
Somatic Copy Number Alterations and Associated Genes in Clear-Cell Renal-Cell Carcinoma in Brazilian Patients

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Study Population—Clinicalpathological Features
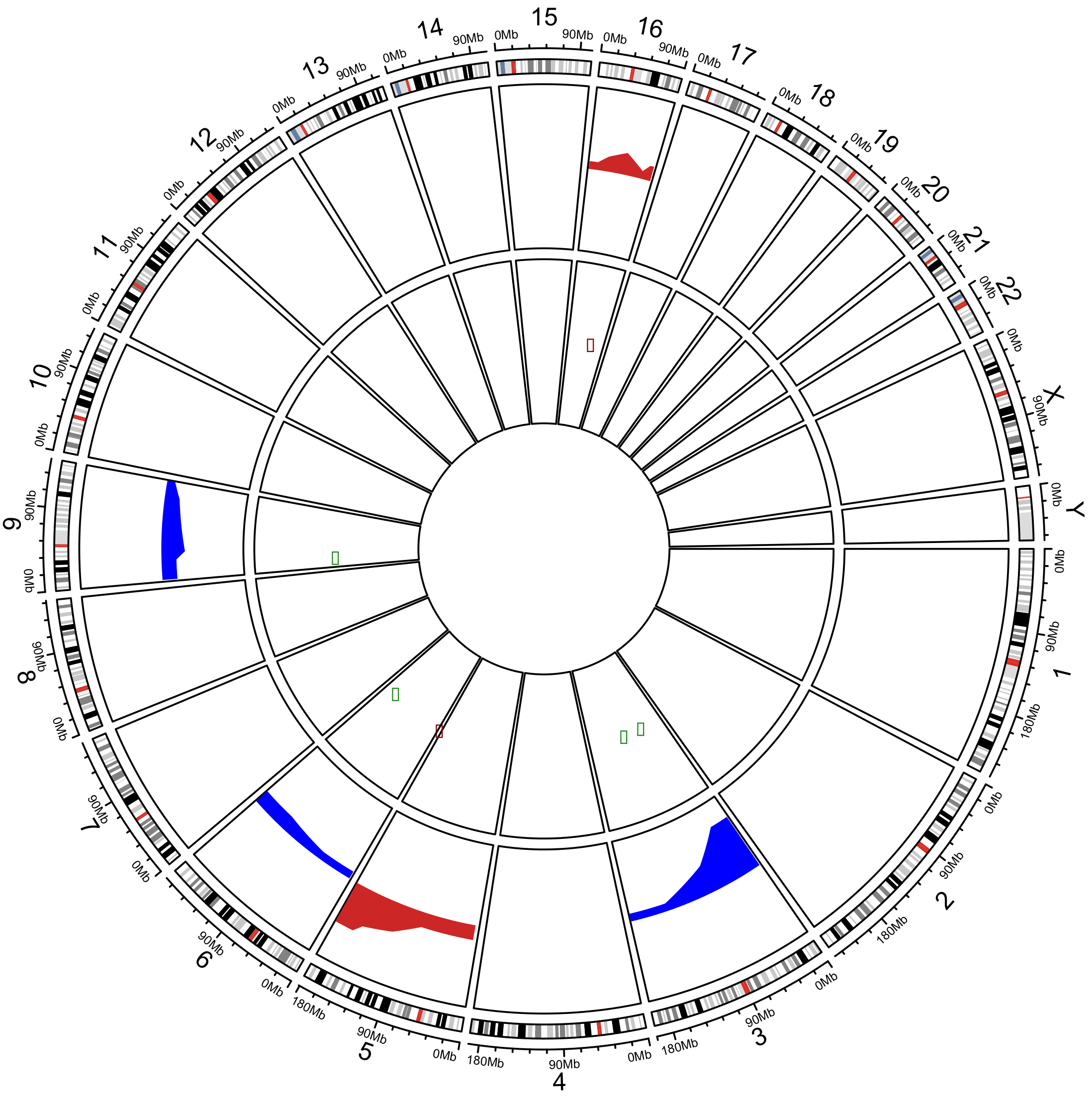
2.2. Analysis of Somatic Copy Number Alterations and Integration with In Silico Transcriptomic Profile
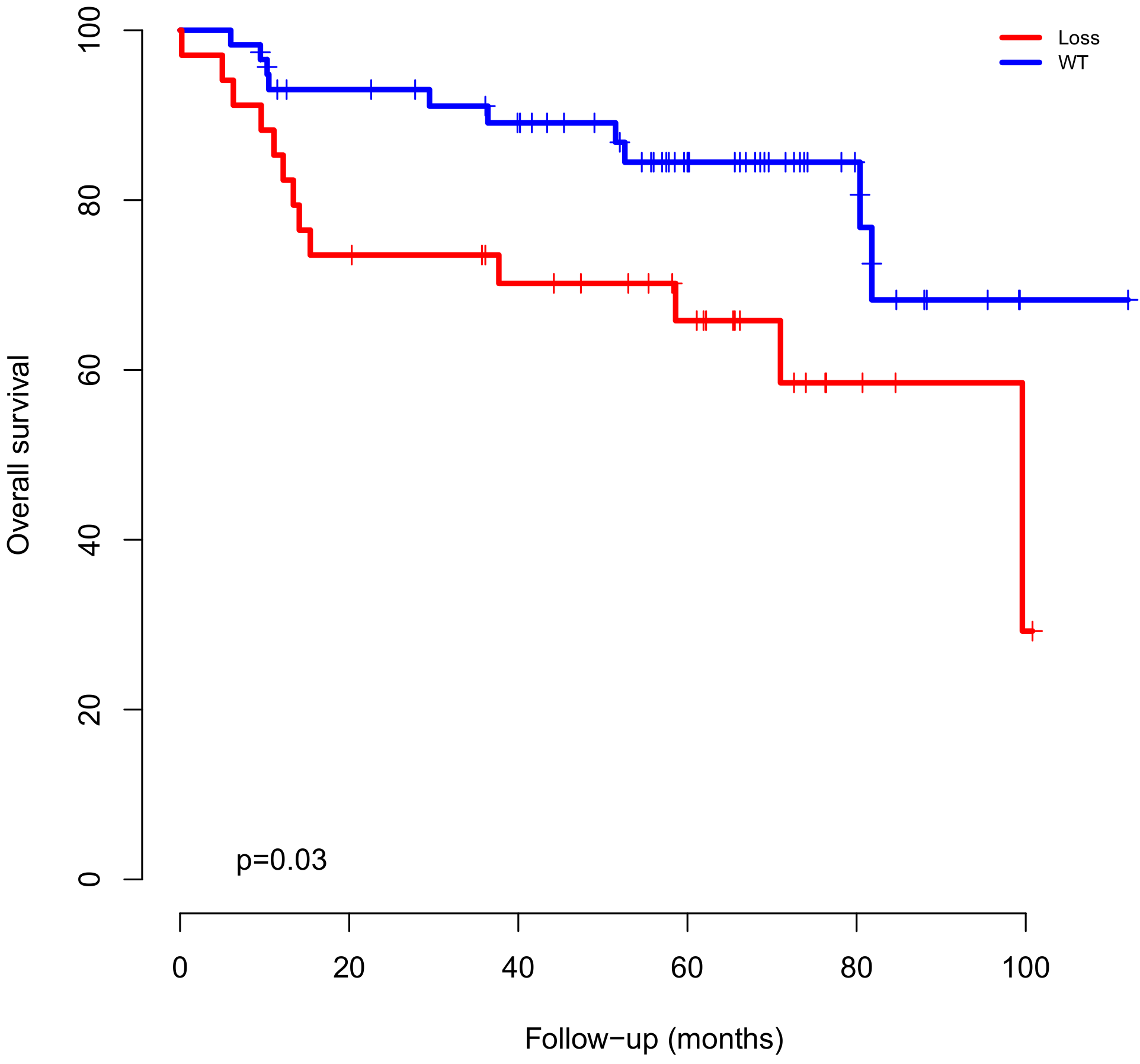
2.3. CNAs and Gene Expression Clinical Impact and Survival Analysis
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. DNA Isolation
4.3. DNA Labeling and Hybridization
4.4. Bioinformatics and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Linehan, W.M.; Srinivasan, R.; Schmidt, L.S. The genetic basis of kidney cancer: A metabolic disease. Nat. Rev. Urol. 2010, 7, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Linehan, W.M. Genetic basis of kidney cancer: Role of genomics for the development of disease-based therapeutics. Genome Res. 2012, 22, 2089–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonasch, E.; Gao, J.; Rathmell, W.K. State of the Art Review: Renal cell carcinoma. BMJ 2014. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.J.; Cubas, A.A.D.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 3698. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries: Global Cancer Statistics 2018. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middleton, R.G. Surgery for metastatic renal cell carcinoma. J. Urol. 1967, 97, 973–977. [Google Scholar] [CrossRef]
- Srigley, J.R.; Delahunt, B.; Eble, J.N.; Egevad, L.; Epstein, J.I.; Grignon, D.; Hes, O.; Moch, H.; Montironi, R.; Tickoo, S.K.; et al. The International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia. Am. J. Surg. Pathol. 2013, 37, 1469–1489. [Google Scholar] [CrossRef]
- Quddus, M.B.; Pratt, N.; Nabi, G. Chromosomal aberrations in renal cell carcinoma: An overview with implications for clinical practice. Urol. Ann. 2019, 11, 6–14. [Google Scholar] [CrossRef]
- Higgins, J.P. Gene Array Studies in Renal Neoplasia. Sci. World J. 2006, 6, 502–511. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Beroukhim, R.; Schumacher, S.E.; Zhou, J.; Chang, M.; Signoretti, S.; Kaelin, W.G. Genetic and Functional Studies Implicate HIF1A as a 14q Kidney Cancer Suppressor Gene. Cancer Discov. 2011, 1, 222–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klatte, T.; Kroeger, N.; Rampersaud, E.N.; Birkhäuser, F.D.; Logan, J.E.; Sonn, G.; Riss, J.; Rao, P.N.; Kabbinavar, F.F.; Belldegrun, A.S.; et al. Gain of chromosome 8q is associated with metastases and poor survival of patients with clear cell renal cell carcinoma. Cancer 2012, 118, 5777–5782. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.S.; Liu, W.B.; Han, F.; Yang, J.T.; Hao, X.L.; Chen, H.Q.; Jiang, X.; Yin, L.; Ao, L.; Cui, Z.H.; et al. Copy number variations and expression of MPDZ are prognostic biomarkers for clear cell renal cell carcinoma. Oncotarget 2017, 8, 78713–78725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakimi, A.A.; Reznik, E.; Lee, C.H.; Creighton, C.J.; Brannon, A.R.; Luna, A.; Aksoy, B.A.; Liu, E.M.; Shen, R.; Lee, W.; et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 2016, 29, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Yang, F.; Xu, Z.; Luo, M.; Wang, P.; Guo, Y.; Nie, H.; Yao, L.; Jiang, Q. Comprehensive Analysis of Copy Number Variations in Kidney Cancer by Single-Cell Exome Sequencing. Front. Genet. 2019, 10, 1379. [Google Scholar] [CrossRef] [Green Version]
- Klatte, T.; Rao, P.N.; de Martino, M.; LaRochelle, J.; Shuch, B.; Zomorodian, N.; Said, J.; Kabbinavar, F.F.; Belldegrun, A.S.; Pantuck, A.J. Cytogenetic profile predicts prognosis of patients with clear cell renal cell carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 746–753. [Google Scholar] [CrossRef]
- Gunawan, B.; Huber, W.; Holtrup, M.; Heydebreck, A.V.; Efferth, T.; Poustka, A.; Ringert, R.H.; Jakse, G.; Füzesi, L. Prognostic Impacts of Cytogenetic Findings in Clear Cell Renal Cell Carcinoma: Gain of 5q31–qter Predicts a Distinct Clinical Phenotype with Favorable Prognosis. Cancer Res. 2001, 61, 7731–7738. [Google Scholar]
- Kovacs, G.; Frisch, S. Clonal chromosome abnormalities in tumor cells from patients with sporadic renal cell carcinomas. Cancer Res. 1989, 49, 651–659. [Google Scholar]
- Wei, E.Y.; Hsieh, J.J. A river model to map convergent cancer evolution and guide therapy in RCC. Nat. Rev. Urol. 2015, 12, 706–712. [Google Scholar] [CrossRef] [Green Version]
- Moch, H.; Presti, J.C.; Sauter, G.; Buchholz, N.; Jordan, P.; Mihatsch, M.J.; Waldman, F.M. Genetic aberrations detected by comparative genomic hybridization are associated with clinical outcome in renal cell carcinoma. Cancer Res. 1996, 56, 27–30. [Google Scholar]
- Brunelli, M.; Eccher, A.; Gobbo, S.; Ficarra, V.; Novara, G.; Cossu-Rocca, P.; Bonetti, F.; Menestrina, F.; Cheng, L.; Eble, J.N.; et al. Loss of chromosome 9p is an independent prognostic factor in patients with clear cell renal cell carcinoma. Mod. Pathol. 2008, 21, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elfving, P.; Mandahl, N.; Lundgren, R.; Limon, J.; Bak-Jensen, E.; Fernö, M.; Olsson, H.; Mitelman, F. Prognostic implications of cytogenetic findings in kidney cancer. Br. J. Urol. 1997, 80, 698–706. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, D.; Dall’Oglio, M.F.; Reis, S.T.; Zerati, M.; Souza, I.C.; Leite, K.R.; Srougi, M. Chromosome 9p deletions are an independent predictor of tumor progression following nephrectomy in patients with localized clear cell renal cell carcinoma. Urol. Oncol. 2014, 32. [Google Scholar] [CrossRef]
- Watkins, T.B.K.; Lim, E.L.; Petkovic, M.; Elizalde, S.; Birkbak, N.J.; Wilson, G.A.; Moore, D.A.; Grönroos, E.; Rowan, A.; Dewhurst, S.M.; et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020, 587, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, J.; Jia, P.; Li, X.; Pei, G.; Wang, C.; Fang, X.; Zhao, Z.; Cai, Z.; Yi, X.; et al. Clonal architectures predict clinical outcome in clear cell renal cell carcinoma. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.J.; Turajlic, S.; Rowan, A.; Nicol, D.; Farmery, J.H.R.; O’Brien, T.; Martincorena, I.; Tarpey, P.; Angelopoulos, N.; Yates, L.R.; et al. Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell 2018, 173, 611–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrot-Zhang, J.; Chambwe, N.; Damrauer, J.S.; Knijnenburg, T.A.; Robertson, A.G.; Yau, C.; Zhou, W.; Berger, A.C.; Huang, K.L.; Newberg, J.Y.; et al. Comprehensive Analysis of Genetic Ancestry and Its Molecular Correlates in Cancer. Cancer Cell 2020, 37, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Hu, Z.; Mahal, B.A.; Zhao, S.D.; Kensler, K.H.; Pi, J.; Hu, X.; Zhang, Y.; Wang, Y.; Jiang, J.; et al. Integrated Analysis of Genetic Ancestry and Genomic Alterations across Cancers. Cancer Cell 2018, 34, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Kardas, I.; Mrózek, K.; Babinska, M.; Krajka, K.; Hadaczek, P.; Lubinski, J.; Roszkiewicz, A.; Kuziemska, E.; Limon, J. Cytogenetic and molecular findings in 75 clear cell renal cell carcinomas. Oncol. Rep. 2005, 13, 949–956. [Google Scholar] [CrossRef]
- Monzon, F.A.; Alvarez, K.; Peterson, L.; Truong, L.; Amato, R.J.; Hernandez-McClain, J.; Tannir, N.; Parwani, A.V.; Jonasch, E. Chromosome 14q loss defines a molecular subtype of clear-cell renal cell carcinoma associated with poor prognosis. Mod. Pathol. 2011, 24, 1470–1479. [Google Scholar] [CrossRef] [Green Version]
- La Rochelle, J.; Klatte, T.; Dastane, A.; Rao, N.; Seligson, D.; Said, J.; Shuch, B.; Zomorodian, N.; Kabbinavar, F.; Belldegrun, A.; et al. Chromosome 9p deletions identify an aggressive phenotype of clear cell renal cell carcinoma. Cancer 2010, 116, 4696–4702. [Google Scholar] [CrossRef]
- Mogensen, M.M.; Malik, A.; Piel, M.; Bouckson-Castaing, V.; Bornens, M. Microtubule minus-end anchorage at centrosomal and non-centrosomal sites: The role of ninein. J. Cell Sci. 2000, 113, 3013–3023. [Google Scholar]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef]
- Fan, Q.W.; Cheng, C.K.; Nicolaides, T.P.; Hackett, C.S.; Knight, Z.A.; Shokat, K.M.; Weiss, W.A. A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res. 2007, 67, 7960–7965. [Google Scholar] [CrossRef] [Green Version]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [Green Version]
- Pfister, S.; Markkanen, E.; Jiang, Y.; Sarkar, S.; D’Angiolella, V.; Dianov, G.; Ryan, A. A novel synthetic lethal interaction between the histone mark H3K36me3 and checkpoint kinases. Eur. J. Cancer 2014, 50, 71. [Google Scholar] [CrossRef]
- Peña-Llopis, S.; Vega-Rubín-de Celis, S.; Liao, A.; Leng, N.; Pavía-Jiménez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef]
- Yap, T.A.; Walton, M.I.; Hunter, L.J.K.; Valenti, M.; de Haven Brandon, A.; Eve, P.D.; Ruddle, R.; Heaton, S.P.; Henley, A.; Pickard, L.; et al. Preclinical pharmacology, antitumor activity and development of pharmacodynamic markers for the novel, potent AKT inhibitor CCT128930. Mol. Cancer Ther. 2011, 10, 360–371. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Sun, Y.; Ding, G.; Wu, Z.; Jiang, H.; Wang, L.; Ding, Q.; Wen, H. PI3KB inhibitor TGX221 selectively inhibits renal cell carcinoma cells with both VHL and SETD2 mutations and links multiple pathways. Sci. Rep. 2015, 5, 9465. [Google Scholar] [CrossRef] [Green Version]
- Eghtedar, A.; Verstovsek, S.; Estrov, Z.; Burger, J.; Cortes, J.; Bivins, C.; Faderl, S.; Ferrajoli, A.; Borthakur, G.; George, S.; et al. Phase 2 study of the JAK kinase inhibitor ruxolitinib in patients with refractory leukemias, including postmyeloproliferative neoplasm acute myeloid leukemia. Blood 2012, 119, 4614–4618. [Google Scholar] [CrossRef]
- Chase, A.; Bryant, C.; Score, J.; Haferlach, C.; Grossmann, V.; Schwaab, J.; Hofmann, W.K.; Reiter, A.; Cross, N.C.P. Ruxolitinib as potential targeted therapy for patients with JAK2 rearrangements. Haematologica 2013, 98, 404–408. [Google Scholar] [CrossRef]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Redman, B.G.; Kuzel, T.M.; Harrison, M.R.; Vaishampayan, U.N.; Drabkin, H.A.; George, S.; Logan, T.F.; et al. Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. J. Clin. Oncol. 2015, 33, 1430–1437. [Google Scholar] [CrossRef]
- Li, S.Q.; Cheuk, A.T.; Shern, J.F.; Song, Y.K.; Hurd, L.; Liao, H.; Wei, J.S.; Khan, J. Targeting wild-type and mutationally activated FGFR4 in rhabdomyosarcoma with the inhibitor ponatinib (AP24534). PLoS ONE 2013, 8, e76551. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.P.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Treon, S.P.; Tripsas, C.K.; Meid, K.; Warren, D.; Varma, G.; Green, R.; Argyropoulos, K.V.; Yang, G.; Cao, Y.; Xu, L.; et al. Ibrutinib in previously treated Waldenström’s macroglobulinemia. N. Engl. J. Med. 2015, 372, 1430–1440. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Adebiyi, M.G.; Zhang, Y.; Couturier, J.P.; Fan, X.; Zhang, H.; Kellems, R.E.; Lewis, D.E.; Xia, Y. Sphingosine-1-phosphate receptor 1 mediates elevated IL-6 signaling to promote chronic inflammation and multitissue damage in sickle cell disease. FASEB J. 2018, 32, 2855–2865. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Bense, R.D.; Urzúa-Traslaviña, C.G.; de Vries, E.G.E.; van Vugt, M.A.T.M.; Fehrmann, R.S.N. Transcriptional effects of copy number alterations in a large set of human cancers. Nat. Commun. 2020, 11, 715. [Google Scholar] [CrossRef]
- Arai, E.; Ushijima, S.; Tsuda, H.; Fujimoto, H.; Hosoda, F.; Shibata, T.; Kondo, T.; Imoto, I.; Inazawa, J.; Hirohashi, S.; et al. Genetic clustering of clear cell renal cell carcinoma based on array-comparative genomic hybridization: Its association with DNA methylation alteration and patient outcome. Clin. Cancer Res. 2008, 14, 5531–5539. [Google Scholar] [CrossRef] [Green Version]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [Green Version]
- Eble, J.N.; Weltgesundheitsorganisation. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs, Proceedings of the Editorial and Consensus Conference in Lyon, France, 14–18 December 2002, reprint ed.; Cancer, I.A., Ed.; Number 6 in World Health Organization Classification of Tumours; IARC Press: Lyon, France, 2006. [Google Scholar]
- Fuhrman, S.A.; Lasky, L.C.; Limas, C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am. J. Surg. Pathol. 1982, 6, 655–663. [Google Scholar] [CrossRef]
- Bidinotto, L.T.; Torrieri, R.; Mackay, A.; Almeida, G.C.; Viana-Pereira, M.; Cruvinel-Carloni, A.; Spina, M.L.; Campanella, N.C.; Menezes, W.P.D.; Clara, C.A.; et al. Copy Number Profiling of Brazilian Astrocytomas. G3 Genes Genomes Genet. 2016, 6, 1867–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diskin, S.J.; Eck, T.; Greshock, J.; Mosse, Y.P.; Naylor, T.; Stoeckert, C.J.; Weber, B.L.; Maris, J.M.; Grant, G.R. STAC: A method for testing the significance of DNA copy number aberrations across multiple array-CGH experiments. Genome Res. 2006, 16, 1149–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hieronymus, H.; Murali, R.; Tin, A.; Yadav, K.; Abida, W.; Moller, H.; Berney, D.; Scher, H.; Carver, B.; Scardino, P.; et al. Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. eLife 2018, 7, e37294. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Chen, H.; Li, S.; Gong, J.; Li, J.; Zou, J.; Wu, L.; Yu, J.; Han, W.; Sun, H.; et al. Tumor copy-number alterations predict response to immune-checkpoint-blockade in gastrointestinal cancer. J. Immunother. Cancer 2020, 8, e000374. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Brunet, J.P.; Di Napoli, A.; Mertz, K.D.; Seeley, A.; Pires, M.M.; Linhart, D.; Worrell, R.A.; Moch, H.; Rubin, M.A.; et al. Patterns of gene expression and copy-number alterations in VHL disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009, 69, 4674–4681. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Clinical Data | Category | Patients (Frequency) |
|---|---|---|
| Gender | Male | 50 (54.3%) |
| Female | 42 (45.7%) | |
| Alive | 69 (75.0%) | |
| Status follow-up | Deceased | 23 (25.0%) |
| Age | Average (min-max) | 58 (20–81) |
| I/II | 60 (65.2%) | |
| Clinical stage | III/IV | 32 (44.8%) |
| T | T1a-b | 47 (51.1%) |
| T2a-b | 23 (25.0%) | |
| T3a-c | 17 (18.5%) | |
| T4 | 5 (5.4%) | |
| N0 | 77 (83.7%) | |
| N1 | 9 (9.8%) | |
| N | Nx | 6 (6.5%) |
| M | M0 | 75 (81.5%) |
| M1 | 17 (18.5%) | |
| <7 cm | 58 (63.0%) | |
| Tumor size | ≥7 cm | 34 (37.0%) |
| Furhman Grade | 1 | 20 (21.7%) |
| 2 | 49 (53.3%) | |
| 3 | 16 (17.4%) | |
| 4 | 7 (7.6%) | |
| White | 64 (69.6%) | |
| Black | 3 (3.3%) | |
| Yellow | 1 (1.0%) | |
| Brown * | 20 (21.7%) | |
| Self-Declared Ethnicity | Unknown | 4 (4.4%) |
| Systemic Therapy | Sunitinib | 12 (13.0%) |
| Interferon | 4 (4.3%) | |
| Pazopanib | 1 (1.1%) | |
| None | 74 (80.4%) | |
| Other | 1 (1.1%) | |
| Alive without disease | 64 (69.6%) | |
| Alive with disease progression | 2 (2.1%) | |
| Progression and death from the disease | 18 (19.6%) | |
| Disease status (last follow-up) | Died without disease recurrence | 8 (8.7%) |
| Chromosome | Cytoband | Event | Patients Frequency (%) | TCGA Frequency (%) | Cancer Genes |
|---|---|---|---|---|---|
| Chr3 | p21.2–p21.3 | DEL | 83.7 | 72.2 | BAP1, SETD2 |
| Chr3 | p22.2 | DEL | 83.7 | 72.2 | MYD88 |
| Chr3 | p25.2 | DEL | 83.7 | 72.2 | PPARG |
| Chr3 | q11.2-q13.11 | DEL | 20.6 | 25.7 | TFG, CBLB |
| Chr5 | q34-q35.3 | AMP | 59.7 | 47.6 | RANBP17, NPM1, FGFR4, NSD1, FLT4 |
| Chr6 | q23.2–q23.3 | DEL | 29.3 | 19.0 | MYB |
| Chr7 | p21.1 | AMP | 29.3 | 22.9 | ETV1 |
| Chr9 | p21.3 | DEL | 28.2 | 17.9 | MLLT3 |
| Chr9 | p24.1 | DEL | 27.1 | 20.7 | JAK2, CD274 |
| Chr10 | q23.31 | DEL | 25.0 | 13.6 | PTEN |
| Chr14 | q22.1 | DEL | 35.9 | 29.0 | NIN |
| Chr16 | q21 | AMP | 20.6 | 11.9 | CDH11 |
| Chromosome | Cytoband | Event | Clinical Features | p Value | FDR |
|---|---|---|---|---|---|
| Chr3 | p21.2 | DEL | Fuhrman Grade (1-2) | 0.02 | 0.15 |
| Chr3 | p25.2 | DEL | Fuhrman Grade (1–2) | 0.01 | 0.14 |
| Chr5 | q34–q35.1 | AMP | Metastasis | 0.03 | 0.18 |
| Tumor size (≥7 cm) | 0.01 | 0.14 | |||
| Fuhrman Grade (3–4) | 0.01 | 0.14 | |||
| Chr7 | p21.2 | AMP | Tumors T3/T4 | 0.02 | 0.15 |
| Chr9 | p21.3 | DEL | Metastasis | 0.01 | 0.14 |
| Tumor size (≥7 cm) | 0.02 | 0.15 | |||
| Tumors T3–T4 | 0.04 | 0.19 | |||
| Clinical stage (3–4) | 0.01 | 0.14 | |||
| Tumor size (≥7 cm) | 0.04 | 0.19 | |||
| Chr9 | p24.1 | DEL | Tumors T3-T4 | 0.04 | 0.19 |
| Chr14 | q22.1 | DEL | Clinical stage (3–4) | 0.04 | 0.19 |
| Outcome (death) | 0.02 | 0.15 | |||
| Metastasis | 0.03 | 0.19 | |||
| Fuhrman Grade (3–4) | 0.01 | 0.14 |
| Clinical Feature | Region | RR | 95% CI | p Value |
|---|---|---|---|---|
| Tumors T3/T4 | 7p21.2 (AMP) | 3.0 | 1.11–8.04 | 0.02 |
| Tumor size (>7 cm) | 5q35.2–q35.3 (AMP) | 2.9 | 1.08–8.14 | 0.03 |
| 9p24.1 (DEL) | 3.3 | 1.25–9.04 | 0.01 | |
| Clinical Stage (3–4) | 9p21.3 (DEL) | 3.9 | 1.51–10.20 | 0.005 |
| Outcome (Death) | 14q22.1 (DEL) | 2.9 | 1.12–7.84 | 0.02 |
| Fuhrman Grade (3–4) | 7p21.2 (AMP) | 3.4 | 1.28–9.10 | 0.01 |
| Metastasis | 9p21.3 (DEL) | 5.2 | 1.72–16.02 | 0.003 |
| Clinical Feature | Category | RR | 95% CI | p Value |
|---|---|---|---|---|
| Tumor size (≥7 cm) | > 50 CNA | 7.8 | 2.14–28.55 | 0.002 |
| Outcome (death) | > 50 CNA | 3.7 | 1.02–14.02 | 0.04 |
| Fuhrman Grade (3–4) | > 50 CNA | 14.1 | 1.80–111.13 | 0.01 |
| Chr | Cytoband | Gene | CN Event | Expression | Clinical Features | p-Value |
|---|---|---|---|---|---|---|
| 3 | p21.2–p21.3 | BAP1 | Loss | Downregulation | Fuhrman Grade (1–2) | 0.02 |
| 5 | q34–q35.3 | FLT4 | Gain | Upregulation | Tumor size (>7 cm) | 0.007 |
| 5 | q34–q35.3 | NSD1 | Gain | Upregulation | Tumor size (>7 cm) | 0.007 |
| Clinical Stage (3-4) | 0.01 | |||||
| 6 | q23.2–q23.3 | MYB | Loss | Downregulation | Fuhrman Grade (3-4) | 0.02 |
| 9 | p21.3 | MLLT3 | Loss | Downregulation | Metastasis | 0.005 |
| Clinical Stage (3–4) | 0.02 | |||||
| Tumor size (>7 cm) | 0.01 | |||||
| Outcome (death) | 0.02 | |||||
| Fuhrman Grade (3–4) | 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, F.G.; Silveira, H.C.S.; Júnior, J.N.A.; da Silveira, R.A.; Zucca, L.E.; Cárcano, F.M.; Sanches, A.O.N.; Neder, L.; Scapulatempo-Neto, C.; Serrano, S.V.; et al. Somatic Copy Number Alterations and Associated Genes in Clear-Cell Renal-Cell Carcinoma in Brazilian Patients. Int. J. Mol. Sci. 2021, 22, 2265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052265
Fernandes FG, Silveira HCS, Júnior JNA, da Silveira RA, Zucca LE, Cárcano FM, Sanches AON, Neder L, Scapulatempo-Neto C, Serrano SV, et al. Somatic Copy Number Alterations and Associated Genes in Clear-Cell Renal-Cell Carcinoma in Brazilian Patients. International Journal of Molecular Sciences. 2021; 22(5):2265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052265
Chicago/Turabian StyleFernandes, Flávia Gonçalves, Henrique Cesar Santejo Silveira, João Neif Antonio Júnior, Rosana Antunes da Silveira, Luis Eduardo Zucca, Flavio Mavignier Cárcano, André Octavio Nicolau Sanches, Luciano Neder, Cristovam Scapulatempo-Neto, Sergio Vicente Serrano, and et al. 2021. "Somatic Copy Number Alterations and Associated Genes in Clear-Cell Renal-Cell Carcinoma in Brazilian Patients" International Journal of Molecular Sciences 22, no. 5: 2265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052265