
KRASG12C Can Either Promote or Impair Cap-Dependent Translation in Two Different Lung Adenocarcinoma Cell Lines

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
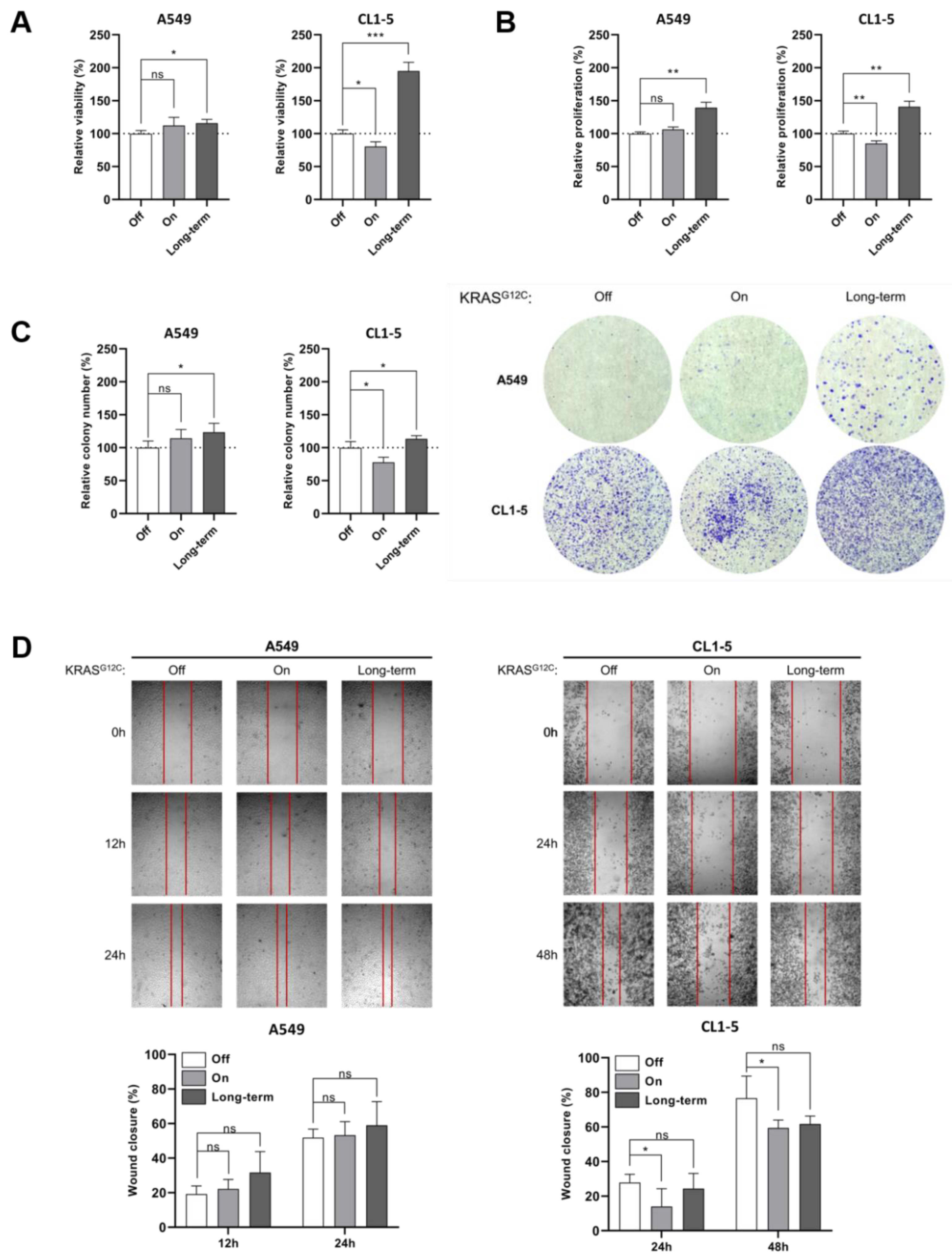
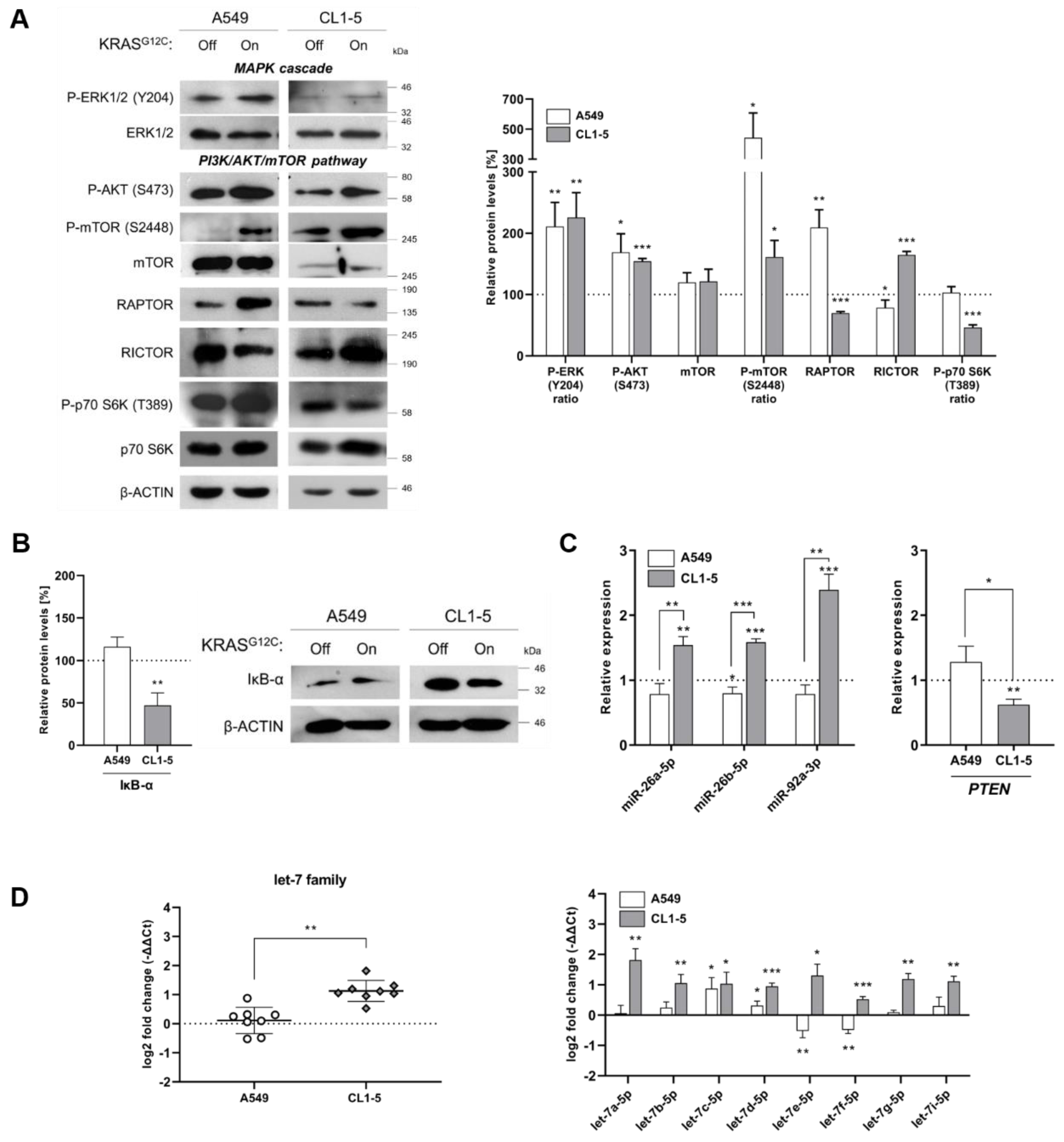
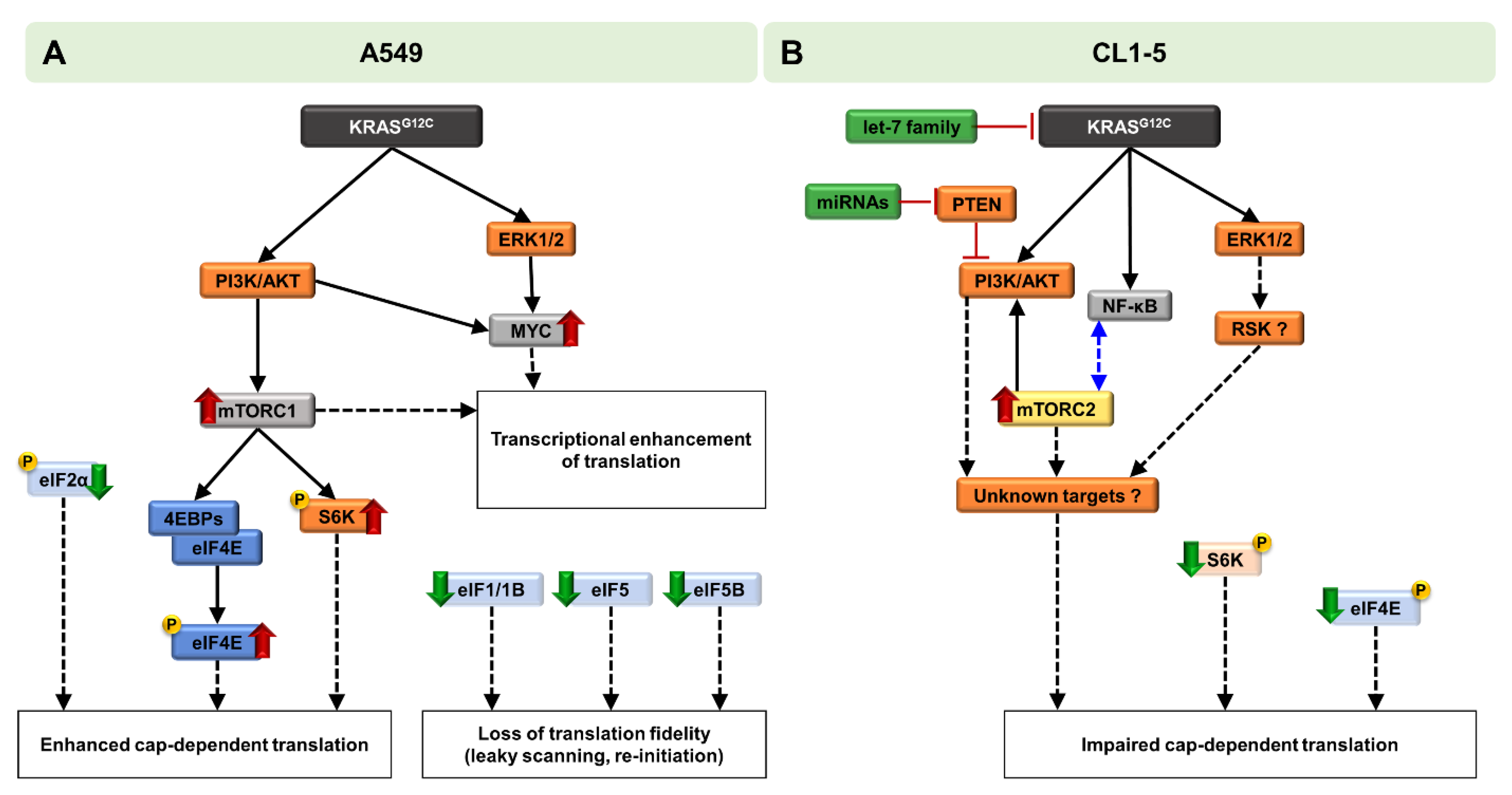
2.1. KRASG12C Signaling Differentially Affects MAPK and PI3K/AKT/mTOR Pathways
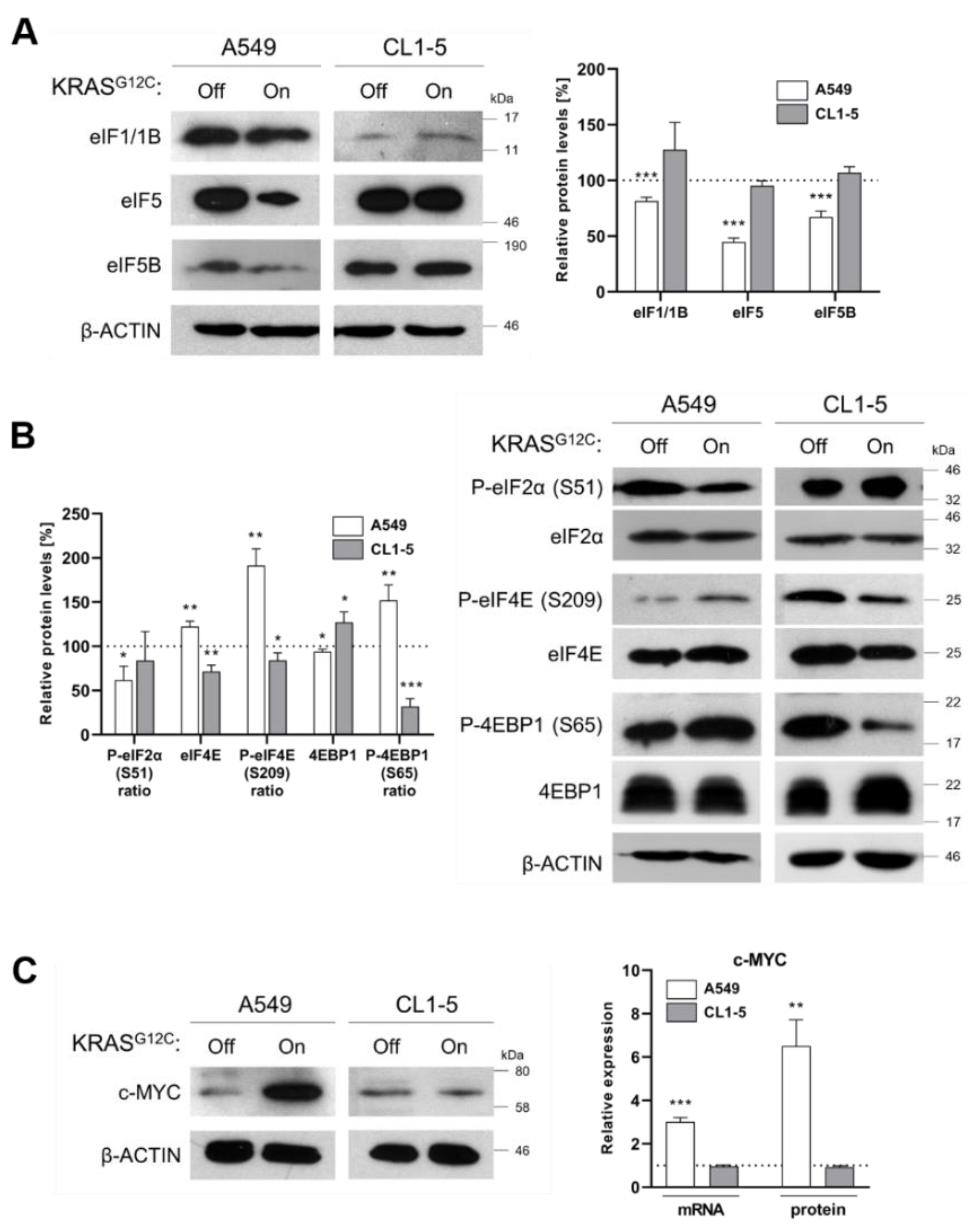
2.2. Reduced Fidelity during mRNA Scanning and Start Codon Selection
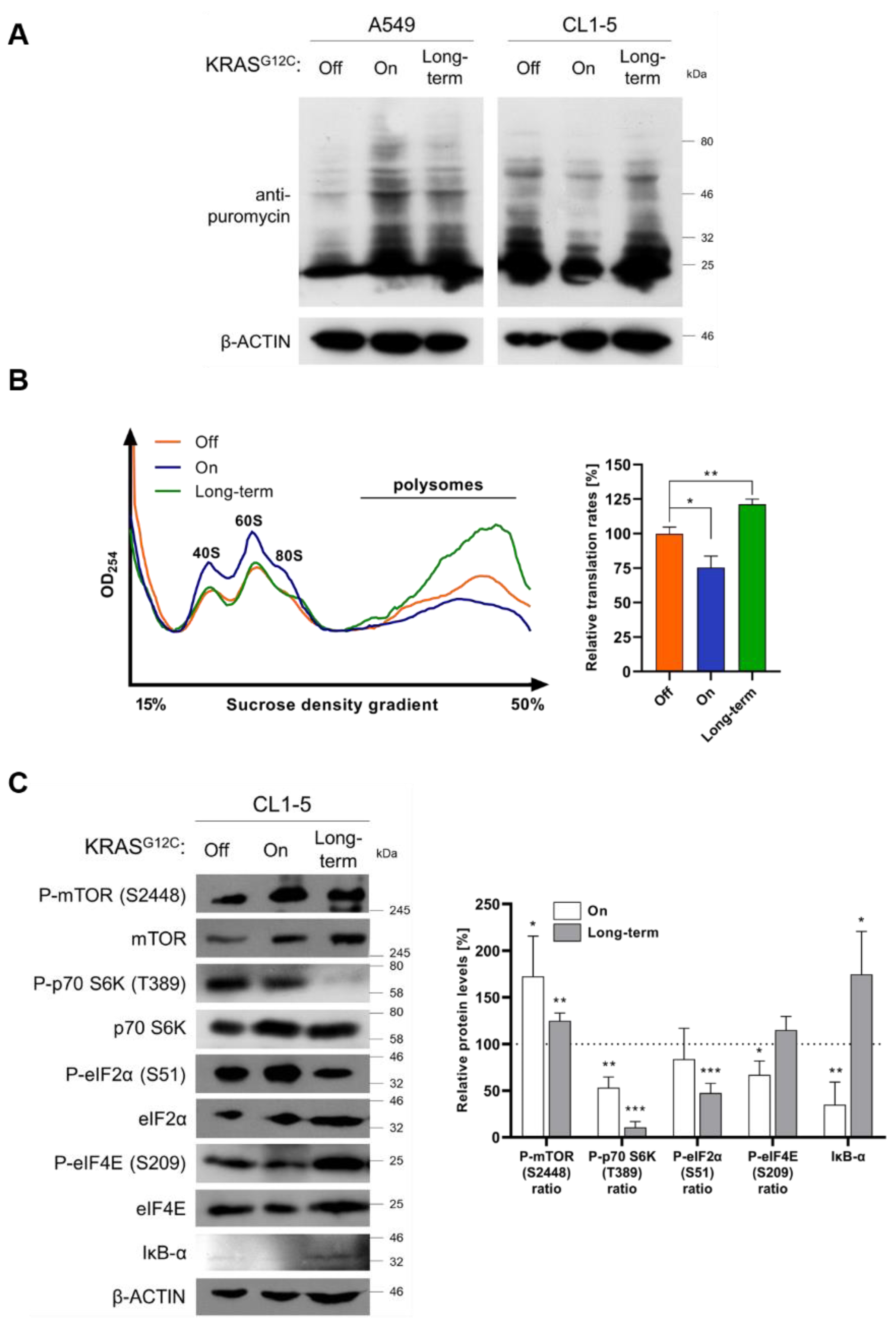
2.3. KRASG12C Modulates Cap-Dependent Translation Initiation
3. Materials and Methods
3.1. Cell Lines and Generation of Tet-Inducible KRASG12C Expression System
3.2. Cell Viability Assay
3.3. Cell Proliferation Assay
3.4. Colony-Formation Assay
3.5. Wound-Healing Assay
3.6. Immunoblotting and Non-Radioactive Measurement of Translation Rates
3.7. Real-Time Quantitative PCR
3.8. Polysome Profile Analysis
3.9. Measurement of Mitochondrial ROS Production Using Flow Cytometry
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F.; et al. Molecular Epidemiology of EGFR and KRAS Mutations in 3,026 Lung Adenocarcinomas: Higher Susceptibility of Women to Smoking-Related KRAS-Mutant Cancers. Clin. Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, I.; Zugazagoitia, J.; Herbertz, S.; John, W.; Paz-Ares, L.; Schmid-Bindert, G. KRAS-Mutant non-small cell lung cancer: From biology to therapy. Lung Cancer 2018, 124, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends in Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS G12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Tahmasebi, S.; Khoutorsky, A.; Mathews, M.B.; Sonenberg, N. Translation deregulation in human disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 791–807. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, P.P.; Topisirovic, I. Signaling pathways involved in the regulation of mRNA translation. Mol. Cell. Biol. 2018, 38, e00070-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef]
- Mandegar, M.A.; Huebsch, N.; Frolov, E.B.; Shin, E.; Truong, A.; Olvera, M.P.; Chan, A.H.; Miyaoka, Y.; Holmes, K.; Spencer, C.I.; et al. CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell 2016, 18, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Chen, L.; Fu, H.; Guo, L.; Guo, H.; Zhang, N. Upregulation of RICTOR gene transcription by the proinflammatory cytokines through NF-κB pathway contributes to the metastasis of renal cell carcinoma. Tumour Biol. 2016, 37, 4457–4466. [Google Scholar] [CrossRef]
- Tanaka, K.; Babic, I.; Nathanson, D.; Akhavan, D.; Guo, D.; Gini, B.; Dang, J.; Zhu, S.; Yang, H.; De Jesus, J.; et al. Oncogenic EGFR Signaling Activates an mTORC2-NF- B Pathway That Promotes Chemotherapy Resistance. Cancer Discov. 2011, 1, 524–538. [Google Scholar] [CrossRef] [Green Version]
- Sfikas, A.; Batsi, C.; Tselikou, E.; Vartholomatos, G.; Monokrousos, N.; Pappas, P.; Christoforidis, S.; Tzavaras, T.; Kanavaros, P.; Gorgoulis, V.G.; et al. The canonical NF-κB pathway differentially protects normal and human tumor cells from ROS-induced DNA damage. Cell. Signal. 2012, 24, 2007–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; Frias, M.A.; Chatterjee, A.; Yellen, P.; Foster, D.A. The Enigma of Rapamycin Dosage. Mol. Cancer Ther. 2016, 15, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Saqcena, M.; Chatterjee, A.; Garcia, A.; Frias, M.A.; Foster, D.A. Reciprocal Regulation of AMP-activated Protein Kinase and Phospholipase D. J. Biol. Chem. 2015, 290, 6986–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; Chatterjee, A.; Kogan, D.; Patel, D.; Foster, D.A. 5-Aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) enhances the efficacy of rapamycin in human cancer cells. Cell Cycle 2015, 14, 3331–3339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Manning, B.D. The TSC1–TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, H.; Liu, Y.; Wu, K.; Luo, H.; Cui, L. AMPK activation overcomes anti-EGFR antibody resistance induced by KRAS mutation in colorectal cancer. Cell Commun. Signal. 2020, 18, 115. [Google Scholar] [CrossRef]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS Is Regulated by the let-7 MicroRNA Family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Markopoulos, G.S.; Roupakia, E.; Tokamani, M.; Vartholomatos, G.; Tzavaras, T.; Hatziapostolou, M.; Fackelmayer, F.O.; Sandaltzopoulos, R.; Polytarchou, C.; Kolettas, E. Senescence-associated microRNAs target cell cycle regulatory genes in normal human lung fibroblasts. Exp. Gerontol. 2017, 96, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Kovalski, J.R.; Bhaduri, A.; Zehnder, A.M.; Neela, P.H.; Che, Y.; Wozniak, G.G.; Khavari, P.A. The Functional Proximal Proteome of Oncogenic Ras Includes mTORC2. Mol. Cell 2019, 73, 830–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna, R.E.; Arthanari, H.; Hiraishi, H.; Akabayov, B.; Tang, L.; Cox, C.; Markus, M.A.; Luna, L.E.; Ikeda, Y.; Watanabe, R.; et al. The Interaction between Eukaryotic Initiation Factor 1A and eIF5 Retains eIF1 within Scanning Preinitiation Complexes. Biochemistry 2013, 52, 9510–9518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fijałkowska, D.; Verbruggen, S.; Ndah, E.; Jonckheere, V.; Menschaert, G.; Van Damme, P. eIF1 modulates the recognition of suboptimal translation initiation sites and steers gene expression via uORFs. Nucleic Acids Res. 2017, 45, 7997–8013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terenin, I.M.; Akulich, K.A.; Andreev, D.E.; Polyanskaya, S.A.; Shatsky, I.N.; Dmitriev, S.E. Sliding of a 43S ribosomal complex from the recognized AUG codon triggered by a delay in eIF2-bound GTP hydrolysis. Nucleic Acids Res. 2016, 44, 1882–1893. [Google Scholar] [CrossRef] [Green Version]
- Pisareva, V.P.; Pisarev, A.V. eIF5 and eIF5B together stimulate 48S initiation complex formation during ribosomal scanning. Nucleic Acids Res. 2014, 42, 12052–12069. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.G.; Wilusz, J.E. Non-AUG translation: A new start for protein synthesis in eukaryotes. Genes Dev. 2017, 31, 1717–1731. [Google Scholar] [CrossRef] [Green Version]
- Jackson, R.J.; Hellen, C.U.T.T.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Holcik, M. Could the eIF2α-Independent Translation Be the Achilles Heel of Cancer? Front. Oncol. 2015, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Gingras, A.C.; Raught, B.; Gygi, S.P.; Niedzwiecka, A.; Miron, M.; Burley, S.K.; Polakiewicz, R.D.; Wyslouch-Cieszynska, A.; Aebersold, R.; Sonenberg, N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001, 15, 2852–2864. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-J.; Malina, A.; Pelletier, J. c-Myc and eIF4F Constitute a Feedforward Loop That Regulates Cell Growth: Implications for Anticancer Therapy. Cancer Res. 2009, 69, 7491–7494. [Google Scholar] [CrossRef] [Green Version]
- Charitou, T.; Srihari, S.; Lynn, M.A.; Jarboui, M.-A.; Fasterius, E.; Moldovan, M.; Shirasawa, S.; Tsunoda, T.; Ueffing, M.; Xie, J.; et al. Transcriptional and metabolic rewiring of colorectal cancer cells expressing the oncogenic KRASG13D mutation. Br. J. Cancer 2019, 121, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Smit, W.L.; Spaan, C.N.; Johannes de Boer, R.; Ramesh, P.; Martins Garcia, T.; Meijer, B.J.; Vermeulen, J.L.M.; Lezzerini, M.; MacInnes, A.W.; Koster, J.; et al. Driver mutations of the adenoma-carcinoma sequence govern the intestinal epithelial global translational capacity. Proc. Natl. Acad. Sci. USA 2020, 117, 25560–25570. [Google Scholar] [CrossRef] [PubMed]
- Anjum, R.; Blenis, J. The RSK family of kinases: Emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 2008, 9, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lu, W.; Chen, G.; Wang, P.; Chen, Z.; Zhou, Y.; Ogasawara, M.; Trachootham, D.; Feng, L.; Pelicano, H.; et al. K-rasG12Vtransformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012, 22, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Peltier, H.J.; Latham, G.J. Normalization of microRNA expression levels in quantitative RT-PCR assays: Identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA 2008, 14, 844–852. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyriakopoulos, G.; Katopodi, V.; Skeparnias, I.; Kaliatsi, E.G.; Grafanaki, K.; Stathopoulos, C. KRASG12C Can Either Promote or Impair Cap-Dependent Translation in Two Different Lung Adenocarcinoma Cell Lines. Int. J. Mol. Sci. 2021, 22, 2222. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042222
Kyriakopoulos G, Katopodi V, Skeparnias I, Kaliatsi EG, Grafanaki K, Stathopoulos C. KRASG12C Can Either Promote or Impair Cap-Dependent Translation in Two Different Lung Adenocarcinoma Cell Lines. International Journal of Molecular Sciences. 2021; 22(4):2222. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042222
Chicago/Turabian StyleKyriakopoulos, George, Vicky Katopodi, Ilias Skeparnias, Eleni G. Kaliatsi, Katerina Grafanaki, and Constantinos Stathopoulos. 2021. "KRASG12C Can Either Promote or Impair Cap-Dependent Translation in Two Different Lung Adenocarcinoma Cell Lines" International Journal of Molecular Sciences 22, no. 4: 2222. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042222