1. Introduction
Pro-inflammatory cytokines contribute significantly to the initiation and progression of osteoarthritis (OA) via up-regulation of catabolic processes [
1,
2]. In addition, the impairment of the mitochondrial function of chondrocytes is thought to be an important factor in the pathophysiology of OA [
3,
4,
5]. Experiments showed that Interleukin-1β (IL-1β) can impair the activity of mitochondrial respiratory chain enzyme complexes [
6]. However, the mechanism by which IL-1β modulates mitochondrial respiration remains unclear. IL-1β induces upregulation of inducible NO synthase (iNOS) and the production of nitric oxide (NO). This mediator regulates numerous putative pathogenic processes in the cartilage (see below), and may also alter mitochondrial respiration and ATP production in chondrocytes [
3]. However, whether IL-1β-induced NO production is causally responsible for mitochondrial impairment and whether inhibition of iNOS can prevent IL-1β-induced mitochondrial dysfunction has not yet been reported.
An increased iNOS expression was found in OA cartilage and synovial tissue [
7]. Although NO produced by constitutive NO synthase at low concentration can reduce OA pain probably through the promotion of blood flow, thus improving oxygen supply and reducing ischemic pain [
8], NO produced by the cytoplasmic iNOS at high concentration is a pro-inflammatory factor and contributes to OA pathogenesis [
9], and can induce cell damage. In chondrocytes, NO increases the release of pro-inflammatory mediators and inhibits the synthesis of cartilage matrix components and increases the activity of matrix-degrading enzymes such as matrix metalloproteinases (MMP) [
10]. Exogenous cytokines can increase iNOS expression and NO release of OA cartilage and cultured chondrocytes [
11,
12]. In addition, NO at high concentration affects cytochrome c oxidase in mitochondria, induces caspase 3, and is presumably responsible for the initiation of apoptosis [
13]. It has been reported that mitochondrial dysfunction also increases the responsiveness of chondrocytes for cytokines [
14]. An interesting question is, therefore, whether NO-induced mitochondrial dysfunction also increases the IL-1β-induced release of pro-inflammatory mediators.
The expression and activity of iNOS are regulated by various signaling pathways such as cyclic adenosine monophosphate (cAMP)- or AMP-activated protein kinase (AMPK)-pathway, and the effect of activation or inhibition of these pathways differs between cell types. It has been reported that the signaling molecule cAMP can increase iNOS expression in adipocytes, smooth muscle cells, and skeletal muscle cells, whereas in hepatocytes and astrocytes cAMP suppressed iNOS expression [
15,
16]. Tissue-specific gene expression and an alteration of cell signaling pathways are thought to be responsible for these opposite effects of cAMP [
16]. Another regulator of iNOS expression is the AMPK, which is an important regulator of cellular energy metabolism. A high AMP/ATP ratio activates AMPK, which inhibits ATP-consuming pathways and increases ATP production [
17]. In many cell types, AMPK exerts anti-inflammatory effects and reduces iNOS expression [
18]. In contrast, in hepatocytes, activation of AMPK upregulates the cytokine-induced expression of iNOS and NO production [
19]. Presumably, the signaling molecule cAMP and the regulator AMPK are part of two independent pathways for the regulation of iNOS expression. Whether cAMP and AMPK modulate IL-1β-induced NO release and mitochondrial function in chondrocytes is unclear.
The current study aimed to evaluate whether increased NO production is responsible for IL-1β-induced mitochondrial dysfunction in human OA chondrocytes obtained from knee joints during arthroplasty. Of particular interest is to evaluate whether blockade of iNOS activity prevents the negative effects of IL-1β on chondrocytes. Additionally, we analyzed the role of cAMP and AMPK in the regulation of NO production and release from these cells and in IL-1β-induced mitochondrial dysfunction. To understand the regulation of iNOS activity in chondrocytes is important to move forward with the development of OA therapies based on iNOS as a target for OA treatment.
3. Discussion
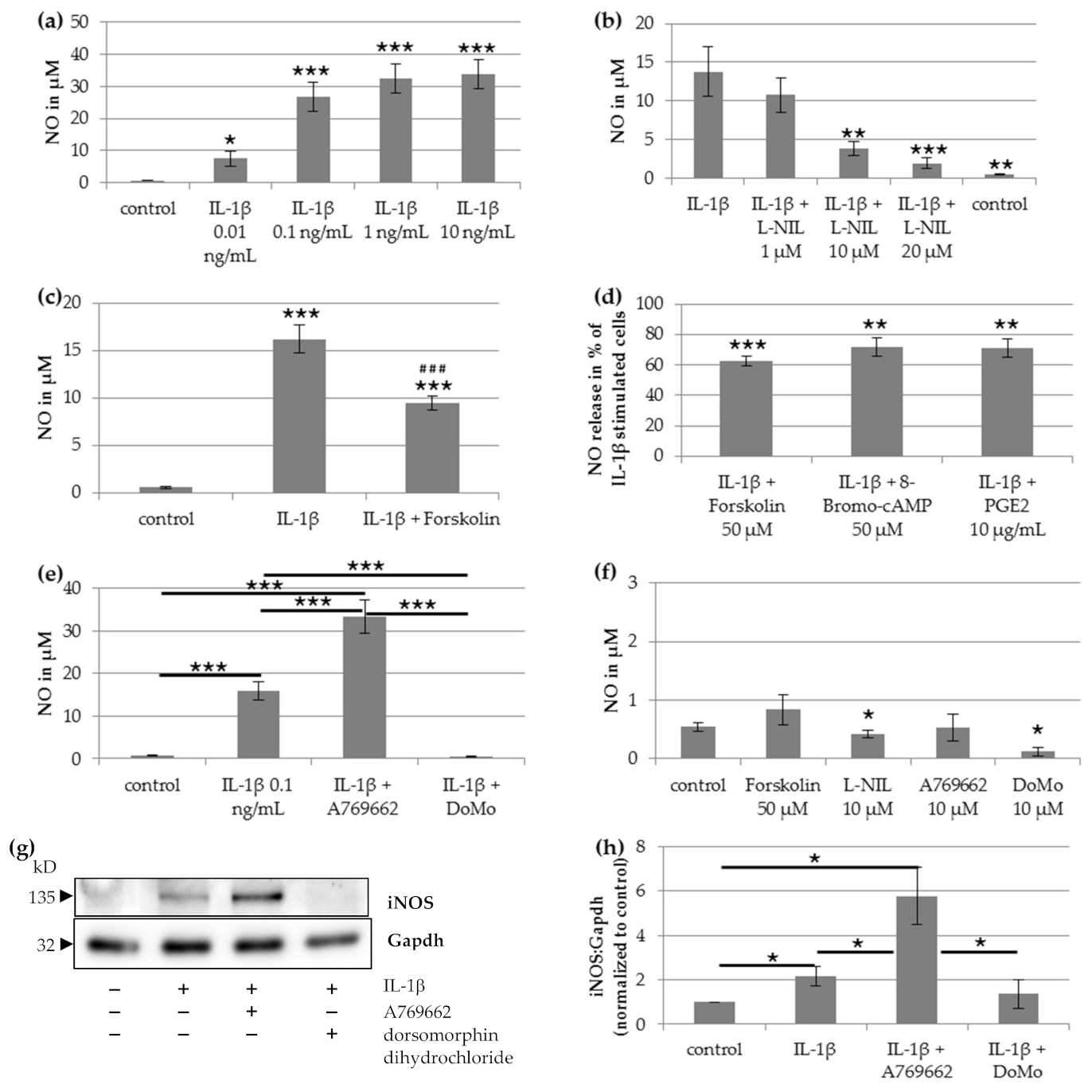
The results of the current study provide evidence that increased NO production in human chondrocytes is responsible for IL-1β-induced mitochondrial impairment. Inhibition of iNOS by L-NIL prevented the IL-1β-induced reduction of mitochondrial respiration and ATP production. The application or induction of cAMP reduced the IL-1β-induced NO release and impairment of mitochondrial function. Furthermore, the results show that AMPK is an important regulator for the IL-1β-induced NO production in chondrocytes. The activation of AMPK increased the IL-β-induced NO release and, therefore, increased the negative impact of IL-1β on mitochondrial function. The inhibition of AMPK resulted in a strong reduction of NO release and prevented IL-1β-induced impairment of mitochondrial respiration.
Previous studies showed that both IL-1β and NO impaired the activity of respiratory chain enzyme complexes and thus affect mitochondrial function in normal human chondrocytes [
6,
20]. Therefore, it was supposed that IL-1β-induced NO production may be responsible for the IL-1β-induced mitochondrial dysfunction. Our data now reveal that the effects of IL-1β and NO on mitochondrial function are causally linked. First, the effects of IL-1β on NO release, mitochondrial respiration, and ATP production were prevented by the iNOS inhibitor L-NIL. Second, IL-1β enhanced iNOS expression and NO release in chondrocytes. Third, downregulation of IL-1β-induced NO release by cAMP or the AMPK inhibitor dorsomorphin dihydrochloride was accompanied by a significant improvement of IL-1β-induced mitochondrial impairment.
NO production and iNOS expression were shown to be modulated by cAMP, which activates protein kinases and regulates gene transcription via transcription factors such as CREB. Interestingly, it seems to depend on the cell type whether cAMP increases or decreases iNOS expression and NO production. In our experiment, the stimulation of cAMP synthesis in chondrocytes decreased IL-1β-induced NO production, similar as in hepatocytes and astrocytes, but in cardiac myocytes, macrophages, and vascular smooth muscle cells, NO production was enhanced by cAMP elevation [
15,
21]. The decrease of NO production by cAMP in hepatocytes and astrocytes was explained by decreasing iNOS mRNA expression, iNOS protein expression, and alteration of iNOS promoter activity. Here, we show for the first time that stimulation of cAMP production by forskolin prevents mitochondrial dysfunction induced by IL-1β, similar to iNOS inhibition, suggesting that the increase of cAMP protects against the negative effect of IL-1β on mitochondrial function by iNOS inhibition or reduced iNOS expression. Since cAMP elevation may also be caused by inflammatory mediators such as PGE
2, this mechanism may limit the mitochondrial impairment of IL-1β in the inflammatory setting.
Activation of AMPK can also influence iNOS activity. AMPK is an important energy-sensing molecule that can switch off ATP-consuming pathways and switch on pathways for ATP production. AMPK is activated by a high AMP/ATP ratio. The effect of AMPK on iNOS activation also depends on the cell type. In myocytes, adipocytes, and macrophages, pharmacological activation of AMPK significantly inhibited iNOS under pro-inflammatory conditions, primarily resulting from post-transcriptional regulation of the iNOS protein [
22]. In hepatocytes, however, AMPK activation increased cytokine-induced iNOS expression and NO production [
19]. In human chondrocytes, we found a significant increase of IL-1β-induced NO production and iNOS expression after AMPK activation by A769662, and a marked reduction of IL-1β-induced NO production and iNOS expression after AMPK inhibition by dorsomorphin dihydrochloride, thus resembling the effects in hepatocytes. Additionally, our data show that A769662 aggravates IL-1β-induced mitochondrial impairment, whereas dorsomorphin dihydrochloride prevents IL-1β-induced mitochondrial effects. Since the effect of A769662 on NO production and mitochondrial function in chondrocytes was only observed in combination with IL-1β, the mechanism by which AMPK regulates iNOS should be related to the IL-1β pathways. In primary hepatocytes, AMPK affected cytokine-induced NO production and iNOS expression through Akt, c-Jun N-terminal kinase, and NF-kB signaling pathways [
19]. Given that the effects of AMPK in hepatocytes and chondrocytes seem to be similar, we assumed analogous regulatory pathways in chondrocytes. IL-1β reduces mitochondrial ATP production through NO production, therefore, it should activate the cytosolic AMPK. As activation of AMPK increased the IL-1β-mediated NO release in chondrocytes, a vicious circle may be produced if AMPK is not inhibited by other mechanisms. Such a mechanism could be the inhibition of phosphodiesterase (PDE), which hydrolyzes cAMP to AMP. Inhibition of AMP production by PDE inhibitors leads to reduced AMPK activity. In human OA chondrocytes, Tenor et al. found that inhibition of PDE4 decreased IL-1β-induced NO production by reducing iNOS protein expression [
23], thus resembling the effect of AMPK inhibition observed in the present study. Importantly, our data show that the modulation of NO synthesis by AMPK signaling also affects IL-1β-induced mitochondrial impairment.
In addition to mitochondrial respiration, AMPK regulates glucose uptake and ROS production, protein synthesis, promotor activity, or receptor activity. Thus the AMPK mechanisms in chondrocytes should be further investigated to evaluate the impact on OA mechanisms.
Since chondrocytes mainly utilize glycolysis for ATP production, which does not involve mitochondrial activity, the relative importance of oxidative phosphorylation for ATP supply in chondrocytes has been discussed. However, several studies reported that mitochondrial dysfunction is involved in pathophysiological processes, which include oxidative stress, apoptosis, production of inflammatory mediators, matrix catabolism, and calcification of cartilage matrix [
24]. Inhibition of mitochondrial respiratory complexes increased the expression of cyclooxygenase 2 and the level of PGE
2 in normal human chondrocytes [
25] as well as the inflammatory responsiveness to cytokines [
14]. Furthermore, mitochondrial dysfunction induced apoptosis by inducing ROS and mtDNA damage [
24]. Thus, intact mitochondrial respiration is thought to be crucial for the homeostasis and survival of chondrocytes. Since NO induces mitochondrial dysfunction, the regulation of NO production and iNOS expression is of particular interest in this context.
While we identified significant negative NO effects on mitochondrial function, our data did not provide evidence that NO is critically involved in the IL-1β-induced production and release of IL-6, PGE
2, and MMP3. Besides the fact that PGE
2, IL-6, and MMP3 are important molecules in OA processes, several studies found that mitochondrial dysfunction affects the production of these mediators [
14,
24,
25,
26]. In human chondrocytes, mitochondrial dysfunction induced by inhibitors of mitochondrial complexes increased the production of PGE
2 and MMP3 [
25,
26] and the inflammatory response to IL-1β [
14]. According to these studies, we expected a reduced release of IL-6, PGE
2, and MMP3 after incubation with L-NIL and prevention of NO-induced mitochondrial impairment. However, we found that the mediators PGE
2, IL-6, and MMP3 were significantly more released upon stimulation with IL-1β, but the IL-1β-induced release was not affected by L-NIL at concentrations that inhibited the effects of IL-1β on NO production, mitochondrial respiration, and ATP production. Thus, while IL-1β alone reduced mitochondrial function in a NO-dependent manner, this effect was not crucial for the IL-1β-induced release of IL-6, PGE
2, and MMP3 in our experiments. In the study of Vaamonde-Garcia et al., mitochondrial impairment induced by oligomycin alone already enhanced the basal release of pro-inflammatory mediators, oligomycin combined with IL-1β led to an additional increase of these mediators [
14]. Thus the mitochondrial dysfunction was directly initiated by inhibitors additionally to the IL-1β-induced mitochondrial impairment. It seems that mitochondrial dysfunction induced by inhibitors of mitochondrial respiration chain complexes results in a slightly different reaction compared to the inhibitory effect of NO on mitochondrial respiration. In addition, anti-inflammatory effects of NO in chondrocytes were described in the literature. In one study, inhibition of NO synthesis enhanced the IL-1β-induced IL-6 and PGE
2 production [
27]. Concerning the different mentioned effects of NO and inhibitors, several pathways might converge on mitochondrial respiration and result in different responsiveness to cytokines.
Some limitations of this study should be noted. We did not observe the negative effects of IL-1β on the vitality of chondrocytes upon exposure to IL-1β for two days. Thus mitochondrial dysfunction by IL-1β may not cause rapid cell death. Our study showed putative mechanisms that may protect against IL-1β-induced mitochondrial malfunction. Induction of apoptosis and cytotoxic effects may be observed after a longer stimulation period or higher IL-1β concentrations. Concerning modulation of mitochondrial function by cAMP and AMPK, most experiments were performed with a single concentration of the used mediators according to preliminary experiments and literature. The described effects of cAMP and AMPK could be stronger or weaker with other concentrations or incubation times. In general, in-vitro models presumably do not reflect exactly the in-vivo situation in OA cartilage, but chondrocytes cultured in the monolayer are the most widely used in-vitro model to study the effect of cytokines on molecular pathways of chondrocytes [
28].
In summary, our data demonstrate the importance of NO for the IL-1β-induced negative effects on the mitochondrial function in chondrocytes. It supports the concept that inhibition of iNOS could be a beneficial treatment of OA. Treatment with iNOS inhibitors showed chondroprotective effects in animal and human studies [
10] and significantly reduced OA progression in an experimental animal model [
29]. The chondroprotective effects of iNOS inhibition may result at least in part from the reduction of mitochondrial dysfunction induced by IL-1β.
4. Materials and Methods
4.1. Reagents/Solutions
Human IL-1β was purchased from PeproTech (Rocky Hill, NJ, USA). N6-(1-iminoethyl)-L-lysine hydrochloride (L-NIL), forskolin, 8-Bromoadenosine-3′,5′-cyclic monophosphate sodium salt (8-Bromo-cAMP), dorsomorphin dihydrochloride, and A769662 were all purchased from Tocris Bioscience (Bristol, UK). Forskolin and A769662 were dissolved in DMSO/water (final DMSO dilution 1:200 and 1:5000, respectively), all other substances were dissolved in water. PGE2 was from Cayman Chemical (Ann Arbor, MI, USA) and was dissolved in DMSO/water (final DMSO dilution 1:5000). Pronase E was obtained from Merck KGaA (Darmstadt, Germany), and collagenase P from Roche Diagnostics GmbH (Mannheim, Germany). The chondrocytes culture medium contains Chondrocyte Basal Medium + 10% Chondrocyte Growth Medium SupplementMix (both from PromoCell GmbH, Heidelberg, Germany) + 1% Penicillin/Streptomycin Solution (Life Technologies Europe BV, NN Bleiswijk, Netherlands).
4.2. Isolation of Human Chondrocytes
Human chondrocytes were obtained from 37 patients (16 female/21 male) with end-stage knee OA who underwent knee replacement surgery. Patients were on average 63.6 years old (±10.1 years, standard deviation). Patients were informed about the purpose of tissue sampling and gave written consent after the nature of all examinations was fully explained. The study was approved by the Ethical Committee for Clinical Trials of the Friedrich-Schiller-University of Jena (ethic approval code: 3966-12/13; date of approval: 23 January 2014) and performed in accordance with the Declaration of Helsinki.
Directly after surgical removal of the condyles, cartilage was removed from the condyles and was cut into small pieces. Cartilage was treated with 0.01 mg/mL pronase E in Dulbecco’s modified Eagles’s medium (DMEM) for 30 min at 37 °C following collagenase P (1.3 mg/mL in chondrocyte culture medium) for 16 h at 37 °C. The cells were filtrated, washed, and seeded in cell culture plates.
4.3. Experiments on Release of Mediators
For release experiments, the chondrocytes were plated on 24-well culture plates at a density of 4 × 105 cells/cm2 and cultured in the chondrocyte culture medium. After 3 days of incubation, the medium was renewed. After an additional 2 days, the cells were stimulated with IL-1β (10–0.01 ng/mL) and the iNOS inhibitor L-NIL (1, 10, and 20 µM) at different concentrations for 48 h to determine the lowest efficient concentration. After preliminary experiments with different concentrations of all mediators in combination with IL-1β, we stimulated the chondrocytes in the final experiments with 50 µM forskolin, 10 µg/mL PGE2, 50 µM 8-Bromo-cAMP, 10 µM L-NIL, 10 µM A769662 (an activator of AMPK), and 10 µM dorsomorphin dihydrochloride (an inhibitor of AMPK) alone or in combination with IL-1β for 48 h. The supernatant of all experiments was collected and stored at −80 °C until use. The cells were plated in duplicate for each condition. Experiments were performed with a minimum of 5 biological replicates (donors) to ensure reproducibility.
Griess assay: Concentration of nitrite in the supernatant was measured using the Griess Reagent Kit (#G7921, Invitrogen, Thermo Fisher Scientific Inc., Darmstadt, Germany,) according to manufacturer’s instruction and used as an indicator for NO synthesis of the cultured chondrocytes.
Measurements of IL-6, PGE2, and MMP-3: Concentrations of IL-6, PGE2, and MMP-3 in the supernatant were measured by ELISA using IL-6 human uncoated ELISA Kit (#88-7066-22, Invitrogen), Prostaglandin E2 ELISA (DRG Instruments, Marburg, Germany, #EIA-5811), and RayBio human MMP-3 ELISA Kit (RayBiotech Inc., Norcross, GA, USA, #ELH-MMP3-5).
Measurement of GAG: The amount of glycosaminoglycan (GAG) was measured spectrophotometrically using 1,9-dimethylmethylene blue (DMB, Sigma-Aldrich, Taufkirchen, Germany). A standard curve of bovine chondroitin sulfate (Sigma-Aldrich) was generated to calculate the GAG concentration.
4.4. Vitality of Chondrocytes
Chondrocytes were plated on 96-well culture plates at a density of 4 × 105 cells/cm2. The cells were cultured and stimulated as described above. For testing the impact of mediators used on the viability of chondrocytes, the LIVE/DEAD Viability/Cytotoxicity Kit from Invitrogen (#L3224) was performed according to the manufacturer’s instruction. The assay determined the percentage of living and dead cells after stimulation with different mediators.
4.5. Measurement of Mitochondrial Function
Functional parameters of mitochondrial respiration were measured with the Seahorse XF Cell Mito Stress Test Kit using the Seahorse XF Analyzer (Agilent, Santa Clara, CA, USA). The assay included modulators of mitochondrial respiration to measure basal respiration, ATP-linked respiration, maximal respiration, and non-mitochondrial respiration. These functional parameters were calculated by measuring the oxygen consumption rate (OCR) of the cultured chondrocytes using the data analysis tool Seahorse XF Report Generator (Agilent). Oligomycin, carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), and antimycin A/rotenone were sequentially injected to modulate the mitochondrial respiration.
For this purpose, chondrocytes were plated on Seahorse cell culture plates at a density of 3 × 104 cells/well and cultured in chondrocytes culture medium for 6 days at 37 °C. Thereafter, cells were stimulated with 0.1 ng/mL IL-1β, 50 µM forskolin, 10 µM L-NIL, 10 µM A769662, and 10 µM dorsomorphin dihydrochloride for 24 h. The cells were plated with 8 repetitions for each combination of stimulation. Experiments were performed with a minimum of 5 biological replicates (donors) to ensure reproducibility.
4.6. Western Blot
Isolated chondrocytes were plated on 12-well culture plates at a density of 4 × 105 cells/cm2. The cells were cultured and stimulated as described above. Chondrocytes were lysed on ice using RIPA lysis buffer (catalog #9806, Cell Signaling, Danvers; MA, USA) freshly supplemented with protease inhibitor cocktail tables (Roche, Mannheim, Germany), transferred in Eppendorf tubes, and frozen at −80 °C. After a freeze-thaw cycle, cell lysates were centrifuged at 15,000× g for 10 min to remove cellular debris. Protein extracts were mixed with Laemmli-buffer and separated on 10% polyacrylamide gels at 125 V and transferred to a polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA).
Immunoblotting was performed with antibodies against iNOS (Invitrogen, catalog #PA1-036) and Gapdh (Sigma Aldrich, catalog #G8795) at 4 °C overnight following incubation with HRP-linked secondary antibodies (KPL, Gaithersburg, MD, USA). Protein signals were visualized with a chemiluminescence reaction reagent (catalog #34075, Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s instruction using a CCD camera system (Synoptics, Cambridge, UK). Densitometry of Western blot images was performed using NIH Image J software (available under:
https://imagej.nih.gov/ij/, accessed on 1 January 2021).
4.7. Statistical Analysis
For statistical analyses, the software SPSS statistics 21 (SPSS, Inc, Chicago, IL, USA) was used. Results were expressed as means ± SEM or percent of control. For multiple sample comparison repeated-measures one-way analysis of variance or Friedman test were performed followed by a paired Student’s test or Wilcoxon signed-rank test, when appropriate. When required, Bonferroni adjustment was performed for multiple comparisons. Significance was accepted at p < 0.05.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




