrP plaques in the hippocampus |
| 19. | F | 65 | 5 | VV | 1 | pos. | A1B1C1 | Angiosclerotic encephalopathy | Lacking PrP plaques in the hippocampus |
| 20. | M | 85 | 2 | MM | 1 | neg. | A2B2C2 | Angiosclerotic encephalopathy | Lacking PrP plaques in the hippocampus |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jankovska, N.; Olejar, T.; Matej, R. Extracellular Protein Aggregates Colocalization and Neuronal Dystrophy in Comorbid Alzheimer’s and Creutzfeldt–Jakob Disease: A Micromorphological Pilot Study on 20 Brains. Int. J. Mol. Sci. 2021, 22, 2099. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042099
Jankovska N, Olejar T, Matej R. Extracellular Protein Aggregates Colocalization and Neuronal Dystrophy in Comorbid Alzheimer’s and Creutzfeldt–Jakob Disease: A Micromorphological Pilot Study on 20 Brains. International Journal of Molecular Sciences. 2021; 22(4):2099. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042099
Chicago/Turabian StyleJankovska, Nikol, Tomas Olejar, and Radoslav Matej. 2021. "Extracellular Protein Aggregates Colocalization and Neuronal Dystrophy in Comorbid Alzheimer’s and Creutzfeldt–Jakob Disease: A Micromorphological Pilot Study on 20 Brains" International Journal of Molecular Sciences 22, no. 4: 2099. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042099