
The Interplay between Drivers of Erythropoiesis and Iron Homeostasis in Rare Hereditary Anemias: Tipping the Balance

 , ,
, ,
Abstract
:1. Introduction
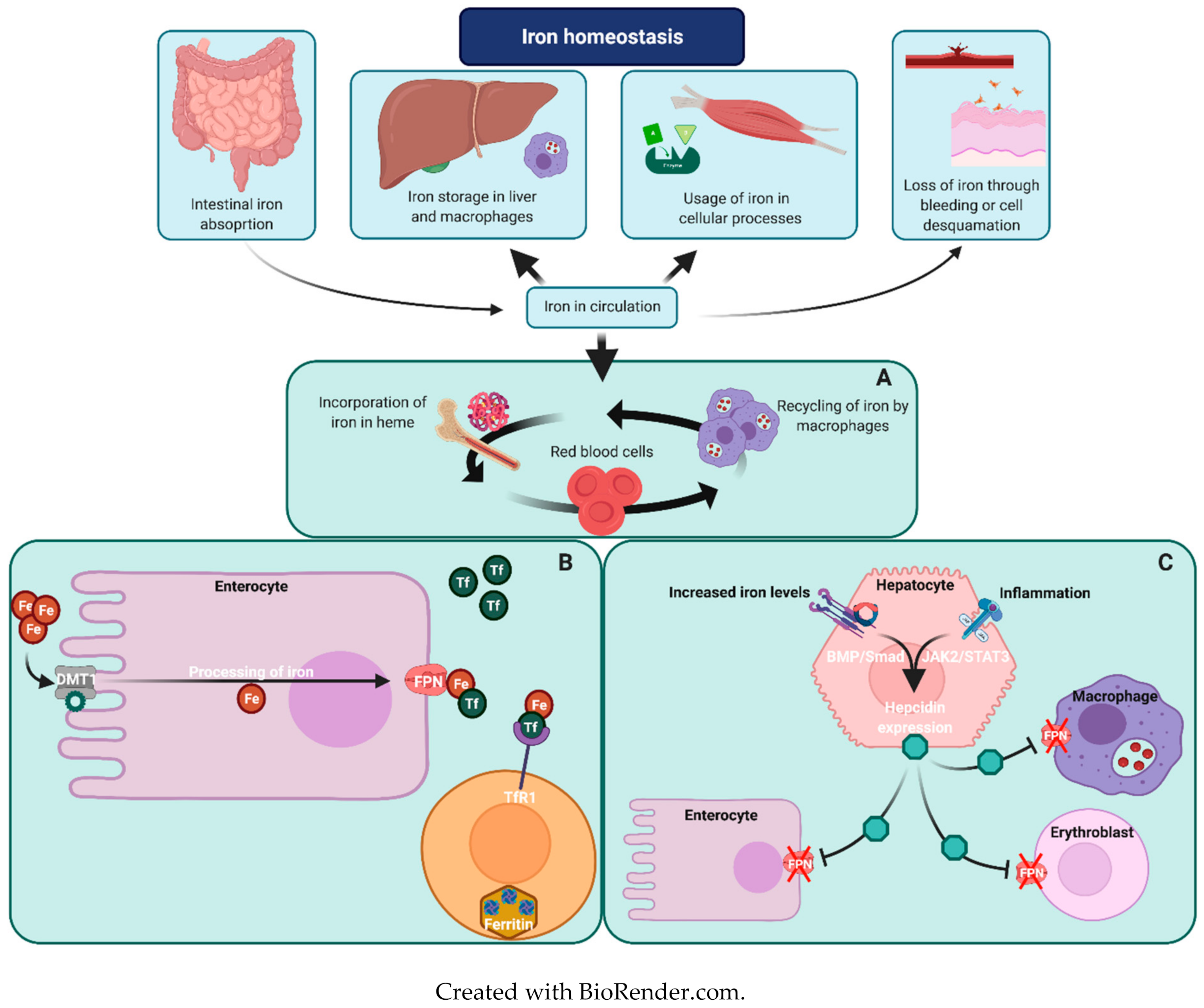
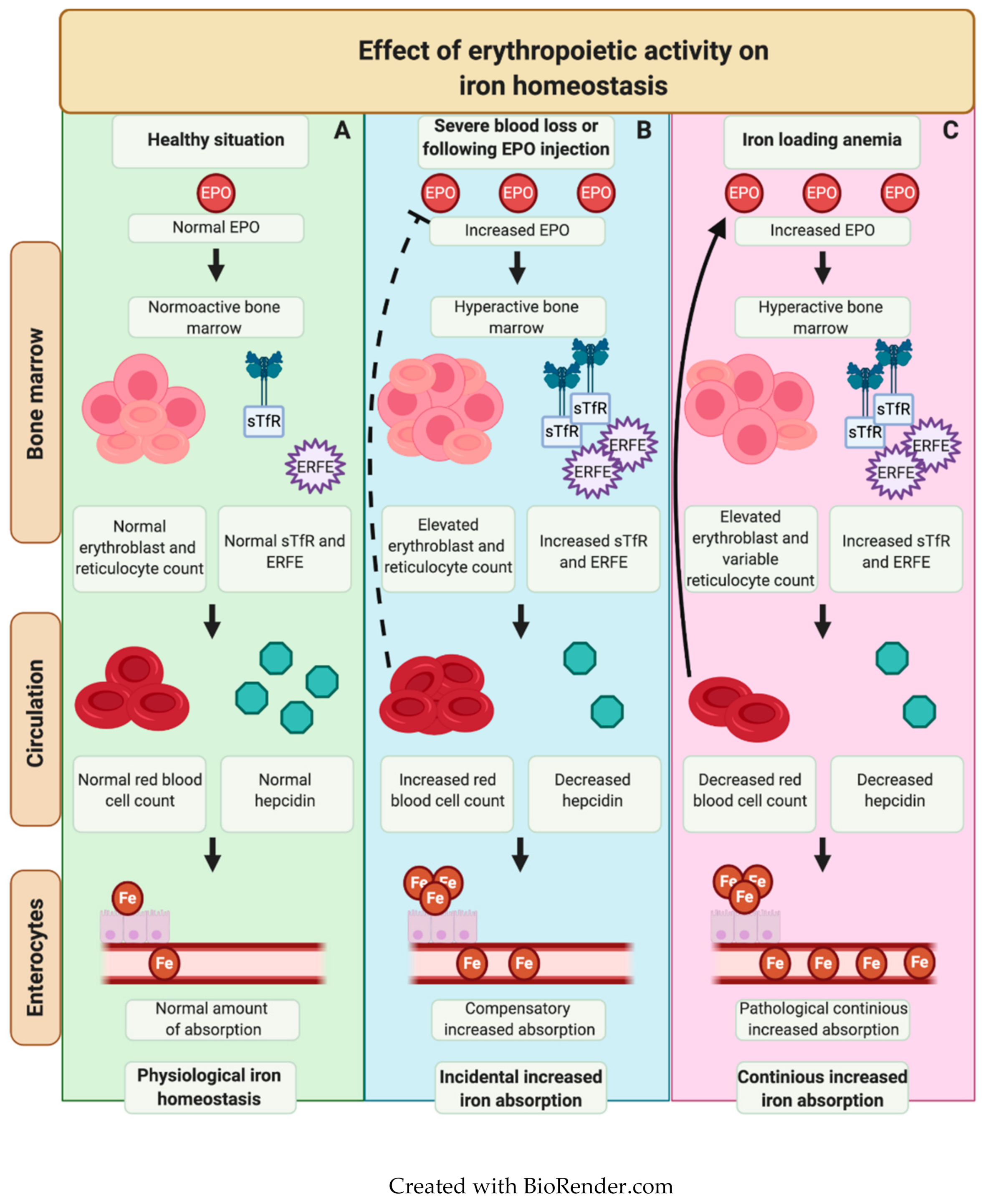
Iron Metabolism in Erythropoiesis
2. Regulation of Iron Overload in RHA
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kassebaum, N.J.; Jasrasaria, R.; Naghavi, M.; Wulf, S.K.; Johns, N.; Lozano, R.; Regan, M.; Weatherall, D.; Chou, D.P.; Eisele, T.P.; et al. A systematic analysis of global anemia burden from 1990 to 2010. Blood 2014, 123, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Modell, B.; Darlison, M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull. World Health Organ. 2008, 86, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E.; Gelbart, T. Estimating the prevalence of pyruvate kinase deficiency from the gene frequency in the general white population. Blood 2000, 95, 3585–3588. [Google Scholar] [CrossRef]
- Gulbis, B.; Eleftheriou, A.; Angastiniotis, M.; Ball, S.; Surrallés, J.; Castella, M.; Heimpel, H.; Hill, A.; Corrons, J.-L.V. Epidemiology of Rare Anaemias in Europe. Adv. Exp. Med. Biol. 2010, 686, 375–396. [Google Scholar] [CrossRef]
- Piperno, A. Classification and diagnosis of iron overload. Haematology 1998, 83, 447–455. [Google Scholar]
- van Straaten, S.; Biemond, B.J.; Kerkhoffs, J.L.; Gitz-Francois, J.; van Wijk, R.; van Beers, E.J. Iron overload in patients with rare hereditary hemolytic anemia: Evidence based suggestion on whom and how to screen. Am. J. Hematol. 2018, 93, E374. [Google Scholar] [CrossRef] [Green Version]
- Brissot, P.; Pietrangelo, A.; Adams, P.C.; de Graaff, B.; McLaren, C.E.; Loréal, O. Haemochromatosis. Nat. Rev. Dis. Primers 2018, 4, 18016. [Google Scholar] [CrossRef]
- Porter, J.B.; Garbowski, M. The Pathophysiology of Transfusional Iron Overload. Hematol. Clin. N. Am. 2014, 28, 683–701. [Google Scholar] [CrossRef]
- Marchetti, M.; De Bei, O.; Bettati, S.; Campanini, B.; Kovachka, S.; Gianquinto, E.; Spyrakis, F.; Ronda, L. Iron Metabolism at the Interface between Host and Pathogen: From Nutritional Immunity to Antibacterial Development. Int. J. Mol. Sci. 2020, 21, 2145. [Google Scholar] [CrossRef] [Green Version]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A Red Carpet for Iron Metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematology 2020, 105, 260–272. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, G.; Viatte, L.; Bennoun, M.; Beaumont, C.; Kahn, A.; Vaulont, S. Hepcidin, a new iron regulatory peptide. Blood Cells Mol. Dis. 2002, 29, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Fleming, R.E.; Ponka, P. Iron Overload in Human Disease. N. Engl. J. Med. 2012, 366, 348–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, J.C. Guidelines for quantifying iron overload. Hematology 2014, 2014, 210–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viprakasit, V.; Ajlan, A.; Aydinok, Y.; Al Ebadi, B.A.A.; Dewedar, H.; Ibrahim, A.S.; Ragab, L.; Trad, O.; Wataify, A.S.; Wong, L.L.L.; et al. MRI for the diagnosis of cardiac and liver iron overload in patients with transfusion-dependent thalassemia: An algorithm to guide clinical use when availability is limited. Am. J. Hematol. 2018, 93, E135–E137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aschemeyer, S.; Qiao, B.; Stefanova, D.; Valore, E.V.; Sek, A.C.; Ruwe, T.A.; Vieth, K.R.; Jung, G.; Casu, C.; Rivella, S.; et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood 2018, 131, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Porter, J.B.; Viprakasit, V.; Kattamis, A.; Chuncharunee, S.; Sutcharitchan, P.; Siritanaratkul, N.; Origa, R.; Karakas, Z.; Habr, D.; et al. Defining serum ferritin thresholds to predict clinically relevant liver iron concentrations for guiding deferasirox therapy when MRI is unavailable in patients with non-transfusion-dependent thalassaemia. Br. J. Haematol. 2014, 168, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, H. Transferrin and transferrin receptors update. Free. Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef]
- Kohgo, Y.; Torimoto, Y.; Kato, J. Transferrin Receptor in Tissue and Serum: Updated Clinical Significance of Soluble Receptor. Int. J. Hematol. 2002, 76, 213–218. [Google Scholar] [CrossRef]
- Skikne, B.S. Serum transferrin receptor. Am. J. Hematol. 2008, 83, 872–875. [Google Scholar] [CrossRef]
- Fertrin, K.Y.; Lanaro, C.; Franco-Penteado, C.F.; De Albuquerque, D.M.; De Mello, M.R.B.; Pallis, F.R.; Bezerra, M.A.C.; Hatzlhofer, B.L.D.; Olbina, G.; Saad, S.T.O.; et al. Erythropoiesis-driven regulation of hepcidin in human red cell disorders is better reflected through concentrations of soluble transferrin receptor rather than growth differentiation factor 15. Am. J. Hematol. 2014, 89, 385–390. [Google Scholar] [CrossRef]
- Kuhrt, D.; Wojchowski, D.M. Emerging EPO and EPO receptor regulators and signal transducers. Blood 2015, 125, 3536–3541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kautz, L.; Jung, G.; Du, X.; Gabayan, V.; Chapman, J.; Nasoff, M.; Ganz, T. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia. Blood 2015, 126, 2031–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, G.; Chauvet, C.; Viatte, L.; Danan, J.L.; Bigard, X.; Devaux, I.; Vaulont, S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Investig. 2002, 110, 1037–1044. [Google Scholar] [CrossRef]
- Pak, M.; Lopez, M.A.; Gabayan, V.; Ganz, T.; Rivera, S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood 2006, 108, 3730–3735. [Google Scholar] [CrossRef] [PubMed]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Seldin, M.M.; Peterson, J.M.; Byerly, M.S.; Wei, Z.; Wong, G.W. Myonectin (CTRP15), a Novel Myokine That Links Skeletal Muscle to Systemic Lipid Homeostasis. J. Biol. Chem. 2012, 287, 11968–11980. [Google Scholar] [CrossRef] [Green Version]
- Arezes, J.; Foy, N.; McHugh, K.; Sawant, A.; Quinkert, D.; Terraube, V.; Brinth, A.; Tam, M.; LaVallie, E.R.; Taylor, S.; et al. Erythroferrone inhibits the induction of hepcidin by BMP6. Blood 2018, 132, 1473–1477. [Google Scholar] [CrossRef] [Green Version]
- Ganz, T. Drugging erythroferrone to treat anemias. Blood 2020, 135, 516–518. [Google Scholar] [CrossRef]
- Tanno, T.; Bhanu, N.V.; A Oneal, P.; Goh, S.-H.; Staker, P.; Lee, Y.T.; Moroney, J.W.; Reed, C.H.; Luban, N.L.C.; Wang, R.-H.; et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat. Med. 2007, 13, 1096–1101. [Google Scholar] [CrossRef]
- Tanno, T.; Noel, P.; Miller, J.L. Growth differentiation factor 15 in erythroid health and disease. Curr. Opin. Hematol. 2010, 17, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Xiang, J.; Wu, D.-C.; Fraser, J.W.; Ruan, B.; Cai, J.; Patterson, A.D.; Lai, Z.-C.; Paulson, R.F. Gdf15 regulates murine stress erythroid progenitor proliferation and the development of the stress erythropoiesis niche. Blood Adv. 2019, 3, 2205–2217. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, G.; Spasić, M.V.; Casu, C.; Rivella, S.; Strelau, J.; Unsicker, K.; Muckenthaler, M.U. The murine growth differentiation factor 15 is not essential for systemic iron homeostasis in phlebotomized mice. Haematology 2012, 98, 444–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanno, T.; Rabel, A.; Lee, Y.T.; Yau, Y.Y.; Leitman, S.F.; Miller, J.L. Expression of growth differentiation factor 15 is not elevated in individuals with iron deficiency secondary to volunteer blood donation. Transfusion 2010, 50, 1532–1535. [Google Scholar] [CrossRef] [Green Version]
- Kanda, J.; Mizumoto, C.; Kawabata, H.; Tsuchida, H.; Tomosugi, N.; Matsuo, K.; Uchiyama, T. Serum hepcidin level and erythropoietic activity after hematopoietic stem cell transplantation. Haematology 2008, 93, 1550–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamary, H.; Shalev, H.; Perez-Avraham, G.; Zoldan, M.; Levi, I.; Swinkels, R.W.; Tanno, T.; Miller, J.L. Elevated growth differentiation factor 15 expression in patients with congenital dyserythropoietic anemia type I. Blood 2008, 112, 5241–5244. [Google Scholar] [CrossRef] [Green Version]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassaemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef]
- Bou-Fakhredin, R.; Bazarbachi, A.-H.; Chaya, B.; Sleiman, J.; Cappellini, M.D.; Taher, A.T. Iron Overload and Chelation Therapy in Non-Transfusion Dependent Thalassemia. Int. J. Mol. Sci. 2017, 18, 2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brissot, P.; Troadec, M.-B.; Loréal, O.; Brissot, E. Pathophysiology and classification of iron overload diseases; update 2018. Transfus. Clin. Et Biol. 2019, 26, 80–88. [Google Scholar] [CrossRef]
- Huang, Y.; Lei, Y.; Liu, R.; Liu, J.; Yang, G.; Xiang, Z.; Liang, Y.; Lai, Y. Imbalance of erythropoiesis and iron metabolism in patients with thalassemia. Int. J. Med. Sci. 2019, 16, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Camaschella, C.; Pagani, A.; Nai, A.; Silvestri, L. The mutual control of iron and erythropoiesis. Int. J. Lab. Hematol. 2016, 38, 20–26. [Google Scholar] [CrossRef]
- van Vuren, A.J.; Eisenga, M.F.; van Straaten, S.; Glenthoj, A.; Gaillard, C.; Bakker, S.J.L.; De Borst, M.H.; Van Wijk, R.; Van Beers, E.J. Interplay of erythropoietin, fibroblast growth factor 23, and erythroferrone in patients with hereditary hemolytic anemia. Blood Adv. 2020, 4, 1678–1682. [Google Scholar] [CrossRef] [Green Version]
- Arezes, J.; Foy, N.; McHugh, K.; Quinkert, D.; Benard, S.; Sawant, A.; Frost, J.N.; Armitage, A.E.; Pasricha, S.-R.; Lim, P.J.; et al. Antibodies against the erythroferrone N-terminal domain prevent hepcidin suppression and ameliorate murine thalassemia. Blood 2020, 135, 547–557. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br. J. Haematol. 2016, 172, 512–523. [Google Scholar] [CrossRef] [Green Version]
- Grace, R.F.; Bianchi, P.; Van Beers, E.J.; Eber, S.W.; Glader, B.; Yaish, H.M.; Despotovic, J.M.; Rothman, J.A.; Sharma, M.; McNaull, M.M.; et al. Clinical spectrum of pyruvate kinase deficiency: Data from the Pyruvate Kinase Deficiency Natural History Study. Blood 2018, 131, 2183–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grace, R.F.; Zanella, A.; Neufeld, E.J.; Morton, D.H.; Eber, S.; Yaish, H.; E Glader, B. Erythrocyte pyruvate kinase deficiency: 2015 status report. Am. J. Hematol. 2015, 90, 825–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grace, R.F.; Layton, D.M.; Barcellini, W. How we manage patients with pyruvate kinase deficiency. Br. J. Haematol. 2019, 184, 721–734. [Google Scholar] [CrossRef] [PubMed]
- Finkenstedt, A.; Bianchi, P.; Theurl, I.; Vogel, W.; Witcher, D.R.; Wroblewski, V.J.; Murphy, A.T.; Zanella, A.; Zoller, H. Regulation of iron metabolism through GDF15 and hepcidin in pyruvate kinase deficiency. Br. J. Haematol. 2009, 144, 789–793. [Google Scholar] [CrossRef]
- Grace, R.F.; Glader, B. Red Blood Cell Enzyme Disorders. Pediatr. Clin. N. Am. 2018, 65, 579–595. [Google Scholar] [CrossRef]
- Aizawa, S.; Harada, T.; Kanbe, E.; Tsuboi, I.; Aisaki, K.-I.; Fujii, H.; Kanno, H. Ineffective erythropoiesis in mutant mice with deficient pyruvate kinase activity. Exp. Hematol. 2005, 33, 1292–1298. [Google Scholar] [CrossRef]
- Piel, F.B.; Weatherall, D.J. The alpha-thalassemias. N. Engl. J. Med. 2014, 371, 1908–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.E.; Ooi, C.; Ha, S.Y.; Cheung, B.M.Y.; Todd, D.; Liang, R.; Chan, T.K.; Chan, V. Genetic and Clinical Features of Hemoglobin H Disease in Chinese Patients. N. Engl. J. Med. 2000, 343, 544–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamary, H.; Dgany, O. Alpha-Thalassemia; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Eds.; GeneReviews((R)): Seattle, WA, USA, 1993. [Google Scholar]
- Tso, S.C.; Loh, T.T.; Chen, W.W.; Wang, C.C.; Todd, D. Iron overload in thalassaemic patients in Hong Kong. Ann. Acad. Med. Singap. 1984, 13, 487–490. [Google Scholar]
- Origa, R.; Cazzola, M.; Mereu, E.; Danjou, F.; Barella, S.; Giagu, N.; Swinkels, D.W. Differences in the erythropoiesis-hepcidin-iron store axis between hemoglobin H disease and beta-thalassemia intermedia. Haematologica 2015, 100, e169–e171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambale, A.; Iolascon, A.; Andolfo, I.; Russo, R. Diagnosis and management of congenital dyserythropoietic anemias. Expert Rev. Hematol. 2016, 9, 283–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iolascon, A.; Esposito, M.R.; Russo, R. Clinical aspects and pathogenesis of congenital dyserythropoietic anemias: From morphology to molecular approach. Haematology 2012, 97, 1786–1794. [Google Scholar] [CrossRef]
- Iolascon, A.; Heimpel, H.; Wahlin, A.; Tamary, H. Congenital dyserythropoietic anemias: Molecular insights and diagnostic approach. Blood 2013, 122, 2162–2166. [Google Scholar] [CrossRef] [Green Version]
- Heimpel, H.; Matuschek, A.; Ahmed, M.; Bader-Meunier, B.; Colita, A.; Delaunay, J.; Garcon, L.; Gilsanz, F.; Goede, J.; Högel, J.; et al. Frequency of congenital dyserythropoietic anemias in Europe. Eur. J. Haematol. 2010, 85, 20–25. [Google Scholar] [CrossRef]
- Dgany, O.; Avidan, N.; Delaunay, J.; Krasnov, T.; Shalmon, L.; Shalev, H.; Eidelitz-Markus, T.; Kapelushnik, J.; Cattan, D.; Pariente, A.; et al. Congenital Dyserythropoietic Anemia Type I Is Caused by Mutations in Codanin-1. Am. J. Hum. Genet. 2002, 71, 1467–1474. [Google Scholar] [CrossRef] [Green Version]
- Ask, K.; Jasencakova, Z.; Menard, P.; Feng, Y.; Almouzni, G.; Groth, A. Codanin-1, mutated in the anaemic disease CDAI, regulates Asf1 function in S-phase histone supply. EMBO J. 2012, 31, 2013–2023. [Google Scholar] [CrossRef]
- Arbiv, O.; Cuvelier, G.; Klaassen, R.; Fernandez, C.; Robitaille, N.; Steele, M.; Breakey, V.; Abish, S.; Wu, J.; Sinha, R.; et al. Molecular analysis and genotype-phenotype correlation of Diamond-Blackfan anemia. Clin. Genet. 2017, 93, 320–328. [Google Scholar] [CrossRef]
- Shalev, H.; Perez-Avraham, G.; Kapelushnik, J.; Levi, I.; Rabinovich, A.; Swinkels, D.W.; Brasse-Lagnel, C.; Tamary, H. High levels of soluble serum hemojuvelin in patients with congenital dyserythropoietic anemia type I. Eur. J. Haematol. 2012, 90, 31–36. [Google Scholar] [CrossRef]
- Moreno-Carralero, M.-I.; Horta-Herrera, S.; Morado-Arias, M.; Ricard-Andrés, M.-P.; Lemes-Castellano, A.; Abio-Calvete, M.; Cedena-Romero, M.-T.; González-Fernández, F.-A.; Llorente-González, L.; Periago-Peralta, A.-M.; et al. Clinical and genetic features of congenital dyserythropoietic anemia (CDA). Eur. J. Haematol. 2018, 101, 368–378. [Google Scholar] [CrossRef]
- Schwarz, K.; Iolascon, A.; Verissimo, F.; Trede, N.S.; Horsley, W.; Chen, W.; Paw, B.H.; Hopfner, K.-P.; Holzmann, K.; Russo, R.; et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat. Genet. 2009, 41, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, P.; Fermo, E.; Vercellati, C.; Boschetti, C.; Barcellini, W.; Iurlo, A.; Zanella, A. Congenital dyserythropoietic anemia type II (CDAII) is caused by mutations in the SEC23B gene. Hum. Mutat. 2009, 30, 1292–1298. [Google Scholar] [CrossRef] [PubMed]
- Barlowe, C.; Orci, L.; Yeung, T.; Hosobuchi, M.; Hamamoto, S.; Salama, N.; Rexach, M.F.; Ravazzola, M.; Amherd, M.; Schekman, R. COPII: A membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 1994, 77, 895–907. [Google Scholar] [CrossRef]
- Russo, R.; Gambale, A.; Langella, C.; Andolfo, I.; Unal, S.; Iolascon, A. Retrospective cohort study of 205 cases with congenital dyserythropoietic anemia type II: Definition of clinical and molecular spectrum and identification of new diagnostic scores. Am. J. Hematol. 2014, 89, E169–E175. [Google Scholar] [CrossRef]
- Kostaridou, S.; Polychronopoulou, S.; Premetis, E.; Papassotiriou, I.; Stamoulakatou, A.; Haidas, S. Ineffective erythropoiesis underlies the clinical heterogeneity of congenital dyserythropoietic anemia type II (CDA II). Pediatr. Int. 2004, 46, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, G.; Swinkels, D.W.; Altamura, S.; Schwarz, K.; Laarakkers, C.M.; Gross, H.-J.; Wiesneth, M.; Heimpel, H.; Muckenthaler, M.U. Growth differentiation factor 15 in patients with congenital dyserythropoietic anaemia (CDA) type II. J. Mol. Med. 2011, 89, 811–816. [Google Scholar] [CrossRef]
- Russo, R.; Andolfo, I.; Manna, F.; De Rosa, G.; De Falco, L.; Gambale, A.; Bruno, M.; Mattè, A.; Ricchi, P.; Girelli, D.; et al. Increased levels of ERFE-encoding FAM132B in patients with congenital dyserythropoietic anemia type II. Blood 2016, 128, 1899–1902. [Google Scholar] [CrossRef] [Green Version]
- Sandström, H.; Wahlin, A.; Eriksson, M.; Bergström, I.; Wickramasinghe, S.N. Intravascular haemolysis and increased prevalence of myeloma and monoclonal gammopathy in congenital dyserythropoietic anaemia, type III. Eur. J. Haematol. 1994, 52, 42–46. [Google Scholar] [CrossRef]
- Jaffray, J.A.; Mitchell, W.B.; Gnanapragasam, M.N.; Seshan, S.V.; Guo, X.; Westhoff, C.M.; Bieker, J.J.; Manwani, D. Erythroid transcription factor EKLF/KLF1 mutation causing congenital dyserythropoietic anemia type IV in a patient of Taiwanese origin: Review of all reported cases and development of a clinical diagnostic paradigm. Blood Cells Mol. Dis. 2013, 51, 71–75. [Google Scholar] [CrossRef] [Green Version]
- Ravindranath, Y.; Johnson, R.M.; Goyette, G.; Buck, S.; Gadgeel, M.; Gallagher, P.G. KLF1 E325K-associated Congenital Dyserythropoietic Anemia Type IV: Insights into the Variable Clinical Severity. J. Pediatr. Hematol. Oncol. 2018, 40, e405–e409. [Google Scholar] [CrossRef]
- Belgemen-Ozer, T.; Gorukmez, O. A Very Rare Congenital Dyserythropoietic Anemia Variant—Type IV in a Patient With a Novel Mutation in the KLF1 Gene: A Case Report and Review of the Literature. J. Pediatr. Hematol. 2020, 42, e536–e540. [Google Scholar] [CrossRef]
- Crispino, J.D.; Horwitz, M.S. GATA factor mutations in hematologic disease. Blood 2017, 129, 2103–2110. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, M.; Aggarwala, A.; Sharma, P.; Bansal, D.; Maitra, A.; Das, R. Phenotypic and genetic heterogeneity arising from a novel substitution at amino acid position Val205 in GATA1 related X-linked thrombocytopenia with dyserythropoietic anemia. Blood Cells Mol. Dis. 2020, 81, 102391. [Google Scholar] [CrossRef]
- Ducamp, S.; Fleming, M.D. The molecular genetics of sideroblastic anemia. Blood 2019, 133, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Peto, T.E.; Pippard, M.J.; Weatherall, D.J. Iron Overload in Mild Sideroblastic Anaemias. Lancet 1983, 321, 375–378. [Google Scholar] [CrossRef]
- Le Rouzic, M.A.; Fouquet, C.; Leblanc, T.; Touati, M.; Fouyssac, F.; Vermylen, C.; Vannier, J.P. Non syndromic childhood onset congenital sideroblastic anemia: A report of 13 patients identified with an ALAS2 or SLC25A38 mutation. Blood Cells Mol. Dis. 2017, 66, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Donker, A.E.; Raymakers, R.A.; Nieuwenhuis, H.K.; Coenen, M.J.H.; Janssen, M.C.; A Mackenzie, M.; Brons, P.P.T.; Swinkels, D.W. X-linked sideroblastic anaemia due to ALAS? mutations in the Netherlands: A disease in disguise. Neth. J. Med. 2014, 72, 210–217. [Google Scholar] [PubMed]
- Ramirez, J.-M.; Schaad, O.; Durual, S.; Cossali, D.; Docquier, M.; Beris, P.; Descombes, P.; Matthes, T. Growth differentiation factor 15 production is necessary for normal erythroid differentiation and is increased in refractory anaemia with ring-sideroblasts. Br. J. Haematol. 2009, 144, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Santini, V.; Girelli, D.; Sanna, A.; Martinelli, N.; Duca, L.; Campostrini, N.; Cortelezzi, A.; Corbella, M.; Bosi, A.; Reda, G.; et al. Hepcidin Levels and Their Determinants in Different Types of Myelodysplastic Syndromes. PLoS ONE 2011, 6, e23109. [Google Scholar] [CrossRef] [PubMed]
- Zipperer, E.; Post, J.G.; Herkert, M.; Kündgen, A.; Fox, F.; Haas, R.; Gattermann, N.; Germing, U. Serum hepcidin measured with an improved ELISA correlates with parameters of iron metabolism in patients with myelodysplastic syndrome. Ann. Hematol. 2013, 92, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Cuijpers, M.L.H.; Raymakers, R.A.P.; MacKenzie, M.A.; De Witte, T.J.M.; Swinkels, D.W. Recent advances in the understanding of iron overload in sideroblastic myelodysplastic syndrome. Br. J. Haematol. 2010, 149, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C. Recent advances in the understanding of inherited sideroblastic anaemia. Br. J. Haematol. 2008, 143, 27–38. [Google Scholar] [CrossRef]
- Andolfo, I.; Russo, R.; Gambale, A.; Iolascon, A. New insights on hereditary erythrocyte membrane defects. Haematology 2016, 101, 1284–1294. [Google Scholar] [CrossRef] [Green Version]
- Zarychanski, R.; Schulz, V.P.; Houston, B.L.; Maksimova, Y.; Houston, D.S.; Smith, B.; Rinehart, J.; Gallagher, P.G. Mutations in the mechanotransduction protein PIEZO1 are associated with hereditary xerocytosis. Blood 2012, 120, 1908–1915. [Google Scholar] [CrossRef] [Green Version]
- Rapetti-Mauss, R.; Lacoste, C.; Picard, V.; Guitton, C.; Lombard, E.; Loosveld, M.; Nivaggioni, V.; DaSilva, N.; Salgado, D.; Desvignes, J.-P.; et al. A mutation in the Gardos channel is associated with hereditary xerocytosis. Blood 2015, 126, 1273–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, V.; Guitton, C.; Thuret, I.; Rose, C.; Bendelac, L.; Ghazal, K.; Aguilar-Martinez, P.; Badens, C.; Barro, C.; Bénéteau, C.; et al. Clinical and biological features in PIEZO1-hereditary xerocytosis and Gardos channelopathy: A retrospective series of 126 patients. Haematology 2019, 104, 1554–1564. [Google Scholar] [CrossRef] [Green Version]
- Andolfo, I.; Rosato, B.E.; Manna, F.; De Rosa, G.; Marra, R.; Gambale, A.; Girelli, D.; Russo, R.; Iolascon, A. Gain-of-function mutations in PIEZO1 directly impair hepatic iron metabolism via the inhibition of the BMP/SMADs pathway. Am. J. Hematol. 2019, 95, 188–197. [Google Scholar] [CrossRef]
- Archer, N.M.; Shmukler, B.E.; Andolfo, I.; Vandorpe, D.H.; Gnanasambandam, R.; Higgins, J.M.; Rivera, A.; Fleming, M.D.; Sachs, F.; Gottlieb, P.A.; et al. Hereditary xerocytosis revisited. Am. J. Hematol. 2014, 89, 1142–1146. [Google Scholar] [CrossRef] [Green Version]
- Ware, R.E.; de Montalembert, M.; Tshilolo, L.; Abboud, M.R. Sickle cell disease. Lancet 2017, 390, 311–323. [Google Scholar] [CrossRef]
- Pauling, L.; Itano, H.A.; Singer, S.J.; Wells, I.C. Sickle Cell Anemia, a Molecular Disease. Science 1949, 110, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Mariani, R.; Trombini, P.; Pozzi, M.; Piperno, A. Iron metabolism in thalassemia and sickle cell disease. Mediterr. J. Hematol. Infect. Dis. 2009, 1, e2009006. [Google Scholar] [CrossRef] [PubMed]
- Karafin, M.S.; Koch, K.L.; Rankin, A.B.; Nischik, D.; Rahhal, G.; Simpson, P.; Field, J.J. Erythropoietic drive is the strongest predictor of hepcidin level in adults with sickle cell disease. Blood CellsMol. Dis. 2015, 55, 304–307. [Google Scholar] [CrossRef] [Green Version]
- Karafin, M.S.; Fu, X.; D’Alessandro, A.; Thomas, T.; Hod, E.A.; Zimring, J.C.; Field, J.J.; Francis, R.O. The clinical impact of glucose-6-phosphate dehydrogenase deficiency in patients with sickle cell disease. Curr. Opin. Hematol. 2018, 25, 494–499. [Google Scholar] [CrossRef]
- Mangaonkar, A.A.; Thawer, F.; Son, J.; Ajebo, G.; Xu, H.; Barrett, N.J.; Wells, L.G.; Bowman, L.; Clair, B.; Patel, N.; et al. Regulation of iron homeostasis through the erythroferrone-hepcidin axis in sickle cell disease. Br. J. Haematol. 2020, 189, 1204–1209. [Google Scholar] [CrossRef]
- Mangaonkar, A.; Patel, N.; Xu, H.; Natrajan, K.; Clair, B.; Wells, L.G.; Bowman, L.; Barret, N.; Kutlar, A. Plasma Biomarkers of Iron Regulation, Overload, and Inflammation in Sickle Cell Disease. Blood 2014, 124, 1380. [Google Scholar] [CrossRef]
- Lee, N.; Makani, J.; Tluway, F.; Makubi, A.; Armitage, A.E.; Pasricha, S.-R.; Drakesmith, H.; Prentice, A.M.; Cox, S.E. Decreased Hepcidin Levels Are Associated with Low Steady-state Hemoglobin in Children With Sickle Cell Disease in Tanzania. EBioMedicine 2018, 34, 158–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroot, J.J.; Kemna, E.H.; Bansal, S.S.; Busbridge, M.; Campostrini, N.; Girelli, M.; Hider, R.C.; Koliaraki, V.; Mamalaki, A.; Olbina, G.; et al. Results of the first international round robin for the quantification of urinary and plasma hepcidin assays: Need for standardization. Haematology 2009, 94, 1748–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasavda, N.; Gutiérrez, L.; House, M.J.; Drašar, E.; Pierre, T.G.S.; Thein, S.L. Renal iron load in sickle cell disease is influenced by severity of haemolysis. Br. J. Haematol. 2012, 157, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Saraf, S.L.; Zhang, X.; Kanias, T.; Lash, J.P.; Molokie, R.E.; Oza, B.; Lai, C.; Rowe, J.H.; Gowhari, M.; Hassan, J.; et al. Haemoglobinuria is associated with chronic kidney disease and its progression in patients with sickle cell anaemia. Br. J. Haematol. 2014, 164, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Bartels, M.; Bierings, M. How I manage children with Diamond-Blackfan anaemia. Br. J. Haematol. 2018, 184, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Aghalar, J.; Atsidaftos, M.E.; Lipton, J.M.; Vlachos, A. Improved Outcomes in Diamond Blackfan Anemia Treated Via Stem Cell Transplantation Since the Year 2000. Blood 2009, 114, 3202. [Google Scholar] [CrossRef]
- Roggero, S.; Quarello, P.; Vinciguerra, T.; Longo, F.; Piga, A.; Ramenghi, U. Severe iron overload in Blackfan-Diamond anemia: A case-control study. Am. J. Hematol. 2009, 84, 729–732. [Google Scholar] [CrossRef]
- Pospisilova, D.; Holub, D.; Zidova, Z.; Sulovska, L.; Houda, J.; Mihal, V.; Hadacova, I.; Radova, L.; Dzubak, P.; Hajduch, M.; et al. Hepcidin levels in Diamond-Blackfan anemia reflect erythropoietic activity and transfusion dependency. Haematology 2014, 99, e118–e121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, J.B.; Walter, P.B.; Neumayr, L.D.; Evans, P.; Bansal, S.; Garbowski, M.; Weyhmiller, M.G.; Harmatz, P.R.; Wood, J.C.; Miller, J.L.; et al. Mechanisms of plasma non-transferrin bound iron generation: Insights from comparing transfused diamond blackfan anaemia with sickle cell and thalassaemia patients. Br. J. Haematol. 2014, 167, 692–696. [Google Scholar] [CrossRef]
- Berdoukas, V.; Nord, A.; Carson, S.; Puliyel, M.; Hofstra, T.; Wood, J.; Coates, T.D. Tissue iron evaluation in chronically transfused children shows significant levels of iron loading at a very young age. Am. J. Hematol. 2013, 88, E283–E285. [Google Scholar] [CrossRef]
- Tsai, P.H.; Arkin, S.; Lipton, J.M. An intrinsic progenitor defect in Diamond-Blackfan anaemia. Br. J. Haematol. 1989, 73, 112–120. [Google Scholar] [CrossRef]
- Perrotta, S.; Gallagher, P.G.; Mohandas, N. Hereditary spherocytosis. Lancet 2008, 372, 1411–1426. [Google Scholar] [CrossRef]
- Iolascon, A.; Andolfo, I.; Russo, R. Advances in understanding the pathogenesis of red cell membrane disorders. Br. J. Haematol. 2019, 187, 13–24. [Google Scholar] [CrossRef]
- van Vuren, A.; van der Zwaag, B.; Huisjes, R.; Lak, N.; Bierings, M.; Gerritsen, E.; van Wijk, R. The Complexity of Genotype-Phenotype Correlations in Hereditary Spherocytosis: A Cohort of 95 Patients: Genotype-Phenotype Correlation in Hereditary Spherocytosis. Hemasphere 2019, 3, e276. [Google Scholar] [CrossRef]
- Blacklock, H.A.; Meerkin, M. Serum Ferritin in Patients with Hereditary Spherocytosis. Br. J. Haematol. 1981, 49, 117–122. [Google Scholar] [CrossRef]
- O’Mahony, S.; O’Brien, P.A.; Whelton, M.J. Genetic haemochromatosis and congenital spherocytosis. Lancet 1987, 329, 282. [Google Scholar]
- Iolascon, A.; Andolfo, I.; Barcellini, W.; Corcione, F.; Garçon, L.; De Franceschi, L.; Pignata, C.; Graziadei, G.; Pospisilova, D.; Rees, D.C.; et al. Recommendations regarding splenectomy in hereditary hemolytic anemias. Haematology 2017, 102, 1304–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiland, O.K.; Smith, E.B. The Morphology of the Spleen in Congenital Hemolytic Anemia (Hereditary Spherocytosis). Am. J. Clin. Pathol. 1956, 26, 619–629. [Google Scholar] [CrossRef]
- Tole, S.; Amid, A.; Baker, J.; Kuo, K.; Pugi, J.; Carcao, M. Mild Hereditary Spherocytosis without Accompanying Hereditary Haemochromatosis: An Unrecognised Cause of Iron Overload. Acta Haematol. 2019, 141, 256–260. [Google Scholar] [CrossRef]
- Castro, O.; Kato, G.J. Iron restriction in sickle cell anemia: Time for controlled clinical studies. Am. J. Hematol. 2015, 90, E217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rio, S.; Gastou, M.; Karboul, N.; Derman, R.; Suriyun, T.; Manceau, H.; Leblanc, T.; El Benna, J.; Schmitt, C.; Azouzi, S.; et al. Regulation of globin-heme balance in Diamond-Blackfan anemia by HSP70/GATA1. Blood 2019, 133, 1358–1370. [Google Scholar] [CrossRef] [Green Version]
- Ulirsch, J.C.; Lareau, C.; Ludwig, L.S.; Mohandas, N.; Nathan, D.G.; Sankaran, V.G. Confounding in ex vivo models of Diamond-Blackfan anemia. Blood 2017, 130, 1165–1168. [Google Scholar] [CrossRef] [Green Version]
- Donker, A.E.; Galesloot, T.E.; Laarakkers, C.M.; Klaver, S.M.; Bakkeren, D.L.; Swinkels, D.W. Standardized serum hepcidin values in Dutch children: Set point relative to body iron changes during childhood. Pediatr. Blood Cancer 2019, 67, e28038. [Google Scholar] [CrossRef]
- Coffey, R.; Sardo, U.; Kautz, L.; Gabayan, V.; Nemeth, E.; Ganz, T. Erythroferrone is not required for the glucoregulatory and hematologic effects of chronic erythropoietin treatment in mice. Physiol. Rep. 2018, 6, e13890. [Google Scholar] [CrossRef]
- Core, A.B.; Ecanali, S.; Babitt, J.L. Hemojuvelin and bone morphogenetic protein (BMP) signaling in iron homeostasis. Front. Pharm. 2014, 5, 104. [Google Scholar] [CrossRef] [Green Version]
- Ferro, E.; Di Pietro, A.; Visalli, G.; Piraino, B.; Salpietro, C.; La Rosa, M.A. Soluble hemojuvelin in transfused and untransfused thalassaemic subjects. Eur. J. Haematol. 2016, 98, 67–74. [Google Scholar] [CrossRef]
- Wang, C.; Liu, Y.; He, D. Diverse effects of platelet-derived growth factor-BB on cell signaling pathways. Cytokine 2019, 113, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Sonnweber, T.; Nachbaur, D.; Schroll, A.; Nairz, M.; Seifert, M.; Demetz, E.; Haschka, D.; Mitterstiller, A.-M.; Kleinsasser, A.; Burtscher, M.; et al. Hypoxia induced downregulation of hepcidin is mediated by platelet derived growth factor BB. Gut 2014, 63, 1951–1959. [Google Scholar] [CrossRef] [Green Version]
- Ravasi, G.; Pelucchi, S.; Comani, G.B.; Greni, F.; Mariani, R.; Pelloni, I.; Bombelli, S.; Perego, R.; Barisani, D.; Piperno, A. Hepcidin regulation in a mouse model of acute hypoxia. Eur. J. Haematol. 2018, 100, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Mastrogiannaki, M.; Matak, P.; Keith, B.; Simon, M.C.; Vaulont, S.; Peyssonnaux, C. HIF-2α, but not HIF-1α, promotes iron absorption in mice. J. Clin. Investig. 2009, 119, 1159–1166. [Google Scholar] [CrossRef]
- Taylor, M.; Qu, A.; Anderson, E.R.; Matsubara, T.; Martin, A.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-Inducible Factor-2α Mediates the Adaptive Increase of Intestinal Ferroportin During Iron Deficiency in Mice. Gastroenterology 2011, 140, 2044–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wei, Y.; Liu, D.; Liu, F.; Li, X.; Pan, L.; Pang, Y.; Chen, D. Role of growth differentiation factor 11 in development, physiology and disease. Oncotarget 2017, 8, 81604–81616. [Google Scholar] [CrossRef] [Green Version]
- Attie, K.M.; Allison, M.J.; McClure, T.; Boyd, I.E.; Wilson, D.M.; Pearsall, A.E.; Sherman, M.L. A phase 1 study of ACE-536, a regulator of erythroid differentiation, in healthy volunteers. Am. J. Hematol. 2014, 89, 766–770. [Google Scholar] [CrossRef] [Green Version]
- Suragani, R.N.; Cadena, S.M.; Cawley, S.M.; Sako, D.; Mitchell, D.; Li, R.; Davies, M.V.; Alexander, M.J.; Devine, M.L.; Kenneth, S.; et al. Transforming growth factor-beta superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat. Med. 2014, 20, 408–414. [Google Scholar] [CrossRef]
- Fang, Z.; Zhu, Z.; Zhang, H.; Peng, Y.; Liu, J.; Lu, H.; Li, J.; Liang, L.; Xia, S.; Wang, Q.; et al. GDF11 contributes to hepatic hepcidin (HAMP) inhibition through SMURF1-mediated BMP-SMAD signalling suppression. Br. J. Haematol. 2020, 188, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Guerra, A.; Oikonomidou, P.R.; Sinha, S.; Zhang, J.; Lo Presti, V.; Hamilton, C.R. Lack of Gdf11 does not improve anemia or prevent the activity of RAP-536 in a mouse model of beta-thalassemia. Blood 2019, 134, 568–572. [Google Scholar] [CrossRef]
- Tanno, T.; Porayette, P.; Sripichai, O.; Noh, S.-J.; Byrnes, C.; Bhupatiraju, A.; Lee, Y.T.; Goodnough, J.B.; Harandi, O.; Ganz, T.; et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 2009, 114, 181–186. [Google Scholar] [CrossRef]
- Mirciov, C.S.G.; Wilkins, S.J.; Dunn, L.A.; Anderson, G.J.; Frazer, D.M. Characterization of Putative Erythroid Regulators of Hepcidin in Mouse Models of Anemia. PLoS ONE 2017, 12, e0171054. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Test | Cut-Off Values | Advantages | Disadvantages |
|---|---|---|---|
| Serum ferritin | >500 μg/L (for chelation >800μg/L) [17] | Widely available. Inexpensive. | Does not correlate with severity of IO in RHA. Is elevated upon inflammation. |
| TSAT | >60% | Possible in most health centers. Closer relation to extrahepatic IO. | Cannot be interpreted correctly during iron chelation therapy. |
| Liver biopsy | >3 mg/g | Golden-standard for liver IO. | Invasive. Dependent on tissue handling. |
| MRI | LIC >3 mg/g Cardiac T2* <20ms | Not invasive. | Expensive. Not widely available. |
| NTBI | >0 | Relevant (relates closely to organ damage). | Not widely available. High interlaboratory variation. Is decreased upon inflammation. |
| Mechanism | Prevalence of IO | EPO | sTfR | GDF-15 | Hepcidin or Hepcidin/Ferritin Ratio *** | Erythroferrone * | Summary | |
|---|---|---|---|---|---|---|---|---|
| Both ineffective erythropoiesis and hemolysis | NTDT | Very common | Extremely high | Increased | Extremely high | Extremely low | Extremely high | Increased iron loading due to ERFE-induced hepcidin suppression as a result of ineffective erythropoiesis. |
| PKD | Very common | Increased | Presumably increased | Increased | Decreased | Increased | Increased iron loading due to ERFE-induced hepcidin suppression as a result of ineffective erythropoiesis, yet to a lesser extent than in NTDT. | |
| HbH disease | Common | Increased | Increased | Increased | Decreased | Presumably increased | Risk of increased iron loading, presumably due to ERFE-induced hepcidin suppression as a result of increased/ineffective erythropoiesis. | |
| CDA type II | Common | Increased | Increased | Normal—increased | Decreased - normal | Extremely high | Increased iron loading due to ERFE-induced hepcidin suppression as a result of ineffective erythropoiesis, yet to a lesser extent than in NTDT. Other yet unknown factors may play a role. | |
| SCD | Uncommon | Increased | Increased | Increased | Variable | Increased | Iron loading is less profound. Altered iron metabolism due to ERFE-induction as a result of increased/ineffective erythropoiesis, simultaneous to increased hepcidin expression due to inflammation. | |
| Reduced or ineffective erythropoiesis | CDA type I | Common | Increased | Increased | Extremely high | Decreased—normal | Presumably increased | Risk of increased iron loading, presumably due to ERFE-induced hepcidin suppression as a result of increased/ineffective erythropoiesis. Other yet unknown factors may play a role. |
| DBA | Common | Very high ** | Decreased—normal | Variable | Variable | Presumably decreased—normal | Iron loading is less profound. Altered iron metabolism which is poorly understood. Individual assessment of iron status is necessary. | |
| CSA | Common | Increased | Presumably increased | Presumably increased | Presumably decreased | Presumably increased | Risk of increased iron loading, presumably due to ERFE-induced hepcidin suppression as a result of ineffective erythropoiesis. Other yet unknown factors may play a role. | |
| Hemolytic anemia | HS | Uncommon | Normal—increased | Normal—increased | Normal—increased | Decreased—normal | Normal—increased | Iron loading is rare. In specific patient groups with continuous hemolysis and increased/ineffective erythropoiesis, iron loading may occur due to hepcidin suppression. |
| DHS | Common | Increased | Increased | Normal—increased | Decreased | Increased | Risk of increased iron loading, presumably due to ERFE-induced hepcidin suppression as a result of increased/ineffective erythropoiesis. Other factors may play a role, such as PIEZO1-dependent hepcidin suppression. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grootendorst, S.; de Wilde, J.; van Dooijeweert, B.; van Vuren, A.; van Solinge, W.; Schutgens, R.; van Wijk, R.; Bartels, M. The Interplay between Drivers of Erythropoiesis and Iron Homeostasis in Rare Hereditary Anemias: Tipping the Balance. Int. J. Mol. Sci. 2021, 22, 2204. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042204
Grootendorst S, de Wilde J, van Dooijeweert B, van Vuren A, van Solinge W, Schutgens R, van Wijk R, Bartels M. The Interplay between Drivers of Erythropoiesis and Iron Homeostasis in Rare Hereditary Anemias: Tipping the Balance. International Journal of Molecular Sciences. 2021; 22(4):2204. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042204
Chicago/Turabian StyleGrootendorst, Simon, Jonathan de Wilde, Birgit van Dooijeweert, Annelies van Vuren, Wouter van Solinge, Roger Schutgens, Richard van Wijk, and Marije Bartels. 2021. "The Interplay between Drivers of Erythropoiesis and Iron Homeostasis in Rare Hereditary Anemias: Tipping the Balance" International Journal of Molecular Sciences 22, no. 4: 2204. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042204