
Overexpression of miR-1306-5p, miR-3195, and miR-3914 Inhibits Ameloblast Differentiation through Suppression of Genes Associated with Human Amelogenesis Imperfecta

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
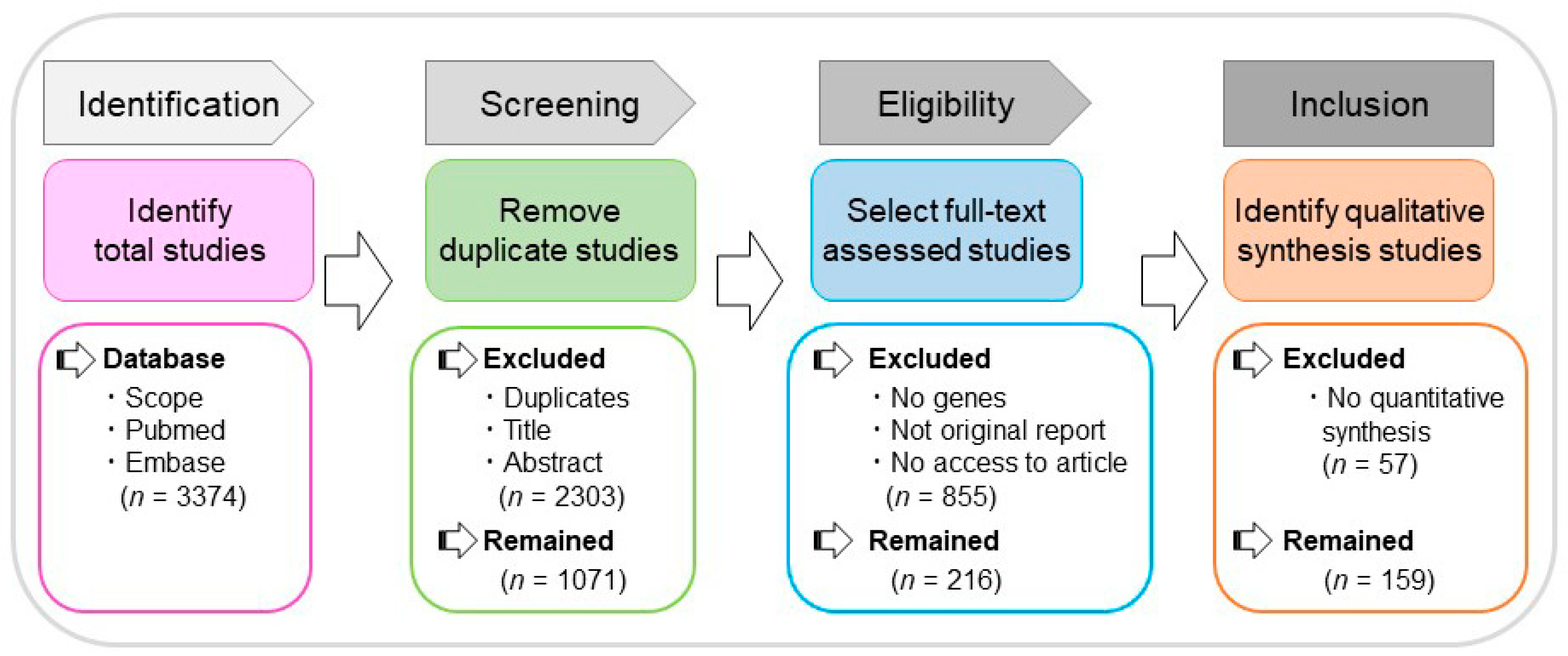
2.1. Literature Search
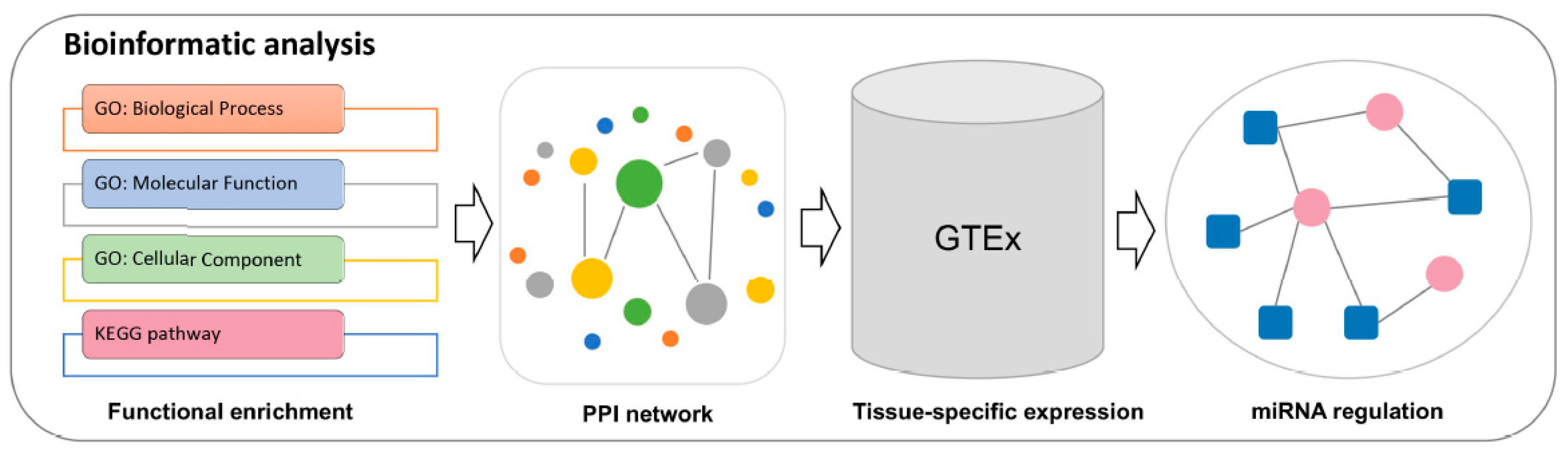
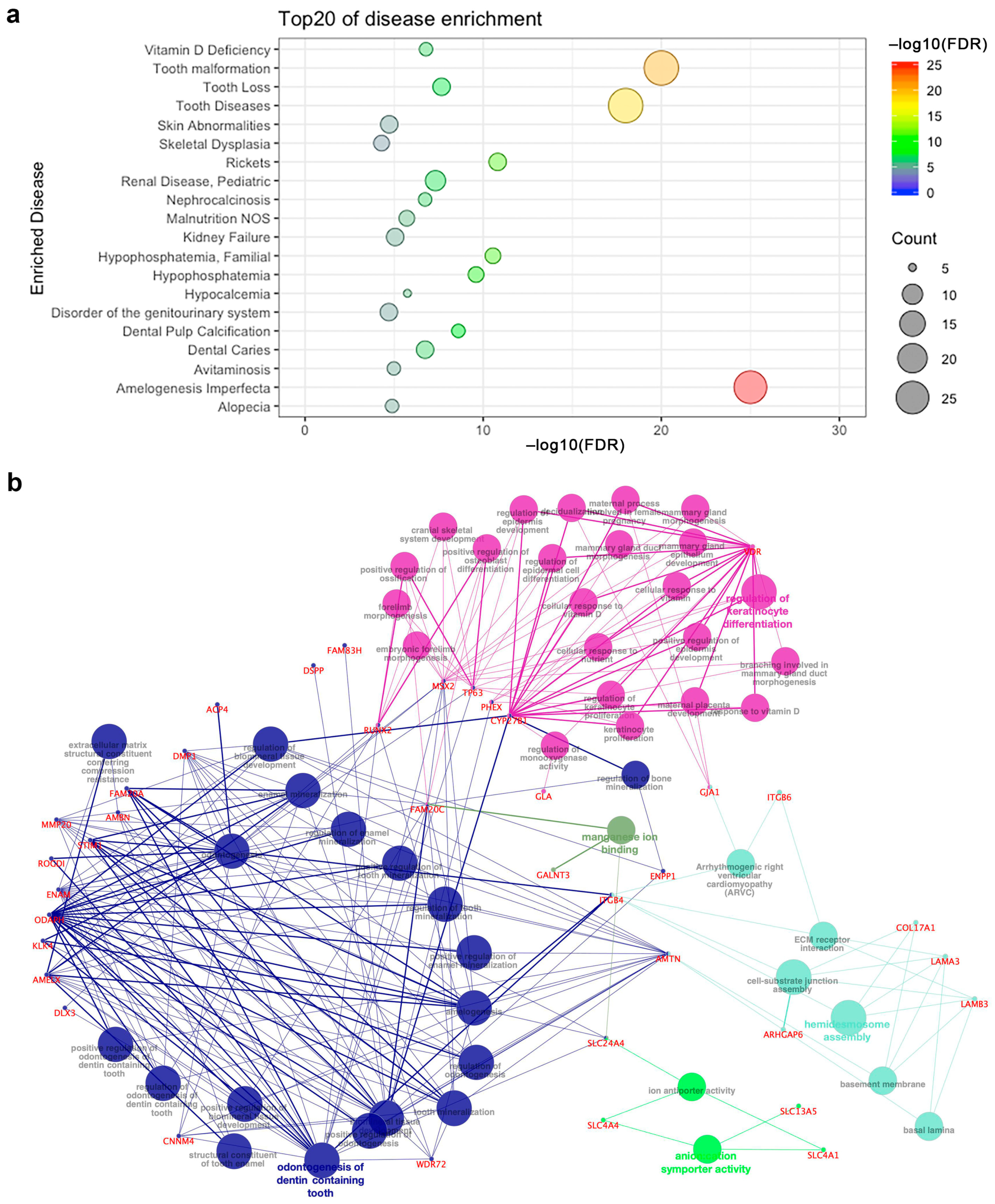
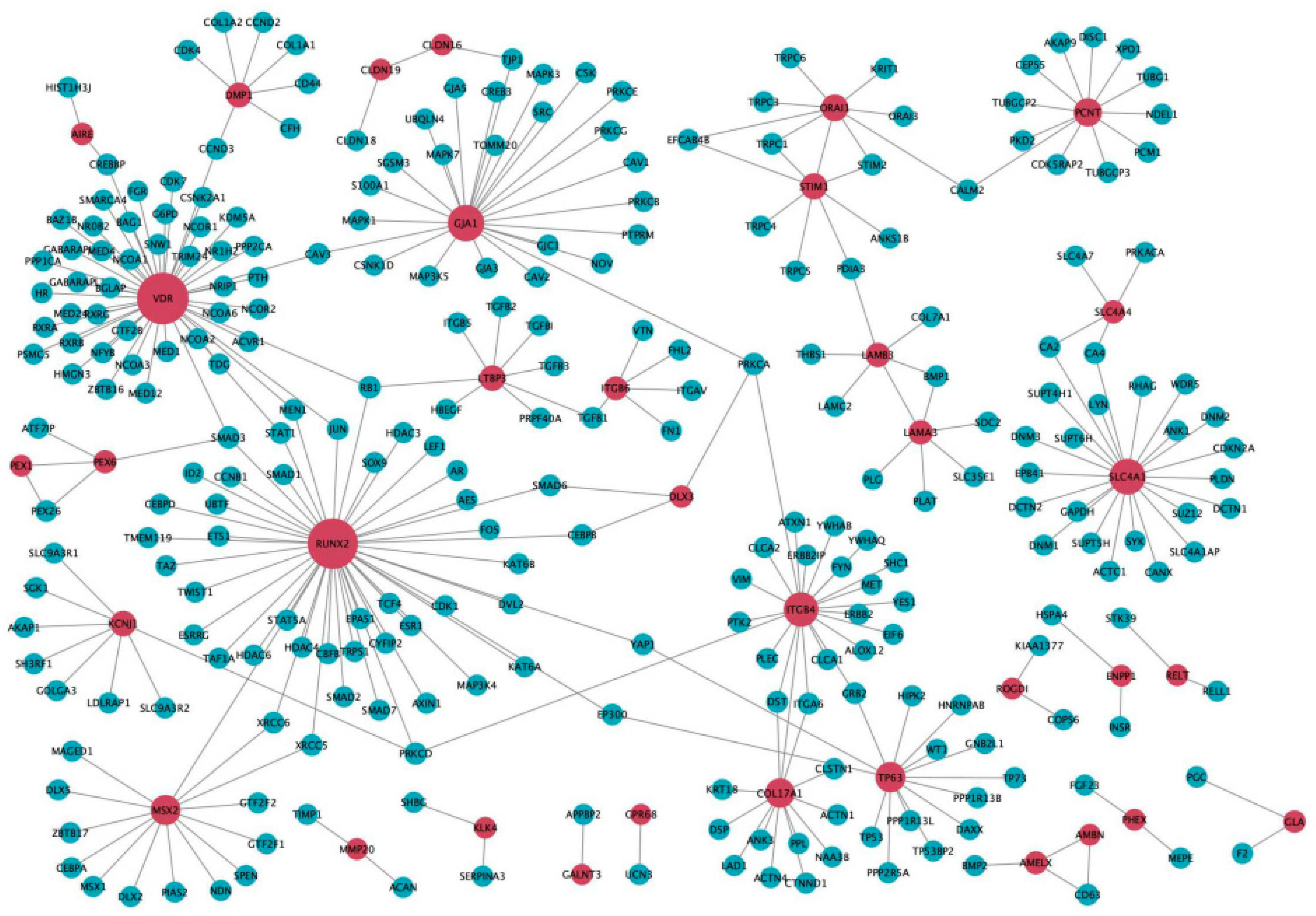
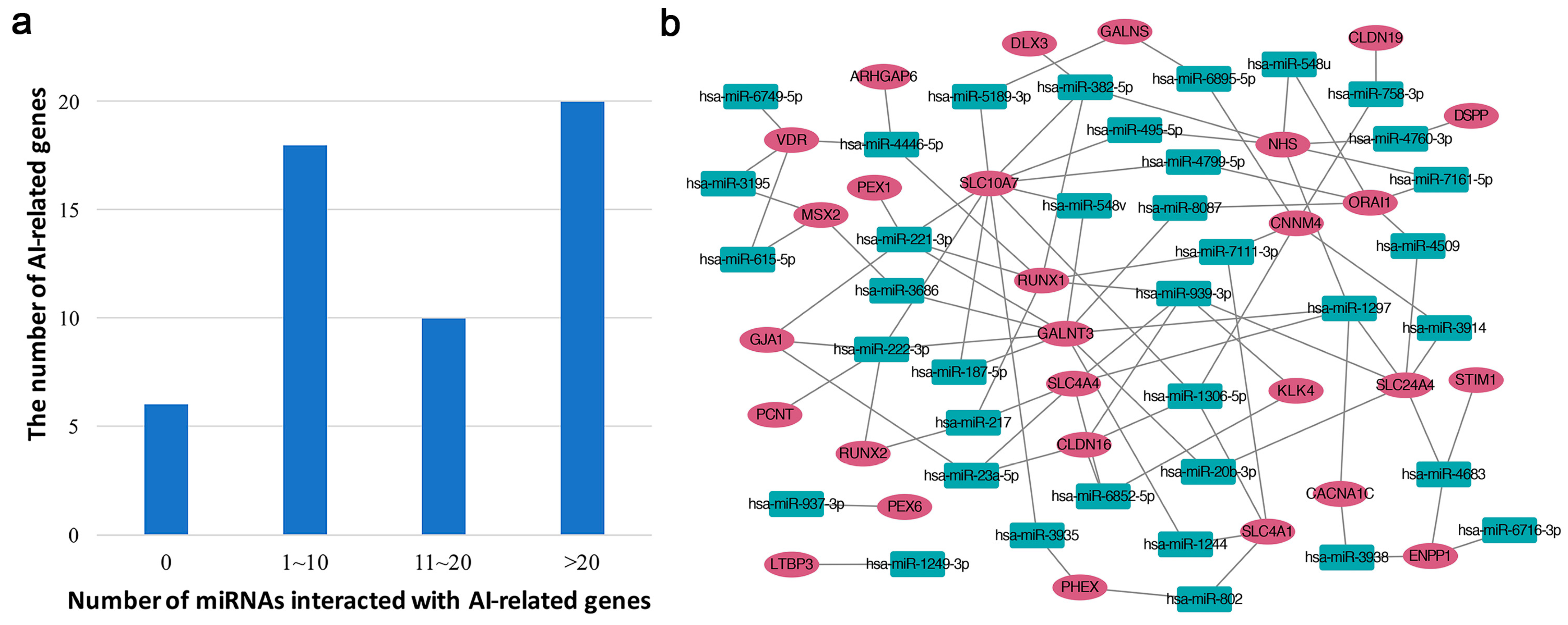
2.2. Bioinformatic Analysis
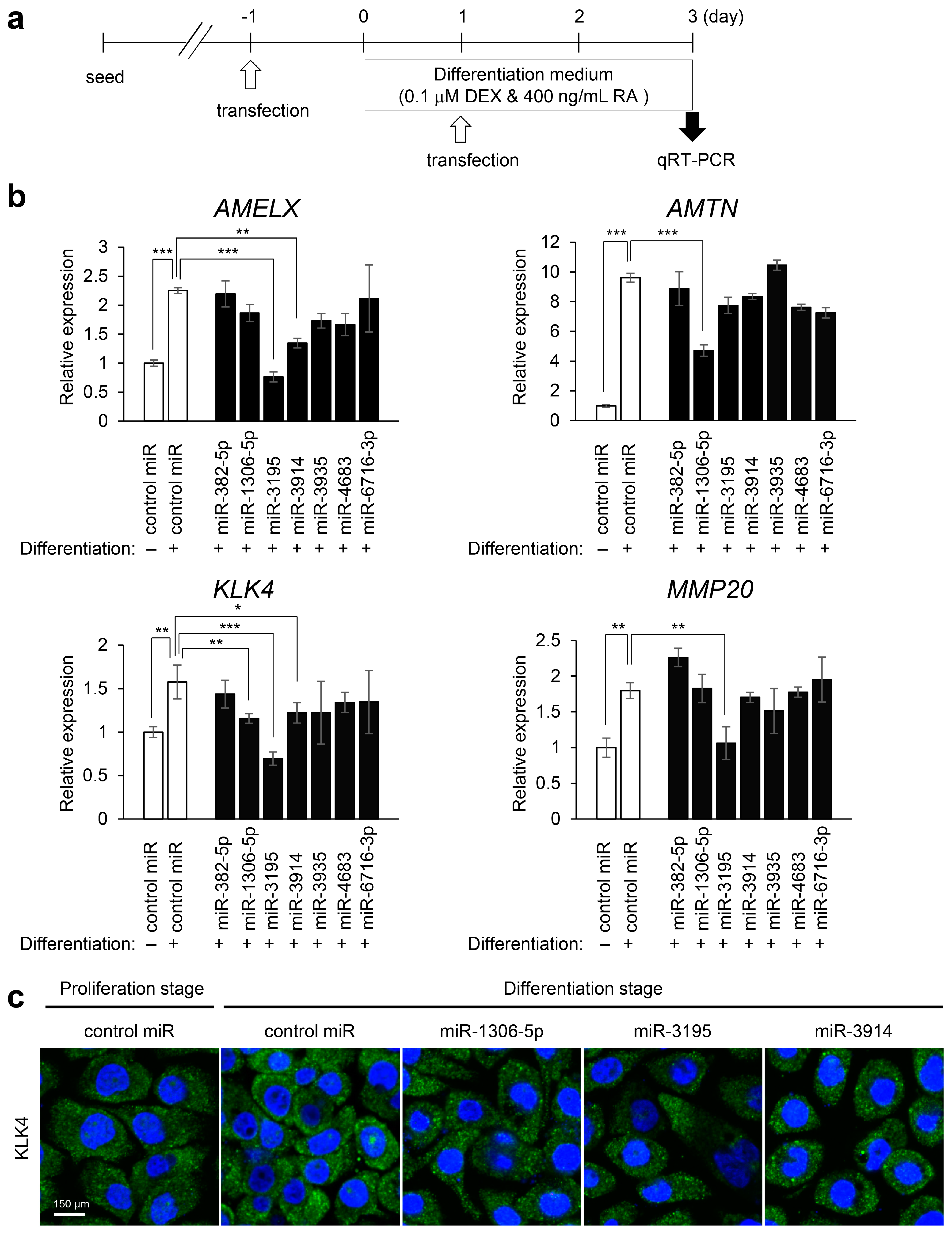
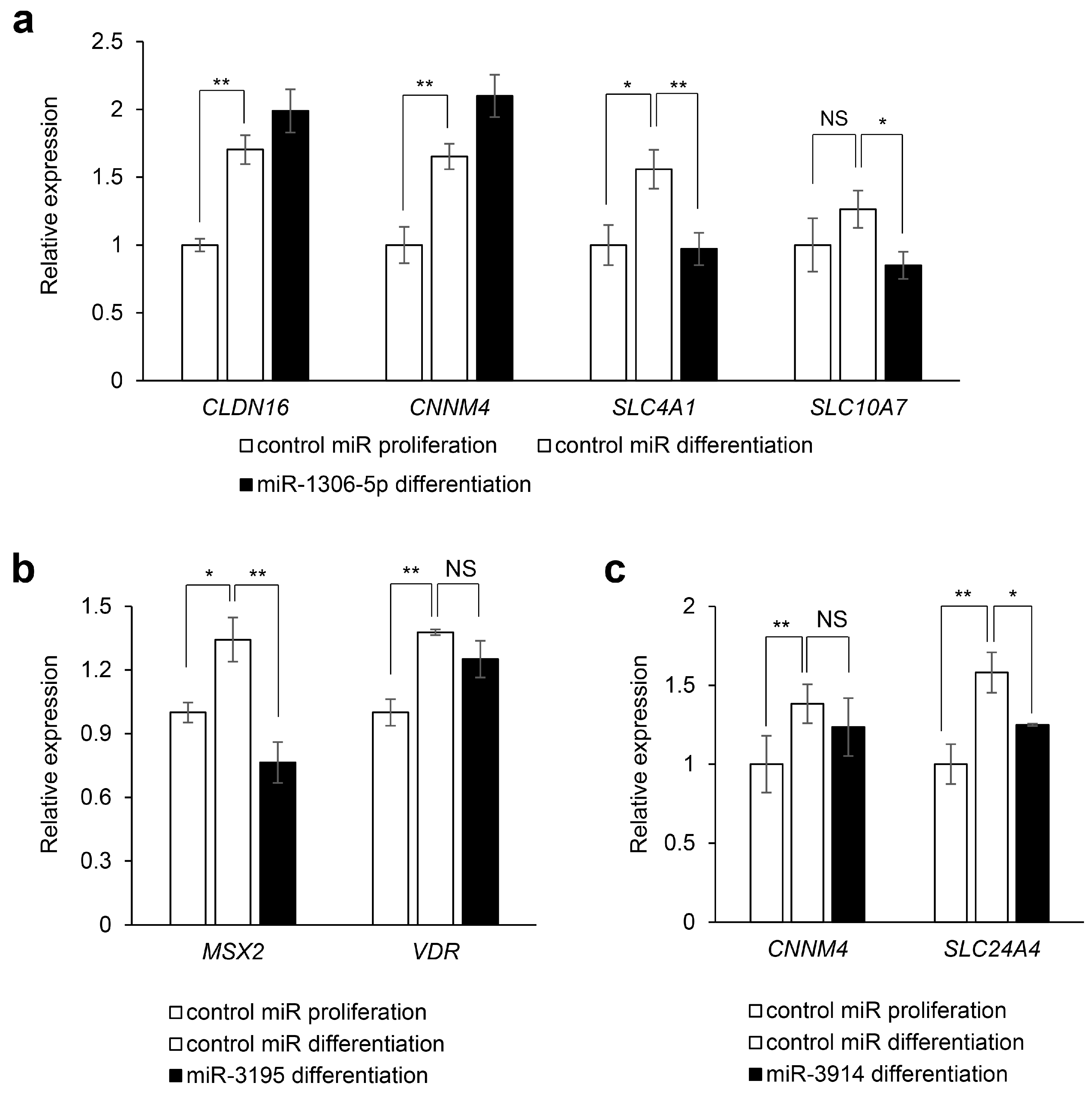
2.3. Experimental Validation
3. Discussion
4. Materials and Methods
4.1. Systematic Review
4.2. Bioinformatic Analyses
4.3. Cell Culture
4.4. Quantitative Revers Transcription Polymerase Chain Reaction (RT-PCR)
4.5. Immunofluorescence Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paine, M.L.; White, S.N.; Luo, W.; Fong, H.; Sarikaya, M.; Snead, M.L. Regulated gene expression dictates enamel structure and tooth function. Matrix Biol. 2001, 20, 273–292. [Google Scholar] [CrossRef]
- Margolis, H.C.; Beniash, E.; Fowler, C.E. Role of macromolecular assembly of enamel matrix proteins in enamel formation. J. Dent. Res. 2006, 85, 775–793. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Papagerakis, P.; Yamakoshi, Y.; Hu, J.C.; Bartlett, J.D.; Simmer, J.P. Functions of KLK4 and MMP-20 in dental enamel formation. Biol. Chem. 2008, 389, 695–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margolis, H.C.; Kwak, S.Y.; Yamazaki, H. Role of mineralization inhibitors in the regulation of hard tissue biomineralization: Relevance to initial enamel formation and maturation. Front. Physiol. 2014, 5, 339. [Google Scholar] [CrossRef] [Green Version]
- Balic, A.; Thesleff, I. Tissue Interactions Regulating Tooth Development and Renewal. Curr. Top. Dev. Biol. 2015, 115, 157–186. [Google Scholar]
- Kwak, S.Y.; Wiedemann-Bidlack, F.B.; Beniash, E.; Yamakoshi, Y.; Simmer, J.P.; Litman, A.; Margolis, H.C. Role of 20-kDa amelogenin (P148) phosphorylation in calcium phosphate formation in vitro. J. Biol. Chem. 2009, 284, 18972–18979. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Xiao, J.; Tagliabracci, V.S.; Wen, J.; Rahdar, M.; Dixon, J.E. A secretory kinase complex regulates extracellular protein phosphorylation. Elife 2015, 4, e06120. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.; Yan, W.; Tian, Y.; He, J.; Brookes, S.J.; Wang, X. The Importance of Serine Phosphorylation of Ameloblastin on Enamel Formation. J. Dent. Res. 2016, 95, 1408–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.J.; Ma, P.; Tian, Y.; Wang, J.Y.; Qin, C.L.; Feng, J.Q.; Wang, X.F. The importance of a potential phosphorylation site in enamelin on enamel formation. Int. J. Oral Sci. 2017, 9, e4. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.Y.; Yamazaki, H.; Beniash, E.; Yang, X.; Margolis, S.S.; Pugach, M.K.; Simmer, J.P.; Margolis, H.C. Amelogenin phosphorylation regulates tooth enamel formation by stabilizing a transient amorphous mineral precursor. J. Biol. Chem. 2020, 295, 1943–1959. [Google Scholar] [CrossRef]
- Bori, E.; Guo, J.; Racz, R.; Burghardt, B.; Foldes, A.; Keremi, B.; Harada, H.; Steward, M.C.; Den Besten, P.; Bronckers, A.L.; et al. Evidence for Bicarbonate Secretion by Ameloblasts in a Novel Cellular Model. J. Dent. Res. 2016, 95, 588–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, K.; Paine, M.L. Bicarbonate Transport During Enamel Maturation. Calcif. Tissue Int. 2017, 101, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.E.; Hong, J.H. The overview of channels, transporters, and calcium signaling molecules during amelogenesis. Arch. Oral Biol. 2018, 93, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Varga, G.; DenBesten, P.; Racz, R.; Zsembery, A. Importance of bicarbonate transport in pH control during amelogenesis—Need for functional studies. Oral Dis. 2018, 24, 879–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paine, M.L.; Snead, M.L. Tooth developmental biology: Disruptions to enamel-matrix assembly and its impact on biomineralization. Orthod. Craniofac. Res. 2005, 8, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Coffield, K.D.; Phillips, C.; Brady, M.; Roberts, M.W.; Strauss, R.P.; Wright, J.T. The psychosocial impact of developmental dental defects in people with hereditary amelogenesis imperfecta. J. Am. Dent. Assoc. 2005, 136, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Aldred, M.J.; Savarirayan, R.; Crawford, P.J. Amelogenesis imperfecta: A classification and catalogue for the 21st century. Oral Dis. 2003, 9, 19–23. [Google Scholar] [CrossRef]
- Witkop, C.J., Jr. Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: Problems in classification. J. Oral Pathol. 1988, 17, 547–553. [Google Scholar] [CrossRef]
- Divaris, K. Precision Dentistry in Early Childhood: The Central Role of Genomics. Dent. Clin. N. Am. 2017, 61, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Strauch, S.; Hahnel, S. Restorative Treatment in Patients with Amelogenesis Imperfecta: A Review. J. Prosthodont. 2018, 27, 618–623. [Google Scholar] [CrossRef]
- Obernosterer, G.; Leuschner, P.J.; Alenius, M.; Martinez, J. Post-transcriptional regulation of microRNA expression. RNA 2006, 12, 1161–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- DeVeale, B.; Swindlehurst-Chan, J.; Blelloch, R. The roles of microRNAs in mouse development. Nat. Rev. Genet. 2021, 1–17. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, X.; Lin, C.; Jia, X.; Zhu, H.; Song, J.; Zhang, Y. Noncoding RNAs regulate alternative splicing in Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 1–14. [Google Scholar] [CrossRef]
- Yin, K.; Hacia, J.G.; Zhong, Z.; Paine, M.L. Genome-wide analysis of miRNA and mRNA transcriptomes during amelogenesis. BMC Genom. 2014, 15, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Yin, K.; Lin, W.; Guo, J.; Sugiyama, T.; Snead, M.L.; Hacia, J.G.; Paine, M.L. MiR-153 Regulates Amelogenesis by Targeting Endocytotic and Endosomal/lysosomal Pathways-Novel Insight into the Origins of Enamel Pathologies. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Wang, J.; Li, X.; Florez, S.; Huang, Z.; Venugopalan, S.R.; Elangovan, S.; Skobe, Z.; Margolis, H.C.; Martin, J.F.; et al. MicroRNAs play a critical role in tooth development. J. Dent. Res. 2010, 89, 779–784. [Google Scholar] [CrossRef]
- Michon, F.; Tummers, M.; Kyyronen, M.; Frilander, M.J.; Thesleff, I. Tooth morphogenesis and ameloblast differentiation are regulated by micro-RNAs. Dev. Biol. 2010, 340, 355–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehic, A.; Risnes, S.; Khuu, C.; Khan, Q.E.; Osmundsen, H. Effects of in vivo transfection with anti-miR-214 on gene expression in murine molar tooth germ. Physiol. Genom. 2011, 43, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Khan, Q.E.; Sehic, A.; Khuu, C.; Risnes, S.; Osmundsen, H. Expression of Clu and Tgfb1 during murine tooth development: Effects of in-vivo transfection with anti-miR-214. Eur. J. Oral Sci. 2013, 121, 303–312. [Google Scholar] [CrossRef]
- Peri, S.; Navarro, J.D.; Kristiansen, T.Z.; Amanchy, R.; Surendranath, V.; Muthusamy, B.; Gandhi, T.K.; Chandrika, K.N.; Deshpande, N.; Suresh, S.; et al. Human protein reference database as a discovery resource for proteomics. Nucleic Acids Res. 2004, 32, D497–D501. [Google Scholar] [CrossRef] [PubMed]
- Chatr-Aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A.; et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017, 45, D369–D379. [Google Scholar] [CrossRef]
- Kerrien, S.; Alam-Faruque, Y.; Aranda, B.; Bancarz, I.; Bridge, A.; Derow, C.; Dimmer, E.; Feuermann, M.; Friedrichsen, A.; Huntley, R.; et al. IntAct--open source resource for molecular interaction data. Nucleic Acids Res. 2007, 35, D561–D565. [Google Scholar] [CrossRef]
- Chatr-aryamontri, A.; Ceol, A.; Palazzi, L.M.; Nardelli, G.; Schneider, M.V.; Castagnoli, L.; Cesareni, G. MINT: The Molecular INTeraction database. Nucleic Acids Res. 2007, 35, D572–D574. [Google Scholar] [CrossRef] [PubMed]
- Xenarios, I.; Salwinski, L.; Duan, X.J.; Higney, P.; Kim, S.M.; Eisenberg, D. DIP, the Database of Interacting Proteins: A research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002, 30, 303–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, G.T. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Pei, G.; Dai, Y.; Zhao, Z.; Jia, P. deTS: Tissue-specific enrichment analysis to decode tissue specificity. Bioinformatics 2019, 35, 3842–3845. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Mitsuyasu, T.; Nakamura, N.; Higuchi, Y.; Toyoshima, K.; Taniguchi, A.; Yasumoto, S. Establishment of ameloblastoma cell line, AM-1. J. Oral Pathol. Med. 1998, 27, 207–212. [Google Scholar] [CrossRef]
- Chen, L.S.; Couwenhoven, R.I.; Hsu, D.; Luo, W.; Snead, M.L. Maintenance of amelogenin gene expression by transformed epithelial cells of mouse enamel organ. Arch. Oral Biol. 1992, 37, 771–778. [Google Scholar] [CrossRef]
- Wang, G.; Yu, J.; Yang, Y.; Liu, X.; Zhao, X.; Guo, X.; Duan, T.; Lu, C.; Kang, J. Whole-transcriptome sequencing uncovers core regulatory modules and gene signatures of human fetal growth restriction. Clin. Transl. Med. 2020, 9, 9. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Gao, Z.; Tian, L.; Liu, M. Expressions of miR-122a and miR-3195 in laryngeal cancer and their effects on the proliferation and apoptosis of laryngeal cancer cell Hep-2. Adv. Clin. Exp. Med. 2020, 29, 525–534. [Google Scholar] [CrossRef]
- Kumar, S.; Sharawat, S.K.; Ali, A.; Gaur, V.; Malik, P.S.; Pandey, M.; Kumar, S.; Mohan, A.; Guleria, R. Differential expression of circulating serum miR-1249-3p, miR-3195, and miR-3692-3p in non-small cell lung cancer. Hum. Cell 2020, 33, 839–849. [Google Scholar] [CrossRef]
- Valera, V.A.; Parra-Medina, R.; Walter, B.A.; Pinto, P.; Merino, M.J. microRNA Expression Profiling in Young Prostate Cancer Patients. J. Cancer 2020, 11, 4106–4114. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Sun, H.; Liu, H.; Ma, L.; Jiang, C.; Liao, H.; Xu, S.; Xiang, J.; Cao, Z. MicroRNA-181b-5p modulates tumor necrosis factor-alpha-induced inflammatory responses by targeting interleukin-6 in cementoblasts. J. Cell. Physiol. 2019, 234, 22719–22730. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, D.; Zheng, J.; Deng, L.; Chen, Z.; Ling, J.; Wu, L. Effect of microRNA-21 on hypoxia-inducible factor-1alpha in orthodontic tooth movement and human periodontal ligament cells under hypoxia. Exp. Ther. Med. 2019, 17, 2830–2836. [Google Scholar]
- Weng, Q.; Yi, F.; Yu, Y.; Ge, S.; Liu, S.; Zhang, Y. Altered miRNA expression profiling in enamel organ of fluoride affected rat embryos. Ecotoxicol. Environ. Saf. 2021, 210, 111876. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef]
- Suzuki, A.; Pelikan, R.C.; Iwata, J. WNT/beta-Catenin Signaling Regulates Multiple Steps of Myogenesis by Regulating Step-Specific Targets. Mol. Cell. Biol. 2015, 35, 1763–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, A.; Ogata, K.; Yoshioka, H.; Shim, J.; Wassif, C.A.; Porter, F.D.; Iwata, J. Disruption of Dhcr7 and Insig1/2 in cholesterol metabolism causes defects in bone formation and homeostasis through primary cilium formation. Bone Res. 2020, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolated vs. Syndromic | Genes |
|---|---|
| Isolated | ACP4, AMBN, AMELX, AMTN, ARHGAP6, DLX3, ENAM, FAM83H, GPR68, ITGB6, KLK4, LAMA3, LAMB3, MMP20, ODAPH, SLC24A4, WDR72 |
| Syndromic | AIRE, ALPL, AMELX, CACNA1C, CLDN16, CLDN19, CNNM4, COL17A1, CYP27B1, DLX3, DMP1, DSPP, ENPP1, FMA20A, FAM20C, GALNS, GALNT3, GJA1, GLA, ITGB4, KCNJ1, LAMA3, LAMB3, LTBP3, MSX2, NHS, ORAI1, PCNT, PEX1, PEX6, PEX26, PHEX, RELT, ROGDI, RUNX1, RUNX2, SLC4A1, SLC4A4, SLC10A7, SLC13A5, STIM1, TP63, VDR, WDR72 |
| Inheritance Type | Genes |
|---|---|
| autosomal recessive | ACP4, AIRE, ALPL, AMBN, CLDN16, CLDN19, CNNM4, CYP27B1, DMP1, ENPP1, FAM20A, FAM20C, GALNS, GALNT3, GPR68, ITGB4, ITGB6, KCNJ1, KLK4, LAMA3, LAMB3, LTBP3, MMP20, ODAPH, ORAI1, PCNT, PEX1, PEX6, PEX26, RELT, ROGDI, SLC4A1, SLC4A4, SLC10A7, SLC13A5, SLC24A4, STIM1, TP63, VDR, WDR72 |
| autosomal dominant | ALPL, AMBN, AMTN, CACNA1C, CNNM4, COL17A1, DLX3, DSPP, ENAM, FAM83H, GJA1, LAMA3, LAMB3, MSX2, RUNX2, SLC4A1 |
| X-linked recessive | ARHGAP6 |
| X-linked dominant | AMELX, GLA, NHS, PHEX |
| Amelogenesis Imperfecta Category | Genes |
|---|---|
| hypoplastic | ACP4, AIRA, ALPL, AMBN, AMELX, CLDN16, CLDN19, CNNM4, COL17A1, CYP27B1, DLX3, DSPP, ENPP1, ENAM, FAM20A, FAM20C, GALNT3, GJA1, GLA, ITGB4, ITGB6, KCNJ1, LAMA3, LAMB3, LTBP3, MMP20, ODAPH, PEX1, PEX6, PEX26, PHEX, ROGDI, RUNX1, RUNX2, SLC4A4, SLC10A7, SLC13A5, TP63, VDR, WDR72 |
| hypomaturation | AMELX, CLDN16, CLDN19, CNNM4, KLK4, MMP20, MSX2, ROGDI, RUNX2, SLC24A4, STIM1, TP63, WDR72 |
| hypomineralized/hypocalcified | AMBN, AMELX, AMTN, CNNM4, DMP1, DSPP, ENAM, FAM83H, GPR68, ITGB6, KLK4, MMP20, ODAPH, ORAI1, PEX1, RELT, ROGDI, SLC10A7, SLC24A4, STIM1, TP63, WDR72 |
| hypoplastic/hypomature enamel with taurodontism | ALPL, DLX3, LAMB3 |
| not specified | ARHGAP6, CACNA1C, GALNS, NHS, PCTN, SLC4A1 |
| miRNAs | Gene Symbol | # Target | # Overlap | p-Value |
|---|---|---|---|---|
| hsa-miR-3195 | MSX2, VDR | 23 | 2 | 0.00287231 |
| hsa-miR-382-5p | DLX3, SLC10A7, NHS, RUNX1 | 186 | 4 | 0.003965 |
| hsa-miR-1306-5p | SLC4A1, CNNM4, SLC10A7, CLDN16 | 199 | 4 | 0.00503641 |
| hsa-miR-4683 | SLC24A4, ENPP1, STIM1 | 108 | 3 | 0.00629218 |
| hsa-miR-6716-3p | ENPP1 | 4 | 1 | 0.01385705 |
| hsa-miR-3914 | SLC24A4, CNNM4 | 56 | 2 | 0.01624945 |
| hsa-miR-3935 | PHEX, SLC10A7 | 56 | 2 | 0.01624945 |
| hsa-miR-23a-5p | SLC4A4, GJA1, CLDN16 | 155 | 3 | 0.0167345 |
| hsa-miR-4509 | SLC24A4, ORAI1 | 57 | 2 | 0.01680318 |
| hsa-miR-1244 | GALNT3, SLC4A1 | 59 | 2 | 0.01793454 |
| hsa-miR-939-3p | KLK4, RUNX1, SLC4A4, SLC24A4, CLDN16 | 439 | 5 | 0.01799166 |
| hsa-miR-3938 | ENPP1, CACNA1C | 65 | 2 | 0.02151565 |
| hsa-miR-802 | PHEX, SLC4A1 | 68 | 2 | 0.0234085 |
| hsa-miR-615-5p | MSX2, VDR | 70 | 2 | 0.02470722 |
| hsa-miR-4760-3p | DSPP, NHS | 71 | 2 | 0.02536746 |
| hsa-miR-221-3p | GALNT3, GJA1, PEX1, RUNX1, SLC10A7 | 489 | 5 | 0.02716289 |
| hsa-miR-1297 | GALNT3, CACNA1C, SLC4A4, SLC24A4, NHS | 496 | 5 | 0.02864777 |
| hsa-miR-6895-5p | GALNS, CNNM4 | 83 | 2 | 0.03383429 |
| hsa-miR-1249-3p | LTBP3 | 10 | 1 | 0.03428995 |
| hsa-miR-3686 | GALNT3, MSX2 | 84 | 2 | 0.03458354 |
| hsa-miR-187-5p | GALNT3, SLC10A7 | 84 | 2 | 0.03458354 |
| hsa-miR-222-3p | GALNT3, PCNT, GJA1, RUNX2, SLC10A7 | 523 | 5 | 0.03485762 |
| hsa-miR-937-3p | PEX6 | 11 | 1 | 0.03765479 |
| hsa-miR-217 | SLC4A4, RUNX2, RUNX1 | 219 | 3 | 0.04067934 |
| hsa-miR-7111-3p | SLC4A1, CNNM4, RUNX1 | 219 | 3 | 0.04067934 |
| hsa-miR-495-5p | NHS, SLC10A7 | 92 | 2 | 0.04080612 |
| hsa-miR-6749-5p | VDR | 12 | 1 | 0.04100813 |
| hsa-miR-5189-3p | GALNS, SLC10A7 | 94 | 2 | 0.04242339 |
| hsa-miR-8087 | GALNT3, ORAI1 | 94 | 2 | 0.04242339 |
| hsa-miR-4799-5p | ORAI1, SLC10A7 | 94 | 2 | 0.04242339 |
| hsa-miR-20b-3p | GALNT3, SLC24A4 | 95 | 2 | 0.04324099 |
| hsa-miR-6852-5p | SLC4A4, KLK4, CLDN16 | 228 | 3 | 0.04494714 |
| hsa-miR-4446-5p | ARHGAP6, RUNX1, VDR | 231 | 3 | 0.04641776 |
| hsa-miR-548u | ORAI1, NHS | 101 | 2 | 0.04826897 |
| hsa-miR-758-3p | CNNM4, CLDN19 | 102 | 2 | 0.04912692 |
| hsa-miR-7161-5p | ORAI1, NHS | 103 | 2 | 0.04999043 |
| hsa-miR-548v | GALNT3, SLC10A7 | 103 | 2 | 0.04999043 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshioka, H.; Wang, Y.-Y.; Suzuki, A.; Shayegh, M.; Gajera, M.V.; Zhao, Z.; Iwata, J. Overexpression of miR-1306-5p, miR-3195, and miR-3914 Inhibits Ameloblast Differentiation through Suppression of Genes Associated with Human Amelogenesis Imperfecta. Int. J. Mol. Sci. 2021, 22, 2202. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042202
Yoshioka H, Wang Y-Y, Suzuki A, Shayegh M, Gajera MV, Zhao Z, Iwata J. Overexpression of miR-1306-5p, miR-3195, and miR-3914 Inhibits Ameloblast Differentiation through Suppression of Genes Associated with Human Amelogenesis Imperfecta. International Journal of Molecular Sciences. 2021; 22(4):2202. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042202
Chicago/Turabian StyleYoshioka, Hiroki, Yin-Ying Wang, Akiko Suzuki, Meysam Shayegh, Mona V. Gajera, Zhongming Zhao, and Junichi Iwata. 2021. "Overexpression of miR-1306-5p, miR-3195, and miR-3914 Inhibits Ameloblast Differentiation through Suppression of Genes Associated with Human Amelogenesis Imperfecta" International Journal of Molecular Sciences 22, no. 4: 2202. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042202