
IL-33 Is Involved in the Anti-Inflammatory Effects of Butyrate and Propionate on TNFα-Activated Endothelial Cells

 , , , and
, , , and 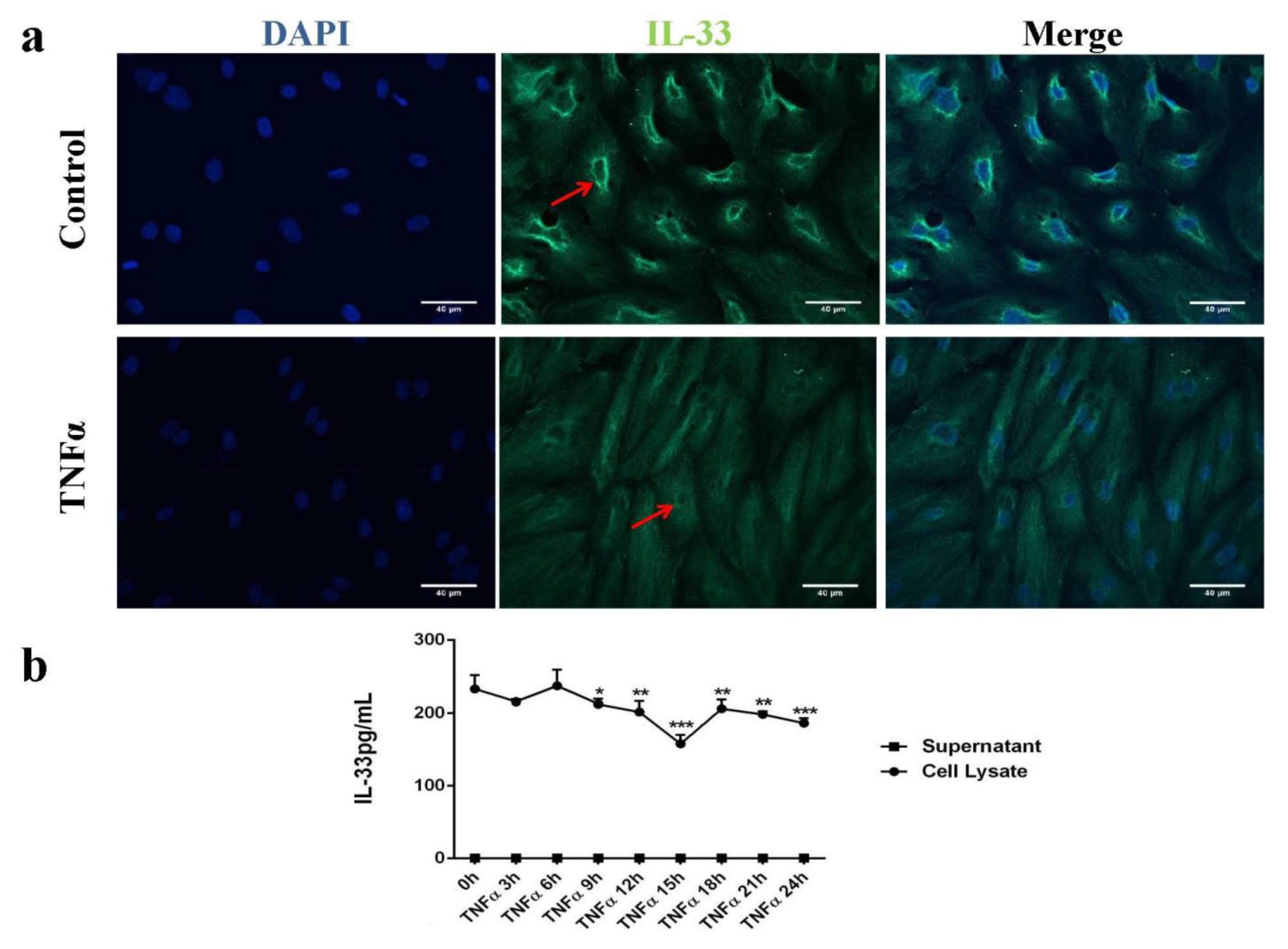{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. IL-33 Was Expressed in the Cytoplasm and Perinuclear in Human Endothelial Cells (HUVECs)
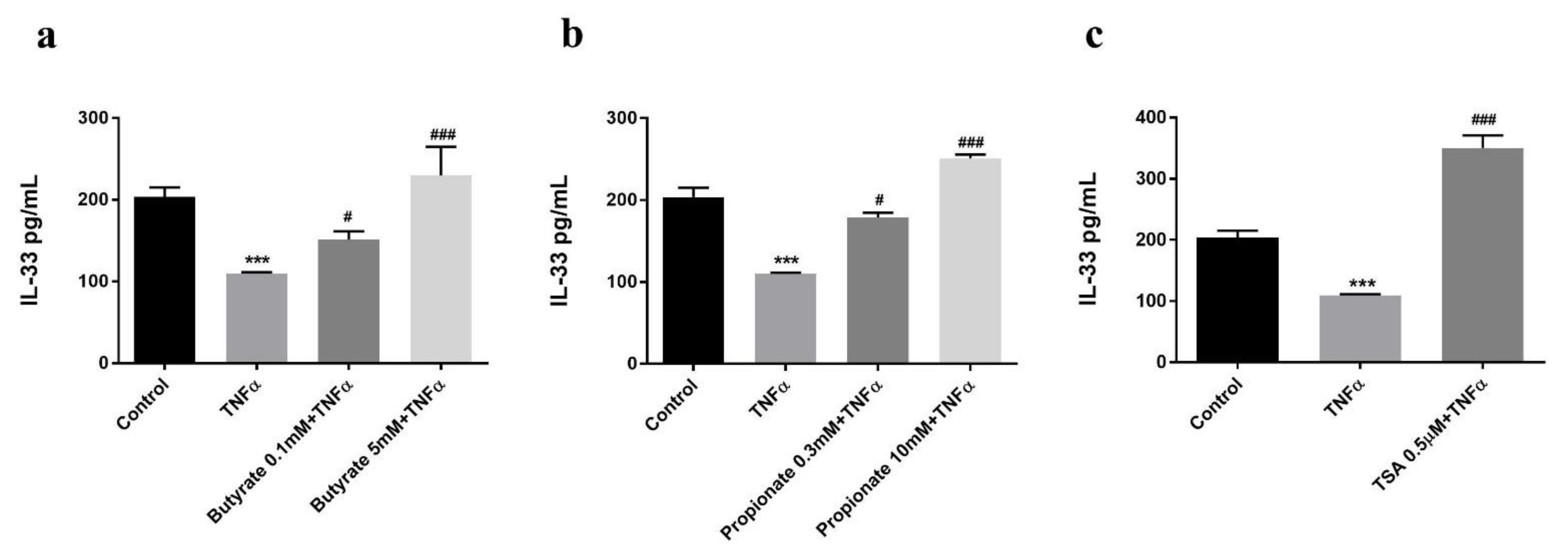
2.2. Butyrate and Propionate Prevented a TNFα-Induced Decrease in the Intracellular IL-33 Level
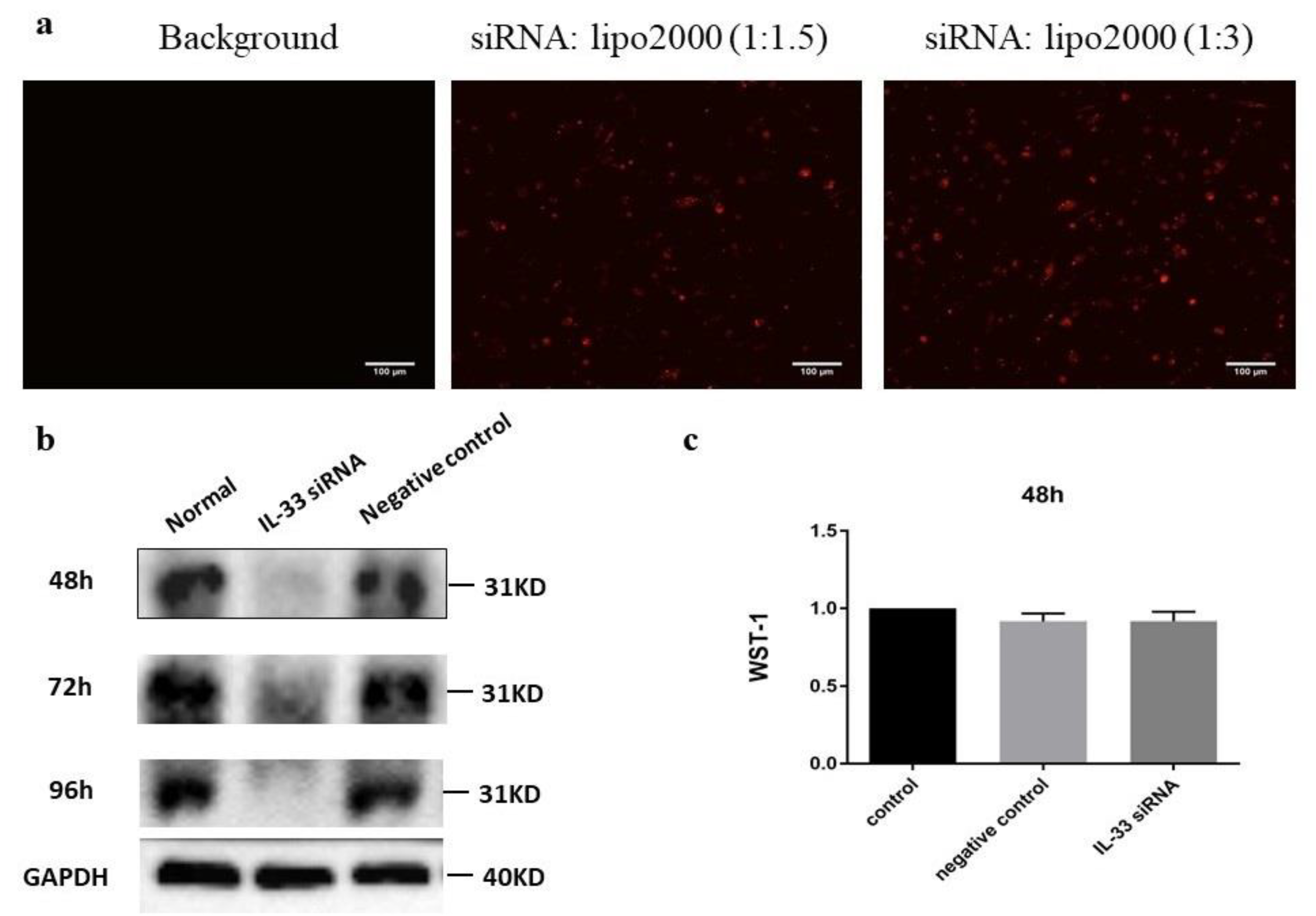
2.3. siIL-33 Transfection
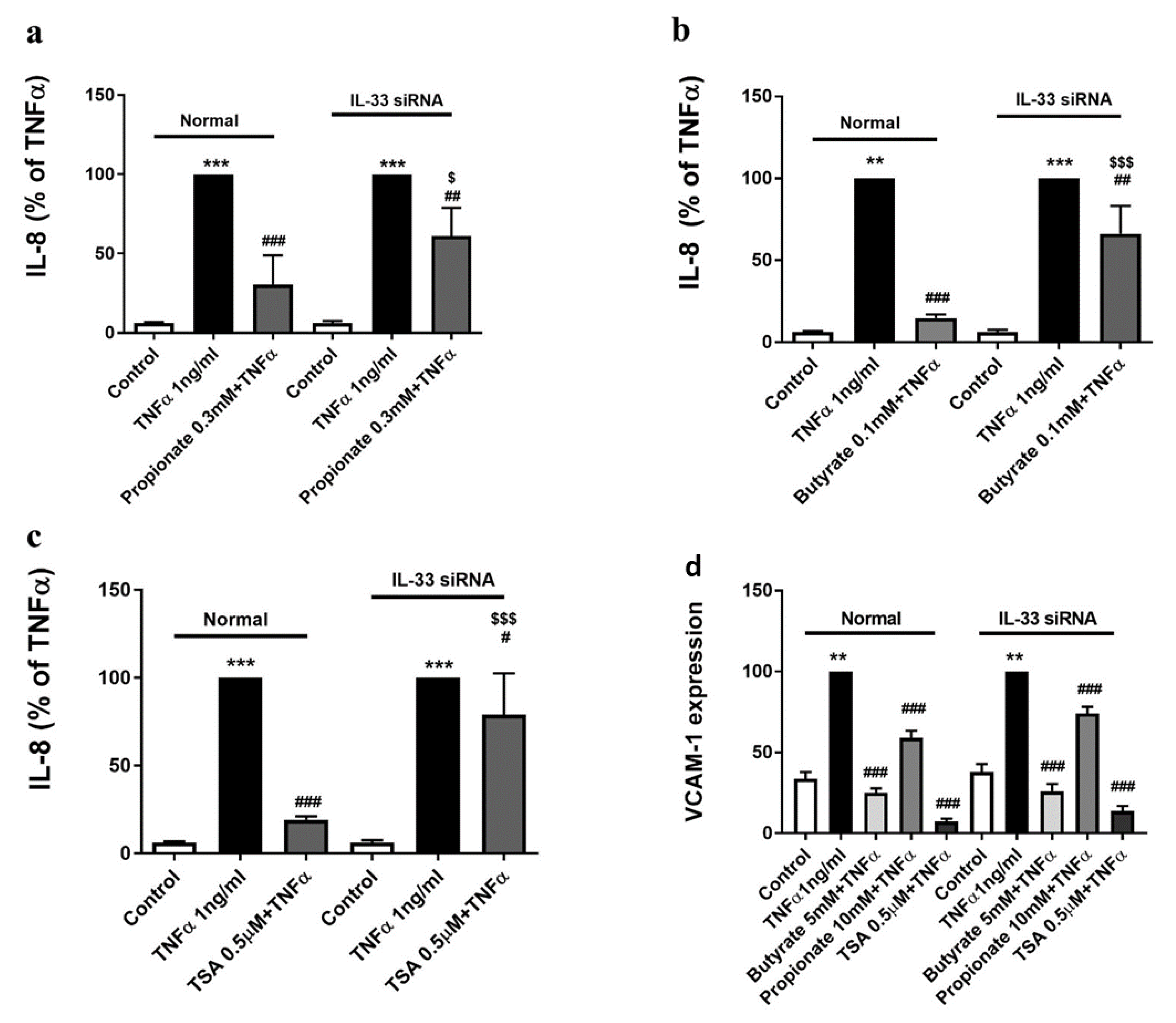
2.4. The Inhibition of TNFα-Induced IL-8 Production by Butyrate and Propionate Is IL-33-Dependent
2.5. The Inhibition of the TNFα-Induced VCAM-1 Expression by Butyrate and Propionate Is IL-33-Independent
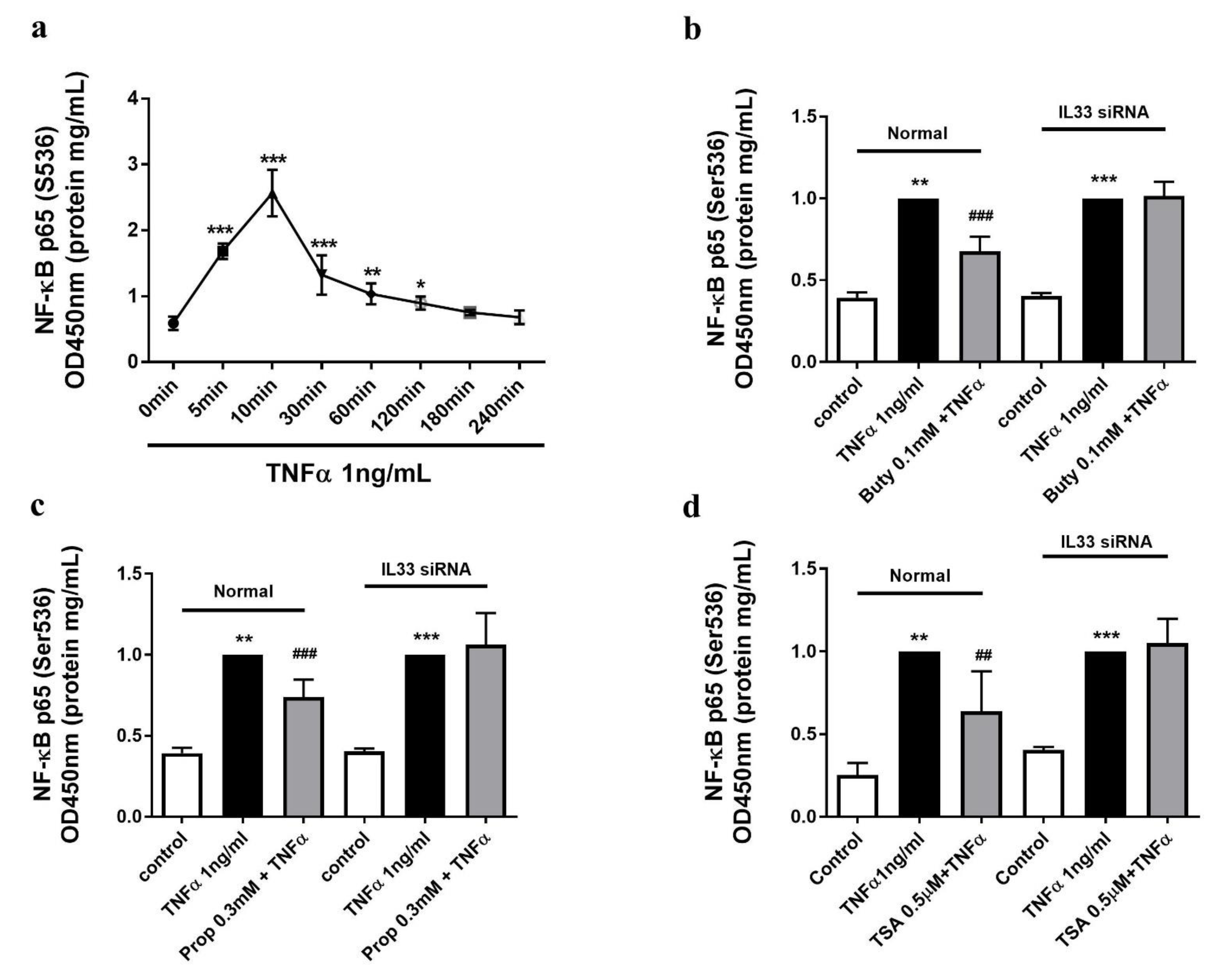
2.6. The Butyrate and Propionate-Inhibited NF-κB Activation Is IL-33 Dependent
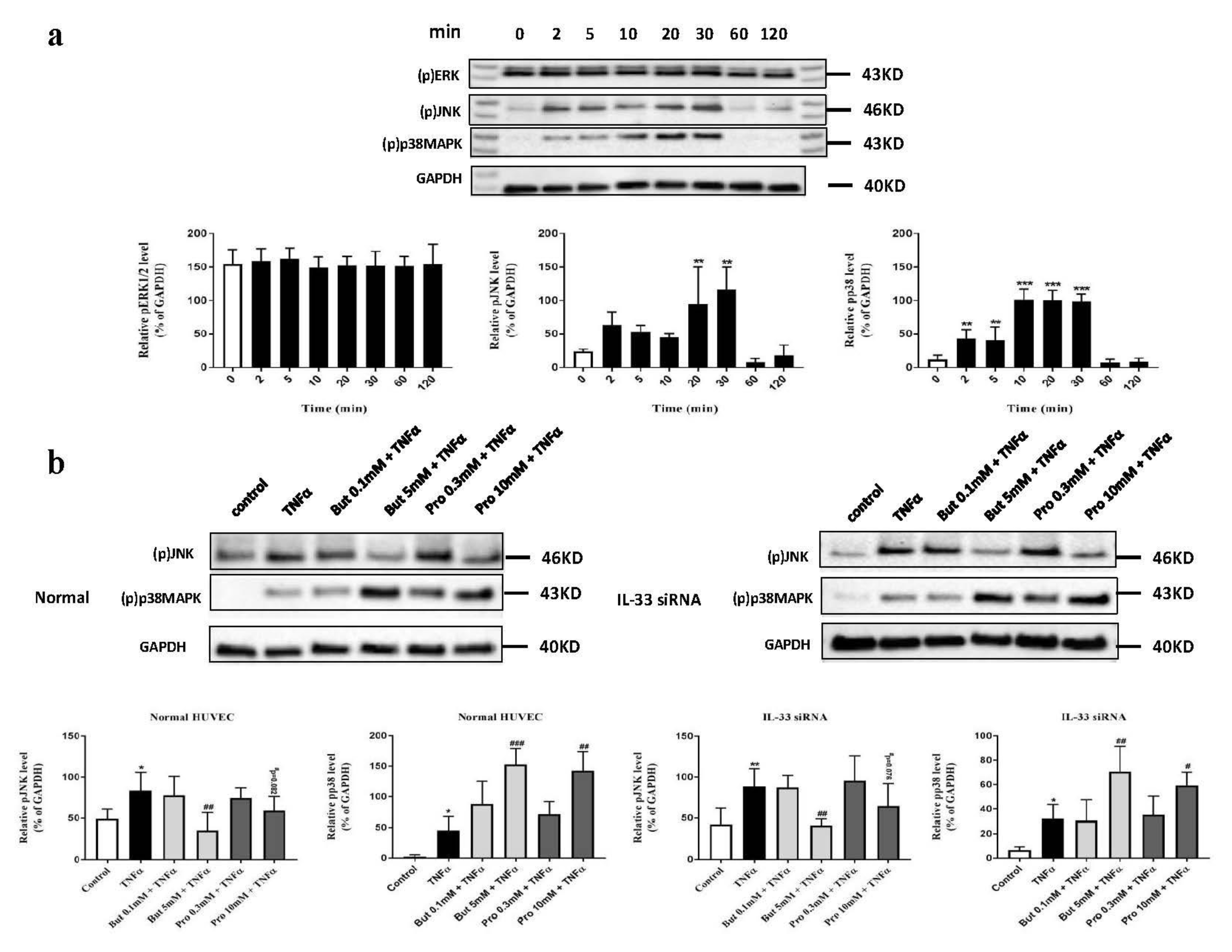
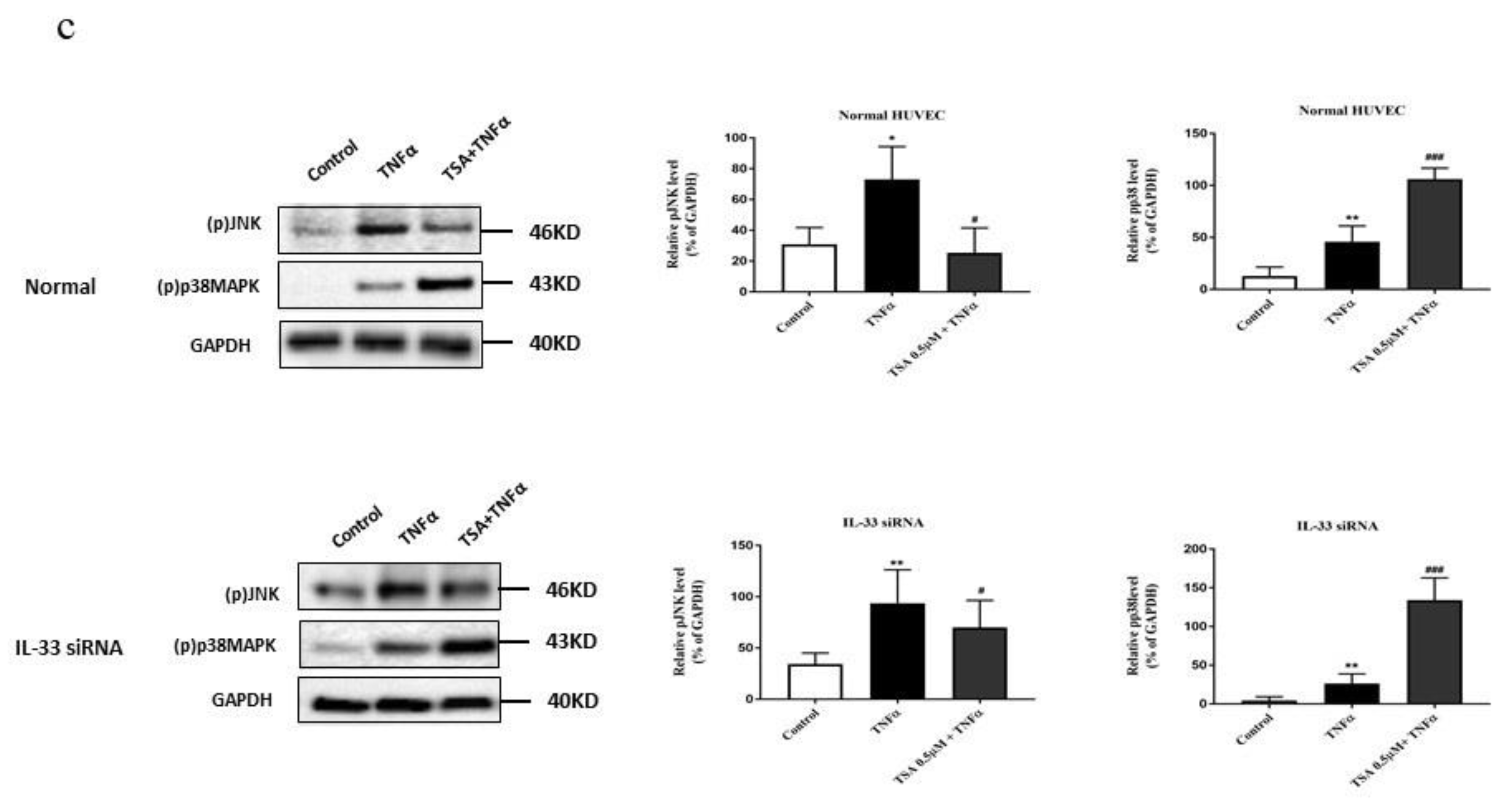
2.7. Butyrate and Propionate Modulated the MAPK Signaling Pathway Independently of IL-33
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. HUVEC Culture
4.3. Small Interfering RNA (siRNA) and Cytotoxicity Test
4.4. Immunocytochemistry
4.5. IL-33 ELISA Assay
4.6. IL-8 Production in siIL-33 Transfected HUVEC
4.7. VCAM-1 Expression in siIL-33 Transfected HUVECs by Flow Cytometry
4.8. Activation of NF-κB P65 in Normal and siIL-33 Transfected HUVEC
4.9. Activation of MAPK Signaling Pathway
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, R.B.; Mengi, A.S.; Xu, Y.-J.; Arneja, A.S.; Dhalla, N.S. Pathogenesis of atherosclerosis: A multifactorial process. Exp. Clin. Cardiol. 2002, 7, 40–53. [Google Scholar]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, Atherosclerosis, and Coronary Artery Disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [Green Version]
- Ohira, H.; Tsutsui, W.; Fujioka, Y. Are Short Chain Fatty Acids in Gut Microbiota Defensive Players for Inflammation and Atherosclerosis? J. Atheroscler. Thromb. 2017, 24, 660–672. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Van Esch, B.C.; Henricks, P.A.J.; Garssen, J.; Folkerts, G. Time and Concentration Dependent Effects of Short Chain Fatty Acids on Lipopolysaccharide- or Tumor Necrosis Factor α-Induced Endothelial Activation. Front. Pharmacol. 2018, 9, 233. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; van Esch, B.C.; Henricks, P.A.J.; Garssen, J.; Folkerts, G. The anti-inflammatory effects of short chain fatty acids on lipopolysaccharide- or tumor necrosis factor α-stimulated endothelial cells via activation of GPR41/43 and inhibi-tion of HDACs. Front. Pharmacol. 2018, 9, 533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuchler, A.M.; Pollheimer, J.; Balogh, J.; Sponheim, J.; Manley, L.; Sorensen, D.R.; De Angelis, P.M.; Scott, H.; Haraldsen, G. Nuclear interleukin-33 is generally expressed in resting endothelium but rapidly lost upon angiogenic or proinflamma-tory activation. Am. J. Pathol. 2008, 173, 1229–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Tossberg, J.T.; Spurlock, C.F.; Yao, S.; Aune, T.M.; Sriram, S. Expression of IL-33 and its epigenetic regulation in multiple sclerosis. Ann. Clin. Transl. Neurol. 2014, 1, 307–318. [Google Scholar] [CrossRef]
- Toki, S.; Goleniewska, K.; Reiss, S.; Zhou, W.; Newcomb, D.C.; Bloodworth, M.H.; Stier, M.T.; Boyd, K.L.; Polosukhin, V.V.; Subramaniam, S.; et al. The histone deacetylase inhibitor trichostatin A suppresses murine innate allergic in-flammation by blocking group 2 innate lymphoid cell (ILC2) activation. Thorax 2016, 71, 633–645. [Google Scholar] [CrossRef] [Green Version]
- Moussion, C.; Ortega, N.; Girard, J.-P. The IL-1-Like Cytokine IL-33 Is Constitutively Expressed in the Nucleus of Endothelial Cells and Epithelial Cells In Vivo: A Novel ‘Alarmin’? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cayrol, C.; Girard, J.-P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Chen, J.; Zhang, H.; Yang, H.; Zhu, P.; Xiong, A.; Xia, Q.; Zheng, F.; Tan, Z.; Gong, F.; et al. Interleukin-33 Ameliorates Experimental Colitis through Promoting Th2/Foxp3+ Regulatory T-Cell Responses in Mice. Mol. Med. 2012, 18, 753–761. [Google Scholar] [CrossRef]
- Gautier, V.; Cayrol, C.; Farache, D.; Roga, S.; Monsarrat, B.; Burlet-Schiltz, O.; de Peredo, A.G.; Girard, J.-P. Extra-cellular IL-33 cytokine, but not endogenous nuclear IL-33, regulates protein expression in endothelial cells. Sci. Rep. 2016, 6, 34255. [Google Scholar] [CrossRef] [PubMed]
- Montanari, E.; Stojkovic, S.; Kaun, C.; Lemberger, C.E.; de Martin, R.; Rauscher, S.; Gröger, M.; Maurer, G.; Neumayer, C.; Huk, I.; et al. Interleukin-33 stimulates GM-CSF and M-CSF production by human endothe-lial cells. Thromb. Haemost. 2016, 116, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.; O’Grady, K.; Lavelle, E.C.; Fallon, P.G. Interleukin 33: An innate alarm for adaptive responses beyond Th2 immunity-emerging roles in obesity, intestinal inflammation, and cancer. Eur. J. Immunol. 2016, 46, 1091–1100. [Google Scholar] [CrossRef]
- Miller, A.M. Role of IL-33 in inflammation and disease. J. Inflamm. 2011, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.S.; Park, J.A.; Kim, J.; Rho, S.S.; Park, H.; Kim, Y.M.; Kwon, Y.G. Nuclear IL-33 is a transcriptional regulator of NF-κB p65 and induces endothelial cell activation. Biochem. Biophys. Res. Commun. 2012, 421, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Demyanets, S.; Konya, V.; Kastl, S.P.; Kaun, C.; Rauscher, S.; Niessner, A.; Pentz, R.; Pfaffenberger, S.; Rychli, K.; Lem-berger, C.E.; et al. Interleukin-33 induces ex-pression of adhesion molecules and inflammatory activation in human endothelial cells and in human atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2080–2089. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.M.; Xu, D.; Asquith, D.L.; Denby, L.; Li, Y.; Sattar, N.; Baker, A.H.; McInnes, I.B.; Liew, F.Y. IL-33 reduces the development of atherosclerosis. J. Exp. Med. 2008, 20, 339–346. [Google Scholar] [CrossRef]
- Ohta, S.; Tago, K.; Funakoshi-Tago, M.; Matsugi, J.; Yanagisawa, K. Intracellular NF-HEV/IL-33 harbors essential roles in Ras-induced cellular transformation by contributing to cyclin D1 protein synthesis. Cell. Signal. 2016, 28, 1025–1036. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, a000034. [Google Scholar] [CrossRef]
- Zheng, X.-X.; Zhou, T.; Wang, X.-A.; Tong, X.-H.; Ding, J.-W. Histone deacetylases and atherosclerosis. Atherosclerosis 2015, 240, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Mohs, A.; Thomas, M.; Klare, J.; Ross, R.; Schmitz, M.L.; Martin, M.U. The dual function cytokine IL-33 interacts with the transcription factor NF-κB to dampen NF-κB-stimulated gene transcription. J. Immunol. 2011, 187, 1609–1616. [Google Scholar] [CrossRef] [Green Version]
- Pietersma, A.; Tilly, B.C.; Gaestel, M.; De Jong, N.; Lee, J.C.; Koster, J.F.; Sluiter, W. P38 Mitogen Activated Protein Kinase Regulates Endothelial VCAM-1 Expression at the Post-transcriptional Level. Biochem. Biophys. Res. Commun. 1997, 230, 44–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, A.K.; Rothlein, R. Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free. Radic. Biol. Med. 2000, 28, 1379–1386. [Google Scholar] [CrossRef]
- Ueno, H.; Pradhan, S.; Schlessel, D.; Hirasawa, H.; Sumpio, B.E. Nicotine enhances human vascular endothelial cell expres-sion of ICAM-1 and VCAM-1 via protein kinase C, p38 mitogen-activated protein kinase, NF-κB, and AP-1. Cardiovasc. Toxicol. 2006, 6, 39–50. [Google Scholar] [CrossRef]
- Li, M.S.; Rafiee, P.; Fisher, P.J.; Lamirand, T.H.; Taras, A.R.; Shidham, V.; Johnson, C.P.; Binion, D.G. Vascular cell adhe-sion molecule-1 (VCAM-1) expression in human intestinal micro-vascular endothelial cells (HIMEC) is mediated by JNK, intracellular oxyradicals and NFκB activation. Gastroenterology 2000, 118, A613. [Google Scholar] [CrossRef]
- Ono, H.; Ichiki, T.; Ohtsubo, H.; Fukuyama, K.; Imayama, I.; Iino, N.; Masuda, S.; Hashiguchi, Y.; Takeshita, A.; Sunagawa, K. cAMP-response element-binding protein mediates tumor necrosis factor-α-induced vascular cell adhesion molecule-1 expression in endothelial cells. Hypertens. Res. 2006, 29, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Roussel, L.; Houle, F.; Chan, C.; Yao, Y.; Bérubé, J.; Olivenstein, R.; Martin, J.G.; Huot, J.; Hamid, Q.; Ferri, L.; et al. IL-17 Promotes p38 MAPK-Dependent Endothelial Activation Enhancing Neutrophil Recruitment to Sites of Inflammation. J. Immunol. 2010, 184, 4531–4537. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.-S.; Li, Y.-J.; Jiang, Z.-A.; Liu, S.-Y.; Guo, B.-Y.; Wang, T. Nicotine-induced ICAM-1 and VCAM-1 expression in mouse cardiac vascular endothelial cell via p38 MAPK signaling pathway. Anal. Quant. Cytopathol. Histopathol. 2014, 36, 258–262. [Google Scholar]
- Jeong, Y.; Du, R.; Zhu, X.; Yin, S.; Wang, J.; Cui, H.; Cao, W.; Lowenstein, C.J. Histone deacetylase isoforms regulate innate immune responses by deacetylating mitogen-activated protein kinase phosphatase. J. Leukoc. Biol. 2014, 95, 651–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Wang, Z.; Zhang, L.; Wang, Y. Roles of Cells from the Arterial Vessel Wall in Atherosclerosis. Mediat. Inflamm. 2017, 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pollheimer, J.; Bodin, J.; Sundnes, O.; Edelmann, R.J.; Skånland, S.S.; Sponheim, J.; Brox, M.J.; Sundlisaeter, E.; Loos, T.; Vatn, M.; et al. Interleukin-33 drives a proinflamma-tory endothelial activation that selectively targets nonquiescent cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, e47–e55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arshad, M.I.; Guihard, P.; Danger, Y.; Noel, G.; Le Seyec, J.; Boutet, M.-A.; Richards, C.D.; L’Helgoualc’H, A.; Genet, V.; Lucas-Clerc, C.; et al. Oncostatin M induces IL-33 expression in liver endothelial cells in mice and expands ST2+CD4+lymphocytes. Am. J. Physiol. Liver Physiol. 2015, 309, G542–G553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boisvert, W.A. The participation of chemokines in atherosclerosis. Discov. Med. 2004, 4, 288–292. [Google Scholar]
- Henricks, P.A.; Nijkamp, F.P. Pharmacological modulation of cell adhesion molecules. Eur. J. Pharmacol. 1998, 344, 1–13. [Google Scholar] [CrossRef]
- Li, M.; Van Esch, B.C.; Wagenaar, G.T.; Garssen, J.; Folkerts, G.; Henricks, P.A. Pro- and anti-inflammatory effects of short chain fatty acids on immune and endothelial cells. Eur. J. Pharmacol. 2018, 831, 52–59. [Google Scholar] [CrossRef]
- Liu, S.F.; Malik, A.B. NF-κB activation as a pathological mechanism of septic shock and inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L622–L645. [Google Scholar] [CrossRef]
- Hsuan, C.-F.; Hsu, H.-F.; Tseng, W.-K.; Lee, T.-L.; Wei, Y.-F.; Hsu, K.-L.; Wu, C.-C.; Houng, J.-Y. Glossogyne tenuifolia ex-tract inhibits TNF-α-induced expression of adhesion molecules in human umbilical vein en dothelial cells via blocking the NF-kB signaling pathway. Molecules 2015, 20, 16908–16923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunsch, C.; Rosen, C.A. NF-kappa B subunit-specific regulation of the interleukin-8 promoter. Mol. Cell. Biol. 1993, 13, 6137–6146. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.-J.; Wang, S.-H.; Chen, Y.-H.; Chang, S.-S.; Hwang, T.-L.; Leu, Y.-L.; Tseng, Y.-C.; Li, C.-Y.; Chen, Y.-L. Viscolin reduces VCAM-1 expression in TNF-α-treated endothelial cells via the JNK/NF-κB and ROS pathway. Free. Radic. Biol. Med. 2011, 51, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, Á.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.A.; Nachman, R.L.; Becker, C.G.; Minick, C.R. Culture of Human Endothelial Cells Derived from Umbilical Veins. Identification by Morphologic and Immunologic Criteria. J. Clin. Investig. 1973, 52, 2745–2756. [Google Scholar] [CrossRef] [PubMed]
- Westenbroek, R.E.; Bischoff, S.; Fu, Y.; Maier, S.K.; Catterall, W.A.; Scheuer, T. Localization of sodium channel subtypes in mouse ventricular myocytes using quantitative immunocytochemistry. J. Mol. Cell. Cardiol. 2013, 64, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, M.J.; Bond, R.A.; Spina, D.; Ahluwalia, A.; Alexander, S.P.A.; Giembycz, M.A.; Gilchrist, A.; Hoyer, D.; Insel, P.A.; Izzo, A.A.; et al. Experimental design and analysis and their reporting: New guidance for publication in BJP. Br. J. Pharmacol. 2015, 172, 3461–3471. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; van Esch, B.C.A.M.; Henricks, P.A.J.; Garssen, J.; Folkerts, G. IL-33 Is Involved in the Anti-Inflammatory Effects of Butyrate and Propionate on TNFα-Activated Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 2447. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052447
Li M, van Esch BCAM, Henricks PAJ, Garssen J, Folkerts G. IL-33 Is Involved in the Anti-Inflammatory Effects of Butyrate and Propionate on TNFα-Activated Endothelial Cells. International Journal of Molecular Sciences. 2021; 22(5):2447. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052447
Chicago/Turabian StyleLi, Meng, Betty C. A. M. van Esch, Paul A. J. Henricks, Johan Garssen, and Gert Folkerts. 2021. "IL-33 Is Involved in the Anti-Inflammatory Effects of Butyrate and Propionate on TNFα-Activated Endothelial Cells" International Journal of Molecular Sciences 22, no. 5: 2447. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052447