
RNA-Binding Proteins and the Complex Pathophysiology of ALS


1
Division of Cosmetic Science and Technology, Daegu Haany University, Hanuidae-ro 1, Gyeongsan, Gyeongbuk 38610, Korea
2
Department of Pharmacology, School of Dentistry, Kyungpook National University, Daegu 41940, Korea
*
Authors to whom correspondence should be addressed.
†
Current Address: Department of Biochemistry, College of Medicine, Gyeongsang National University, Jinju 52828, Korea.
Int. J. Mol. Sci. 2021, 22(5), 2598; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052598
Submission received: 27 January 2021
/
Revised: 26 February 2021
/
Accepted: 1 March 2021
/
Published: 5 March 2021
(This article belongs to the Special Issue Proteins in Human Diseases: Molecular Insights into Drug Targets and Novel Therapeutic Modalities)
Abstract
:Genetic analyses of patients with amyotrophic lateral sclerosis (ALS) have identified disease-causing mutations and accelerated the unveiling of complex molecular pathogenic mechanisms, which may be important for understanding the disease and developing therapeutic strategies. Many disease-related genes encode RNA-binding proteins, and most of the disease-causing RNA or proteins encoded by these genes form aggregates and disrupt cellular function related to RNA metabolism. Disease-related RNA or proteins interact or sequester other RNA-binding proteins. Eventually, many disease-causing mutations lead to the dysregulation of nucleocytoplasmic shuttling, the dysfunction of stress granules, and the altered dynamic function of the nucleolus as well as other membrane-less organelles. As RNA-binding proteins are usually components of several RNA-binding protein complexes that have other roles, the dysregulation of RNA-binding proteins tends to cause diverse forms of cellular dysfunction. Therefore, understanding the role of RNA-binding proteins will help elucidate the complex pathophysiology of ALS. Here, we summarize the current knowledge regarding the function of disease-associated RNA-binding proteins and their role in the dysfunction of membrane-less organelles.
1. RNA-Binding Proteins and ALS
In 1939, the major league baseball player Lou Gehrig was diagnosed with amyotrophic lateral sclerosis (ALS), which is often called Lou Gehrig’s disease. The first descriptions of the disease in 1869 connected the symptoms and underlying neurological problems, and Charcot introduced the term ALS in 1874 [1]. ALS is a progressive neurodegenerative disease that affects nerve cells in the brain and spinal cord [2]. The progressive degeneration of motor neurons in ALS eventually leads to their death. Indeed, ALS is associated with complex pathophysiology including neuron loss, muscle wasting, excitotoxicity, microglial activation, peripheral inflammation, oxidative stress, mitochondrial dysfunction, endoplasmic reticulum stress, and synaptic remodeling [3,4,5,6,7,8,9,10,11]. There is no cure for ALS and disease progression is generally rapid. Eventually, after substantial respiratory and nutritional failure, death occurs in 70–80% of diagnosed individuals within 5 years [12]. The average length of survival from ALS onset to death is 2–4 years, and only 10% of ALS patients survive for more than 10 years [13]. To establish therapeutic strategies for treating ALS, the identification of disease-causing molecules and the unraveling of pathogenic pathways are required. Many putative exogenous factors have been investigated, including exposure to pesticides, viruses, cyanobacterial toxins, magnetic fields, heavy metals, a history of medical conditions, and lifestyle choices [12,14]. However, a definite environmental risk factor has not been clearly identified so far. Since the first identification of superoxide dismutase 1 (SOD1) as an ALS causative gene in 1993 [7], significant research efforts and advanced genetic approaches have identified mutations in more than 30 genes that cause ALS and frontotemporal dementia (FTD) [15,16,17,18]. In addition, 147 different gene mutations found in many different pathways have been shown to contribute to the pathogenesis of ALS [19,20]. Although sporadic ALS cases are much more common than familial ALS cases, a portion can be explained by ALS-causing mutations in genes. Indeed, some cases of ALS previously assumed to be sporadic have been subsequently identified as being caused by pathogenic mutations in ALS genes [17,21,22]. Interestingly, most ALS-causing mutation-harboring genes are closely related to RNA metabolism and form aggregates. Mutations in a series of RNA-binding protein genes, TAR DNA binding protein (TARDBP), FUS RNA-binding protein (FUS), TATA-box binding protein associated factor 15 (TAF15), EWS RNA-binding protein 1 (EWSR1), heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1), HNRNPA2B1, Ataxin 2 (ATXN2), and TIA1 cytotoxic granule associated RNA-binding protein (TIA1), have been shown to cause or influence the disease risk for ALS and/or FTD [23,24,25,26,27,28,29,30,31]. ALS causative genes such as FUS and TARDBP encode RNA-binding proteins and lead to disrupted RNA metabolism [18]. ALS-causative mutations in FUS or TARDBP show abnormal stress granule formation with defects in translation, the formation of pathogenic RNA foci, the dysregulation of nucleocytoplasmic shuttling, as well as other forms of disrupted RNA metabolism [32,33]. The most common ALS- and FTD-causing gene mutation is a GGGGCC hexanucleotide repeat expansion in the first intron region of the chromosome 9 open reading frame 72 (C9orf72) gene [34,35]. ALS and FTD patients display several hundred to a few thousand copies of the GGGGCC repeat in the C9orf72 gene [34], and the C9orf72 repeat expansion’s toxicity increases with age, repeat length, and expression level [19,36]. Repeat-containing transcripts of the mutated C9orf72 gene can form RNA foci enriched with RNA-binding proteins in induced pluripotent stem cell (iPSC)-derived neurons from ALS and FTD patients, as well as in motor neurons of C9orf72-ALS patients [37,38,39,40]. Even though conflicting evidence remains about the mechanisms of toxicity of C9orf72 mutations [19,36,41], RNA foci are believed to sequestrate bound RNA-binding proteins and result in toxicity [42]. Dipeptide repeat (DPR), glycine-alanine (GA), glycine-arginine (GR), proline-arginine (PR), proline-alanine (PA), and glycine-proline (GP) are generated through repeat-associated non-ATG translation from hexanucleotide repeat expansion-containing transcripts [43,44,45,46]. Among the proposed disease-causing mechanisms of C9orf72 mutations, the production and accumulation of DPR is considered to be one of the main contributors to its pathogenesis. Studies demonstrating the mechanisms of toxicity of DPRs in vitro and in Drosophila revealed that poly-GR and poly-PR DPRs interact with RNA-binding proteins or proteins that contain a low complexity sequence domain (LCD), also called the prion-like domain [47,48]. LCD is the key component that induces liquid-liquid phase separation and thereby mediates the formation of membrane-less organelles such as stress granules [23,47]. Indeed, poly-GR and poly-PR DPRs are most clearly associated with the nucleolus, a membrane-less organelle. Interestingly, the expression of poly-GR and poly-PR DPRs impairs the dynamics and function of interacting RNA-binding proteins that associate with numerous membrane-less organelles, including the nucleolus, stress granules, nuclear speckles, and Cajal bodies [47,49]. RNA-binding proteins have diverse functions; they can be involved in the post-transcriptional regulation of target genes and thus regulate their translation, mRNA degradation, and mRNA shuttling. Indeed, many RNA-binding proteins have liquid-liquid phase separation ability to form membrane-less organelles. The dysregulation of liquid-liquid phase separation is closely associated with several diseases such as dementia, ALS, and cancer [50,51,52,53].
Here, we focus on the cellular and functional features of ALS/FTD-related RNA-binding proteins, and discuss the complex pathophysiology of ALS by highlighting the role of RNA-binding proteins (Figure 1).
2. RNA-Binding Proteins and Membrane-Less Organelles
2.1. RNA-Binding Proteins and Disrupted Nucleocytoplasmic Shuttling
A defining characteristic feature of ALS and FTD is the loss of specific RNA-binding proteins from the nucleus and their mislocalization into cytoplasmic aggregates [54,55]. Emerging evidence suggests that impaired nucleocytoplasmic trafficking by the dysregulation of RNA-binding proteins is one of the key mechanisms causing ALS [56,57]. The cytoplasmic mislocalization and aggregation of the RNA-binding protein TARDBP is a common histopathological hallmark of ALS/FTD [58]. TARDBP pathology is present in about 97% of patients with ALS and up to 45% of patients with FTD [54,59]. Motor neurons in ALS show elevated poly(ADP-ribose) polymerase (PARP) activity and accelerated TARDBP aggregation in mammalian cells associated with neuronal death [60]. Aggregated and disease-causing mutations harboring TARDBP trigger the sequestration and disturbed localization of nucleocytoplasmic components such as nucleoporins and transport factors, as well as the disruption of nucleocytoplasmic shuttling [61]. Other nucleocytoplasmic transport factors are also recruited and sequestrated in the stress granules upon stress or treatment with ALS-implicated mutant proteins [62]. Although FUS pathology is uncommon and found in less than 1% of ALS and up to 9% of FTD cases [54,59], ALS-causing mutations in the nuclear localization signals of FUS also impair nucleocytoplasmic transport [57,63,64]. Although wild-type FUS normally localizes primarily in the nucleus, FUS has shown cytoplasmic mislocalization in ALS [65]. ALS-associated mutations in FUS impede its nuclear localization, and the increase in the altered localization pattern of mutant FUS in the cytoplasm correlates with rapid disease progression and a lower age of disease onset [16,66]. It has been proposed that the translocation of the transcript of splicing factor proline and glutamine-rich (SFPQ) accelerates the nuclear export of FUS, which can directly interact with SFPQ mRNA [65]. Mislocalized FUS mutants cause the mislocalization of snRNAs to the cytoplasm, which in turn causes a change in the behavior of the alternative splicing machinery [67]. ALS-associated FUS mutants that show the mislocalization and formation of cytoplasmic aggregates inhibit the splicing of minor introns by the sequestration of U11 and U12 small nuclear ribonucleoprotein (snRNP) [68]. Studies on TARDBP and FUS mislocalization show that either of these can affect the entire nucleo-cytoplasmic transport capability of the cells [19,69]. Several studies have indicated that the physical interaction between repeat RNA and RNA-binding proteins might also lead to the functional sequestration of RNA-binding proteins such as HNRNPA3, which plays a role in the cytoplasmic trafficking of RNA [70,71]. Several studies have reported a direct connection between C9orf72 hexanucleotide repeat mutations and defects in nucleocytoplasmic shuttling [56,72]. These studies identified the co-aggregation of components of nucleocytoplasmic shuttling machinery with pathological protein aggregates or RNA foci. C9orf72 hexanucleotide repeat expansion has been shown to cause nuclear import deficits in Drosophila and iPSC neurons, including an abnormal cytoplasmic accumulation of TARDBP [54]. Several RNA nuclear export factors, including ALYREF and GLE1, have been identified as genetic modifiers in the ALS-associated C9orf72 model system [56]. The ALS-associated RNA-binding protein Matrin-3 (MATR3) was also found to colocalize with GGGGCC RNA foci in patient tissues as well as iPSC-derived motor neurons harboring the C9orf72 mutation. Heaxanucleotide repeat expansion of C9orf72 sequestered and perturbed the subcellular distribution of MATR3 in C9orf72-ALS patient-derived motor neurons [73].
C9orf72-derived DPR poly-GR and poly-PR have been shown to interact with several mediators of nucleocytoplasmic shuttling, including IPO5, IPO7, KPNA2, KPNB1, NUP205, XPO1, and TNPO1, as well as with RNA-binding proteins consisting of nuclear pore complexes, such as PARP1, YBX1, and LBR [47]. C9orf72-patient-iPSC-derived motor neurons have shown the mislocalization of U2 snRNP in the cytoplasm, and poly-GR as well as poly-PR are both associated with U2 snRNP [74]. The DPR-mediated dysfunction of U2 snRNP might explain mis-splicing events in patients with ALS/FTD [74]. Poly-GA also causes the mislocalization of HNRNPA3 by sequestration and leads to DNA damage [75]. The nuclear depletion of HNRNPA3 in fibroblasts derived from patients harboring C9orf72 hexanucleotide repeats leads to an accumulation of pathological dipeptide repeat protein-containing inclusions [76]. ALS-causing mutations in the RNA-binding proteins HNRNPA1 and HNRNPA2B1, especially in the prion-like domains of genes, promote cytoplasmic inclusion formation [26,77]. Several mutations in Profilin1 (PFN1), an actin-binding protein involved in actin cytoskeleton dynamics, have been identified in ALS [78,79], and mutant PFN1 has been shown to alter the distribution of RNA-binding proteins such as TARDBP, FUS, FMRP, and SMN, and disrupt the post-transcriptional regulation of their target mRNAs [80]. The modulation of actin homeostasis rescues nuclear pore instability and the dysfunction of RNA-binding proteins mediated by C9orf72 repeat expansion or mutant PFN1 [80].
2.2. RNA-Binding Protein and Stress Granule Formation
Stress granules are a specific type of RNA granule and are composed of RNA and RNA-binding proteins, including repressed translational complexes. Stress granules are cytoplasmic members of the RNA granule family and are formed during cellular stress, particularly in neurodegenerative diseases and myopathies, resulting in dynamic membrane-less compartments [81]. Defects in both stress granule assembly and disassembly have been linked to neurodegenerative diseases [19,81,82]. Stress granules function as an RNA quality control system, stabilizing and editing mRNA, and arresting translation to prevent the accumulation of aberrant proteins [16,81]. Stress granules allow cells to cope with stress by stalling mRNA translation and moving synthesis towards cytoprotective proteins [23]. However, in disease-related stress conditions, stress granules can become persistent structures and act as a seed for the accumulation of RNA-binding proteins [81]. Stress granules have become one of the pathogenic phenotypes of ALS and FTD, and most ALS-causing mutations harboring RNA-binding proteins have a tendency to increase liquid-liquid phase separation and stress granule formation [23]. TARDBP in ALS has been reported to have a tendency to undergo liquid-liquid phase separation in vivo [33]; it forms amyloid-like aggregates in vitro and shows pathological oligomerization in vivo [33]. Indeed, in ALS and FTD, subsets of TARDBP-containing cytoplasmic inclusions were frequently positive for stress granule markers such as TIA-1, PABP, and eukaryotic initiation factor 3 (IF3) [83]. Although wild-type TARDBP localizes to stress granules under cellular stress conditions [84], the pathological RNA-binding protein TARDBP shows aggregation and accumulation in the spinal cords of patients with ALS [58] and leads to the formation of abnormal stress granules both in vitro and in vivo [16,85]. Conversely, mutations in TIA-1 [28] or ATXN2 [25], which are crucial components of stress granules [86,87], have also been identified as either causing ALS or increasing its risk, and ATXN2 altered the subcellular distribution and toxicity of TARDBP with enhanced mutant FUS toxicity [16,25,88]. Several ALS-related mutations in TIA-1 have been identified [28,89,90], and disease-causing mutations delayed stress granule disassembly and promoted the accumulation of TDP-43 positive stress granules [28,91]. Mutations of FUS and HNRNPA2B1 [26] have also led to the formation of aberrant stress granules. Wild-type FUS normally shows no re-localization from the nucleus to stress granules, but FUS mutation causes FUS to colocalize with stress granules that are larger and more numerous than wild-type FUS-mediated stress granules [92,93]. ALS-associated mutations in HNRNPA1, especially the Gly-rich low-complexity domain of HNRNPA1, have shown increased incorporation into stress granules [26,94]. Hexanucleotide expansions in the C9orf72 gene impair the formation, dynamics, and function of stress granules [47,95], which might originate from the aberrant deposition of RNA-binding protein stress granule components, including phosphorylated TARDBP [96]. Poly-GR and poly-PR DPRs that arise from the C9orf72 mutation can cause reduced stress granule internal dynamics, with HNRNPA1 and TIA1 phase separation [47] leading to translation repression [47,97]. Poly-GR and poly-PR DPR interact or colocalize with most of the stress granule component proteins, including G3BP1, TIA1, PABPC1, PTBP1, TARDBP, SFPQ, YBX1, HNRNPUL1, HNRNPA1, HNRNPA2B1, FXR1, LBR, SYNCRIP, HNRNPH1, and other RNA-binding proteins [47,98,99,100,101,102,103]. Mutations in PFN1 cause not only defects in nucleocytoplasmic shuttling but also impairments in stress granule formation or clearance [80,104]. SOD1 ALS mutations cause G3BP1-positive SOD1 aggregation in spinal cord motor neurons [92,105]. Mutant SOD1 is preferentially recruited to G3BP1 and TIA1-related protein (TIAR)-positive stress granules, and aberrant mutant SOD1-G3BP1 interaction delays stress granule assembly dynamics without direct binding to RNA according to the stress response [105,106,107]. Stress granule-recruited SOD1 induces changes in alternative splicing similar to mutations in FUS that cause ALS [108].
2.3. RNA-Binding Protein with Dysfunction of the Nucleolus and Cajal Bodies
The nucleolus is the largest nonmembrane-bound nuclear compartment and an important region for rRNA synthesis and the assembly of the eukaryotic ribosome [109]. Although the nucleolus is packed with a high density of protein and RNA, it is a dynamic structure with highly mobile constituents that can diffuse in and out of the nucleoplasm [110]. The role of liquid-liquid phase separation information of the nucleoli is increasingly recognized [47,110,111]. The nucleolus is one of the cellular stress sensors, and nucleolar stress, which disrupts nucleolar integrity by the dislocation of nucleolar proteins or abnormalities in the synthesis and processing of rRNA, is closely related to neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and ALS with DNA damage [109,110,112,113,114]. The disruption of the nucleolus or stress leads to cell cycle arrest, apoptosis, differentiation, or senescence [110]. Cellular analysis in different patients with ALS-FTD has shown altered nucleolar morphology and function, including swollen nucleoli and mislocalized nucleoproteins [109]. In the context of ALS-FTD, poly-GR and poly-PR DPRs generated from C9orf72 mutations preferentially accumulate in the nucleoli and disrupt the transport of ribosomal proteins, rRNA processing, and ribosome assembly, leading to cell death [115,116,117]. These dipeptides, poly-GR, and poly-PR colocalize and interact with nucleolar proteins such as nucleolin (NCL), nucleophosmin 1 (NPM1), and fibrillarin (FBL) [47,109,118,119]. NPM1 translocates to the nucleoplasm depending on poly-GR and poly-PR and C9orf72 transcripts [118,120], and NPM1 could act as an integrator of stress signals and determine cell fate according to diverse stresses [109]. NPM1 nucleoplasmic translocation and nucleolar stress induced by poly-GR or poly-PR expression have been shown to impair the dynamics of the nucleolus and decrease rRNA levels with translation efficiencies [47,109,118]. In particular, poly PR disperses and sequesters NPM1 from nucleoli and dissolved droplets. Poly-PR also entraps rRNA in static condensates [115]. Another nucleolar protein, NCL, is sequestered by GGGGCC repeated RNA originating from the C9orf72 mutation and shows an altered distribution pattern, eventually inducing nucleolar stress [121,122,123]. Several studies have indicated that the physical interaction between repeat RNA and RNA-binding proteins might lead to the functional sequestration of RNA-binding proteins such as NCL, ADARB2, HNRNPA3, and HNRNPH [38]. In addition to the RNA-mediated sequestration of NCL, poly-GR and poly-PR from ALS-causing mutations of C9orf72 also interact with NCL RNA-binding protein and disrupt its molecular dynamics in the nucleolus [47].
The nuclear bodies termed Cajal bodies are regions within the nucleus that are enriched in proteins and RNAs involved in mRNA processing. They are the main sites for the assembly of snRNPs. These Cajal bodies show frequent close proximity to the nucleolus but are structurally distinct from the nucleolus, although it can be difficult to clearly delimit the border between the two structures [124].
Toxic DPR poly-GR and poly-PR interact with Cajal body components such as RNA-binding proteins SRRM2 and XPO, which do not bind mRNA directly; however, other adaptor proteins act as a bridge to the interaction between XPO and mRNAs [47]. In live cells, the expression of poly-GR and poly-PR DPRs has been shown to reduce the mobile fractions of proteins that are associated with membrane-less organelles, including Cajal bodies [47,49]. DPRs, especially poly-GR, poly-PR, and poly-GA, have been associated with a strong change in the number of Cajal bodies [69]. ALS-linked FUS mutants show a decreased number of Cajal bodies [69], and FUS mutations induce the cytoplasmic accumulation of snRNAs and associated Sm proteins, leading to a decrease in the nuclear distribution of snRNPs in Cajal bodies [68,69]. The activation of the integrated stress response induces a profound reorganization of Cajal bodies, which is associated with the cytoplasmic assembly of stress granules and disturbance in the nuclear import of U-rich snRNPs [69]. Taken together, ALS-causing mutations and stress might disturb the nucleocytoplasmic shuttling of spliceosome components of snRNP and lead to abnormalities in RNA modification and ALS pathogenesis, with the disruption of telomerase biogenesis [125].
2.4. RNA-Binding Protein-Associated Dysregulation of Nuclear Speckle and Paraspeckle
Nuclear speckles, also known as interchromatin granule clusters, are membrane-less nuclear compartments enriched in pre-mRNA splicing and transcription factors located in the interchromatin regions of the nucleoplasm of cells [126]. Nuclear speckles are sites for splicing factor storage and modification with transcription, mRNA maturation, and export [127,128,129,130,131]. Alteration of the function of the composition of nuclear speckles leads to changes in alternative pre-mRNA splicing events as well as the disruption of diverse nuclear gene expression regulations [127]. Indeed, ALS and FTD are associated with a general disruption of nuclear structures and the disruption of nuclear speckles [127].
The self-organizing structure of nuclear speckles has dynamic liquid-like properties [47,127]. DPRs, especially poly-GR and poly-PR generated from C9orf72 mutations, have also been shown to accumulate in nuclear speckles and disrupt their dynamics [47,97]. Poly-GR and poly-PR DPRs interact with splicing factor and the nuclear speckle component SRSF7, and significantly impact the dynamics of SRSF7 in nuclear speckles [47]. The alteration of liquid-like properties in nuclear speckles by DPRs induces specific splicing alterations in cells [132]. SRSF2, which interacts with TARDBP, FUS, and the hexanucleotide repeat expansion in C9orf72, has been shown to co-localize with C9orf72 antisense RNA foci in C9orf72 ALS patients [38,133]. SRSF2 also displays a nuclear punctate staining pattern with strong and larger speckles [133]. TARDBP protein has been shown to serve as a cellular scaffold for multiple nuclear bodies, including Cajal bodies and nuclear speckles [127,134]. In ALS, TARDBP forms intracellular inclusions that are mostly localized within the cytoplasm, leading to significant disruption of the nuclear structure and the function of nuclear compartments including nuclear speckles [127,134,135]. The dysregulation of TARDBP levels also impairs splicing events and especially disturbs the splicing of disease-associated transcripts [136]. The depletion of TARDBP decreases the expression of the nuclear speckle-specific noncoding RNA, MALAT1 [137]. FUS is also found in nuclear speckles, and so mutations in FUS are expected to disrupt the structure and function of nuclear speckles [127].
Paraspeckles are mammalian-specific nuclear bodies that contain a long non-coding RNA, NEAT1, which acts as a seed for recruiting RNA-binding proteins and building a paraspeckle as well as more than 60 paraspeckle proteins [138,139]. Paraspeckles are involved in various aspects of the regulation of gene expression, and their role in many pathologies has been addressed [140]. A number of paraspeckle-enriched RNA-binding proteins, including SFPQ, FUS, EWSR1, TAF15, TARDBP, SS18L1, and HNRNPA1, are mutated in familial cases of ALS as well as other neurodegenerative diseases [17,24,141,142,143,144]. In spinal motor neurons during the early phase of ALS, TARDBP and FUS are enriched in paraspeckles and bound to NEAT1 RNA [145]. Spinal neurons and glial cells in both sporadic and familial ALS with TARDBP pathology show enhanced paraspeckle formation. The loss and mutation of TARDBP accelerates paraspeckle assembly [146]. ALS-associated mutations in TARDBP diminished NEAT1 non-coding RNA-mediated TARDBP liquid-liquid phase separation and resulted in a specific defect in the nuclear body and paraspeckle [147]. FUS is required for paraspeckle assembly [148], and ALS-associated mutations in FUS, especially in the low complexity domain, lead to the redistribution of FUS to the cytoplasm [149]. Mutation-harboring FUS also enhances dysfunctional paraspeckle formation through the accumulation of NEAT1 non-coding RNA in cells [146,149]. GGGGCC repeat RNA-mediated RNA foci from C9orf72 mutations are predominantly associated with paraspeckle proteins SFPQ, NONO, RBM14, FUS, and HNRNPH in cells and brain tissue in FTD [150]. ALS-associated poly-GR and poly-PR DPR interact with paraspeckle components such as HNRNPF, RNRPH1, HNRNPM, SFPQ, NONO, FAM98, RBM14, and MATR3 as well as NEAT1 non-coding RNA [138,151]. Poly-PR DPR upregulates NEAT1 expression and suppresses the function of HNRNPF and HNRNPH1, leading to increased paraspeckle formation and neuronal toxicity [138].
3. Conclusions and Future Directions
Although a limitation of this study includes the fact that we were not able to include all identified studies in the manuscript due to space restrictions, we have reviewed RNA-binding proteins in the context of ALS and highlighted how they are involved in many diverse membrane-less organelles. Most components of membrane-less organelles and ALS-related genes also have common features. As mentioned in this paper, many RNA-binding proteins have been directly or indirectly implicated in ALS and FTD.
Several membrane-less organelles have distinct function, composition, and localization but appear to be closely connected. It has been shown that the activation of the integrated stress response induces the accumulation of several factors into stress granules that are crucial mediators of nucleocytoplasmic transport, including import and export factors such as importin-β1, -β2, -α1, and Exportin-1, as well as nucleoporins, the core subunits of the nuclear pore complex [62,69]. Consequently, the induction of stress granule formation has a tendency to lead to the accumulation of nucleocytoplasmic shutting-related factors, which might lead to defects in nucleocytoplasmic trafficking.
With regard to the components of membrane-less organelles, one example, TARDBP, is one of the protein components of paraspeckles but is also found in stress granules, Cajal bodies, and in the nucleoplasm. Therefore, many RNA-binding proteins are components of several membrane-less organelles simultaneously. The interactome of ALS-associated genes shares somewhat similar RNA-binding proteins. Each interactome of the hnRNP family or key pathological genes is associated with ALS, including TARDBP, C9orf72, and FUS [151]. In the case of FUS, several hnRNPs, including HNRNPA1, R, and SYNCRIP, have been shown to co-accumulate in FUS-positive pathological inclusions [151,152]. Many other hnRNPs, including HNRNPD, L, and I, have also been found within FUS-negative pathological inclusions [151]. RNA foci derived from C9orf72 hexanucleotide repeat expansion have been associated with HNRNPH1, HNRNPH3, HNRNPF, HNRNPA1, HNRNPA3, and other nuclear speckle related RNA-binding proteins [151,153]. The poly-GR and poly-PR interactome also somewhat similarly overlaps with other interactomes of ALS-related proteins. Additionally, RNA-binding proteins implicated in ALS might act co-operatively. Cytoplasmic inclusions of TARDBP increase HNRNPA1B protein levels, which harbors an elongated prion-like domain by the alteration of HNRNPA1 pre-mRNA splicing and eventually leads to HNRNPA1B cytoplasmic accumulation [154]. We have summarized membrane-less organelles and the corresponding RNA-binding proteins that are components or have a functional relationship in Table 1.
Given this connectivity, a precise and systematic understanding of RNA binding might be important. Despite structural heterogeneity in RNA-binding proteins, many RNA-binding proteins have common functional roles. RNA-binding proteins can form dynamic and cooperative complexes with other RNA-binding proteins to fulfill their role in membrane-less organelles and in diverse RNA regulatory steps. Therefore, it is not at all surprising that so many RNA-binding proteins have been linked directly or indirectly to the pathogenesis of ALS. The molecular pathogenesis of ALS appears to be driven by various and complex inter-connected RNA metabolic processes that cannot be solely attributed to any one dysfunctional event. The dysregulation of one RNA-binding protein might disrupt the structure or function of a specific complex of RNA-binding proteins. This might disrupt the dynamics and function of membrane-less organelles that harbor a dysfunctional complex of RNA-binding proteins. An abnormality in one RNA-binding protein will also lead to an unusual RNA-binding protein complex that harbors an abnormal RNA-binding protein; this will eventually lead to the dysfunction of other membrane-less organelles as well as the malfunction of RNA metabolism, known as the domino or snowball effect. Given the diverse functional and structural role of RNA-binding proteins, the dysregulation of specific RNA-binding proteins might lead to diverse functional abnormalities and complex pathophysiology. The complex and various pathophysiologies of ALS might be linked directly or indirectly to each other because of RNA-binding proteins in some but not all cases.
For a better understanding of the complex and linked pathophysiology of ALS, the functional role of RNA-binding proteins could be important. In terms of the functional role of RNA-binding proteins, they generally bind to miscellaneous RNA transcripts and exhibit a widespread distribution in cells. The relationship between target mRNA and RNA-binding protein should also be considered in order to understand the exact function and importance of RNA-binding proteins in ALS. Knockdown or overexpression of specific RNA-binding proteins often shows exactly the opposite effect according to target mRNAs [91,151]. For example, SYNCRIP, which functions as a transacting factor for posttranscriptional regulation, has been shown to rhythmically accelerate mRNA degradation and increase non-canonical translation of several target genes [156,157]. PTBP1 is also known to be one of the components of the pre-mRNA splicing machinery, which is a multi-component complex necessary for the splicing step [158]. In addition to splicing regulation, PTBP1 also activates mRNA degradation but increases the translation of specific target genes [159,160]. Therefore, the dysfunction of specific RNA-binding proteins can lead to reduced mRNA degradation of target mRNA, but also more rapid mRNA degradation of the other target mRNA. Translation initiation, mRNA degradation, the nucleocytoplasmic shuttling of RNA, splicing, and many other RNA metabolism statuses could differ according to the target mRNA. Therefore, it is usually difficult to define the role of specific RNA-binding proteins as being singular. To understand these complex functional roles of RNA-binding proteins, one needs to consider the context of RNA-binding proteins such as target mRNA and status-dependent RNA-binding protein complex. In addition to target RNA of RNA-binding proteins, the opposite and various functions of RNA-binding proteins are based on the context of interacting proteins. These RNA-binding proteins usually form complexes with other RNA-binding proteins and RNAs for specific roles, and various functions of RNA-binding proteins are based on the context of the interacting proteins. RNA-dependent regulation can be modulated by multiple RNA and RNA-binding protein complexes that exchange their binding partners in response to the cellular environment. RNA-binding proteins generally bind to target RNAs and recruit their interacting proteins in different ways based on the cellular context. Context-dependent interactions of RNA-binding proteins with target transcripts lead to diverse consequences, resulting in functional divergence. To elucidate the roles of the given RNA-binding protein, context-dependent approaches should be considered.
Sporadic ALS cases are still more common than familial ALS cases, although some sporadic ALS cases have been identified as being caused by pathogenic mutations. It is thought that a complex and various interaction between genetic and environmental factors might be involved in sporadic ALS. Although RNA-binding protein abnormalities cannot account for all sporadic ALS cases, RNA binding proteins such as TARDBP, FUS, TAF15, EWSR1, HNRNPA1, HNRNPA2B1, MATR3, and TIA1 are strongly linked with sporadic ALS [17,24,77,89,90,91,161,162]. Furthermore, the global dysregulation of non-coding RNAs has been reported in motor neurons from people with sporadic ALS, and the generation and regulation of non-coding RNA are mediated by RNA binding proteins including ALS-associated RNA binding proteins [163,164]. There is also a possible association between some ALS-causing environmental factors and the resulting abnormal function in RNA binding proteins. Therefore, further studies on not only the ALS-causative mutation but also the functional abnormality of the RNA binding proteins according to the environmental determinants of ALS are necessary.
In the field of ALS, the reason why there is a selective affection of particular types of neurons in ALS is one of the more perplexing conundrums. Accumulating evidence demonstrates that neuronal stimulation and signal transmission require the trafficking and local translation of mRNA for proper neural function [165,166]. The upper and lower motor neurons are the longest cells in the body, and the transport of RNA and the localized translation of target mRNA are crucial for their proper function. RNA binding proteins, including ALS-associated RNA binding proteins, are involved in RNA transport and local translation. The relationship between the unique physiology of motor neurons and the function of RNA binding proteins might be one of the reasons for the susceptibility of motor neurons in ALS. Moreover, the brain shows unusually high levels of alternative splicing, including testis and liver [167]. Since RNA binding proteins discussed in this review are directly or indirectly involved in RNA splicing, the dysregulation of RNA binding proteins could mediate the improper function of the central nervous system by disrupting the diversity of gene expression in the neuron.
This information indicates that the context-dependent complex of RNA-binding proteins may warrant further study; it may be the key to understanding the complex, linked, and entangled pathophysiology of ALS. The elucidated map of RNA-binding proteins in ALS will shed light on strategies for the development of therapeutics for ALS.
Author Contributions
Conceptualization, K.-H.L.; writing, K.-H.L. and D.-Y.K.; visualization, K.-H.L. and W.K.; editorial assistance, W.K.; funding acquisition, K.-H.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the National Research Foundation of Korea (NRF) funded by the Korea government (MSIT) (2017R1D1A1B03033241, 2018R1A5A2025272).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rowland, L.P. How Amyotrophic Lateral Sclerosis Got Its Name. Arch. Neurol. 2001, 58, 512–515. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Berg, L.H.V.D. Amyotrophic Lateral Sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef]
- Swinnen, B.; Robberecht, W. The Phenotypic Variability of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Morgan, S.; Orrell, R.W. Pathogenesis of Amyotrophic Lateral Sclerosis. Br. Med. Bull. 2016, 119, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Ebrites, D.; Vaz, A.R. Microglia Centered Pathogenesis in Als: Insights in Cell Interconnectivity. Front. Cell. Neurosci. 2014, 8, 117. [Google Scholar] [CrossRef]
- Valori, C.F.; Brambilla, L.; Martorana, F.; Rossi, D. The Multifaceted Role of Glial Cells in Amyotrophic Lateral Sclerosis. Cell. Mol. Life Sci. 2013, 71, 287–297. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.C.; Hentati, A.; Donaldson, D.H.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn Superoxide Dismutase Gene Are Associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Palomo, G.M.; Manfredi, G. Exploring New Pathways of Neurodegeneration in ALS: The Role of Mitochondria Quality Control. Brain Res. 2015, 1607, 36–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 Misplacing and Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis Pathogenesis. Front. Cell. Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadic, V.; Prell, T.; Lautenschlaeger, J.; Grosskreutz, J. The ER Mitochondria Calcium Cycle and ER Stress Response as Therapeutic Targets in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2014, 8, 147. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; Del Tredici, K. Amyotrophic Lateral Sclerosis—a Model of Corticofugal Axonal Spread. Nat. Rev. Neurol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Filippini, T.; Fiore, M.; Tesauro, M.; Malagoli, C.; Consonni, M.; Violi, F.; Arcolin, E.; Iacuzio, L.; Conti, G.O.; Cristaldi, A.; et al. Clinical and Lifestyle Factors and Risk of Amyotrophic Lateral Sclerosis: A Population-Based Case-Control Study. Int. J. Environ. Res. Public Health 2020, 17, 857. [Google Scholar] [CrossRef] [Green Version]
- Hobson, E.V.; McDermott, E.V.H.C.J. Supportive and Symptomatic Management of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 2016, 12, 526–538. [Google Scholar] [CrossRef]
- Oggiano, R.; Pisano, A.; Sabalic, A.; Farace, C.; Fenu, G.; Lintas, S.; Forte, G.; Bocca, B.; Madeddu, R. An Overview on Amyotrophic Lateral Sclerosis and Cadmium. Neurol. Sci. 2021, 42, 531–537. [Google Scholar] [CrossRef]
- Renton, E.A.; Chiò, A.; Traynor, B.J. State of Play in Amyotrophic Lateral Sclerosis Genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef]
- Ito, D.; Hatano, M.; Suzuki, N. RNA Binding Proteins and the Pathological Cascade in ALS/FTD Neurodegeneration. Sci. Transl. Med. 2017, 9, eaah5436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From Genes to Mechanism. Nat. Cell Biol. 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, Y.C.; Ng, C.S.; Xiang, P.; Liu, H.; Zhang, K.; Mohamud, Y.; Luo, H. Dysregulation of RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2020, 13, 78. [Google Scholar] [CrossRef] [PubMed]
- Dudman, J.; Qi, X. Stress Granule Dysregulation in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2020, 14, 598517. [Google Scholar] [CrossRef] [PubMed]
- Dervishi, I.; Gozutok, O.; Murnan, K.; Gautam, M.; Heller, D.; Bigio, E.; Ozdinler, P.H. Protein-Protein Interactions Reveal Key Canonical Pathways, Upstream Regulators, Interactome Domains, and Novel Targets in ALS. Sci. Rep. 2018, 8, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Gamez, J.; Corbera-Bellalta, M.; Nogales, G.; Raguer, N.; García-Arumí, E.; Badia-Canto, M.; Lladó-Carbó, E.; Álvarez-Sabín, J. Mutational Analysis of the Cu/Zn Superoxide Dismutase Gene in a Catalan ALS Population: Should All Sporadic ALS Cases Also Be Screened for SOD1? J. Neurol. Sci. 2006, 247, 21–28. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Hewitt, C.; Highley, J.R.; Brockington, A.; Milano, A.; Man, S.; Martindale, J.; Hartley, J.; Walsh, T.; Gelsthorpe, C.; et al. Clinico-Pathological Features in Amyotrophic Lateral Sclerosis with Expansions in C9ORF72. Brain 2012, 135, 751–764. [Google Scholar] [CrossRef]
- Baradaran-Heravi, Y.; Van Broeckhoven, C.; van der Zee, J. Stress Granule Mediated Protein Aggregation and Underlying Gene Defects in the FTD-ALS Spectrum. Neurobiol. Dis. 2020, 134, 104639. [Google Scholar] [CrossRef] [PubMed]
- Couthouis, J.; Hart, M.P.; Erion, R.; King, O.D.; Diaz, Z.; Nakaya, T.; Ibrahim, F.; Kim, H.-J.; Mojsilovic-Petrovic, J.; Panossian, S.; et al. Evaluating the Role of the FUS/TLS-Related Gene EWSR1 in Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2012, 21, 2899–2911. [Google Scholar] [CrossRef] [PubMed]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 Intermediate-Length Polyglutamine Expansions Are Associated with Increased Risk for ALS. Nat. Cell Biol. 2010, 466, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, N.C.; Wang, Y.-D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in Prion-Like Domains in hnRNPA2B1 and hnRNPA1 Cause Multisystem Proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS Gene on Chromosome 16 Cause Familial Amyotrophic Lateral Sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A.; et al. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron 2017, 95, 808–816.e9. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Bentmann, E.; Dormann, D.; Jawaid, A.; DeJesus-Hernandez, M.; Ansorge, O.; Roeber, S.; Kretzschmar, H.A.; Munoz, D.G.; Kusaka, H.; et al. FET Proteins TAF15 and EWS are Selective Markers that Distinguish FTLD with FUS Pathology from Amyotrophic Lateral Sclerosis with FUS Mutations. Brain 2011, 134, 2595–2609. [Google Scholar] [CrossRef] [Green Version]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Van Langenhove, T.; Van Der Zee, J.; Sleegers, K.; Engelborghs, S.; Vandenberghe, R.; Gijselinck, I.; Broeck, M.V.D.; Mattheijssens, M.; Peeters, K.; De Deyn, P.P.; et al. Genetic Contribution of FUS to Frontotemporal Lobar Degeneration. Neurology 2010, 74, 366–371. [Google Scholar] [CrossRef]
- Ishigaki, S.; Sobue, G. Importance of Functional Loss of FUS in FTLD/ALS. Front. Mol. Biosci. 2018, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-Nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef]
- Sareen, D.; O’Rourke, J.G.; Meera, P.; Muhammad, A.K.M.G.; Grant, S.; Simpkinson, M.; Bell, S.; Carmona, S.; Ornelas, L.; Sahabian, A.; et al. Targeting RNA Foci in iPSC-Derived Motor Neurons from ALS Patients with a C9ORF72 Repeat Expansion. Sci. Transl. Med. 2013, 5, 208ra149. [Google Scholar] [CrossRef] [Green Version]
- Cooper-Knock, J.; Higginbottom, A.; Stopford, M.J.; Highley, J.R.; Ince, P.G.; Wharton, S.B.; Pickering-Brown, S.; Kirby, J.; Hautbergue, G.M.; Shaw, P.J. Antisense RNA Foci in the Motor Neurons of C9ORF72-ALS Patients are Associated with TDP-43 proteinopathy. Acta Neuropathol. 2015, 130, 63–75. [Google Scholar] [CrossRef]
- Zu, T.; Liu, Y.; Bañez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN Proteins and RNA Foci from Antisense Transcripts in C9ORF72 ALS and Frontotemporal Dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef] [Green Version]
- Martier, R.; Liefhebber, J.M.; García-Osta, A.; Miniarikova, J.; Cuadrado-Tejedor, M.; Espelosin, M.; Ursua, S.; Petry, H.; van Deventer, S.J.; Evers, M.M.; et al. Targeting RNA-Mediated Toxicity in C9orf72 ALS and/or FTD by RNAi-Based Gene Therapy. Mol. Ther. Nucleic Acids 2019, 16, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Lin, S.; Staats, K.A.; Li, Y.; Chang, W.-H.; Hung, S.-T.; Hendricks, E.; Linares, G.R.; Wang, Y.; Son, E.Y.; et al. Haploinsufficiency Leads to Neurodegeneration in C9ORF72 ALS/FTD Human Induced Motor Neurons. Nat. Med. 2018, 24, 313–325. [Google Scholar] [CrossRef]
- McEachin, Z.T.; Parameswaran, J.; Raj, N.; Bassell, G.J.; Jiang, J. RNA-Mediated Toxicity in C9orf72 ALS and FTD. Neurobiol. Dis. 2020, 145, 105055. [Google Scholar] [CrossRef]
- Ash, P.E.; Bieniek, K.F.; Gendron, T.F.; Caulfield, T.; Lin, W.-L.; DeJesus-Hernandez, M.; van Blitterswijk, M.M.; Jansen-West, K.; Paul, J.W.; Rademakers, R.; et al. Unconventional Translation of C9ORF72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to c9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.-J.; Jansen-West, K.; Ash, P.E.A.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense Transcripts of the Expanded C9ORF72 Hexanucleotide Repeat form Nuclear RNA Foci and Undergo Repeat-Associated Non-ATG Translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K.; Arzberger, T.; Grässer, F.A.; Gijselinck, I.; May, S.; Rentzsch, K.; Weng, S.-M.; Schludi, M.H.; Van Der Zee, J.; Cruts, M.; et al. Bidirectional Transcripts of the Expanded C9orf72 Hexanucleotide Repeat Are Translated into Aggregating Dipeptide Repeat Proteins. Acta Neuropathol. 2013, 126, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Wang, S.; Zhang, Z.; Morgens, D.W.; Hayes, L.R.; Lee, S.; Portz, B.; Xie, Y.; Nguyen, B.V.; Haney, M.S.; et al. CRISPR-Cas9 Screens Identify the RNA Helicase DDX3X as a Repressor of C9ORF72 (GGGGCC)n Repeat-Associated Non-AUG Translation. Neuron 2019, 104, 885–898.e8. [Google Scholar] [CrossRef]
- Lee, K.-H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Mori, E.; Kato, M.; Xiang, S.; Wu, L.; Kwon, I.; McKnight, S.L. Toxic PR Poly-Dipeptides Encoded by the C9orf72 Repeat Expansion Target LC Domain Polymers. Cell 2016, 167, 789–802.e12. [Google Scholar] [CrossRef] [Green Version]
- Freibaum, B.D.; Taylor, J.P. The Role of Dipeptide Repeats in C9ORF72-Related ALS-FTD. Front. Mol. Neurosci. 2017, 10, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbaum-Garfinkle, S. Matter over Mind: Liquid Phase Separation and Neurodegeneration. J. Biol. Chem. 2019, 294, 7160–7168. [Google Scholar] [CrossRef] [Green Version]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid–Liquid Phase Separation of the Microtubule-Binding Repeats of the Alzheimer-Related Protein Tau. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau Protein Liquid–Liquid Phase Separation Can Initiate Tau Aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef]
- Alberti, S.; Dormann, D. Liquid–Liquid Phase Separation in Disease. Annu. Rev. Genet. 2019, 53, 171–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallini, C.; Khalil, B.; Smith, C.L.; Rossoll, W. Traffic Jam at the Nuclear Pore: All Roads Lead to Nucleocytoplasmic Transport Defects in ALS/FTD. Neurobiol. Dis. 2020, 140, 104835. [Google Scholar] [CrossRef] [PubMed]
- Mandrioli, J.; Mediani, L.; Alberti, S.; Carra, S. ALS and FTD: Where RNA Metabolism Meets Protein Quality Control. Semin. Cell Dev. Biol. 2020, 99, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.-H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC Repeat Expansion in C9orf72 Compromises Nucleocytoplasmic Transport. Nat. Cell Biol. 2015, 525, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Taylor, J.P. Lost in Transportation: Nucleocytoplasmic Transport Defects in ALS and Other Neurodegenerative Diseases. Neuron 2017, 96, 285–297. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging Mechanisms in ALS and FTD: Disrupted RNA and Protein Homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- McGurk, L.; Mojsilovic-Petrovic, J.; Van Deerlin, V.M.; Shorter, J.; Kalb, R.G.; Lee, V.M.; Trojanowski, J.Q.; Lee, E.B.; Bonini, N.M. Nuclear Poly(ADP-Ribose) Activity Is a Therapeutic Target in Amyotrophic Lateral Sclerosis. Acta Neuropathol. Commun. 2018, 6, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, C.-C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 Pathology Disrupts Nuclear Pore Complexes and Nucleocytoplasmic Transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Daigle, J.G.; Cunningham, K.M.; Coyne, A.N.; Ruan, K.; Grima, J.C.; Bowen, K.E.; Wadhwa, H.; Yang, P.; Rigo, F.; et al. Stress Granule Assembly Disrupts Nucleocytoplasmic Transport. Cell 2018, 173, 958–971.e17. [Google Scholar] [CrossRef] [Green Version]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.; Capell, A.; Schmid, B.; et al. ALS-Associated Fused in Sarcoma (FUS) Mutations Disrupt Transportin-Mediated Nuclear Import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef] [Green Version]
- Gal, J.; Zhang, J.; Kwinter, D.M.; Zhai, J.; Jia, H.; Jia, J.; Zhu, H. Nuclear Localization Sequence of FUS and Induction of Stress Granules by ALS Mutants. Neurobiol. Aging 2011, 32, 2323.e27–2323.e40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyzack, E.G.; Luisier, R.; Taha, D.M.; Neeves, J.; Modic, M.; Mitchell, J.S.; Meyer, I.; Greensmith, L.; Newcombe, J.; Ule, J.; et al. Widespread FUS Mislocalization Is a Molecular Hallmark of Amyotrophic Lateral Sclerosis. Brain 2019, 142, 2572–2580. [Google Scholar] [CrossRef] [Green Version]
- Ito, D.; Seki, M.; Tsunoda, Y.; Uchiyama, H.; Suzuki, N. Nuclear Transport Impairment of Amyotrophic Lateral Sclerosis-Linked Mutations in FUS/TLS. Ann. Neurol. 2010, 69, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Gerbino, V.; Carrì, M.T.; Cozzolino, M.; Achsel, T. Mislocalised FUS Mutants Stall Spliceosomal snRNPs in the Cytoplasm. Neurobiol. Dis. 2013, 55, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Reber, S.; Stettler, J.; Filosa, G.; Colombo, M.; Jutzi, D.; Lenzken, S.C.; Schweingruber, C.; Bruggmann, R.; Bachi, A.; Barabino, S.M.; et al. Minor Intron Splicing is Regulated by FUS and Affected by ALS-Associated FUS Mutants. EMBO J. 2016, 35, 1504–1521. [Google Scholar] [CrossRef]
- Rossi, S.; Rompietti, V.; Antonucci, Y.; Giovannini, D.; Scopa, C.; Scaricamazza, S.; Scardigli, R.; Cestra, G.; Serafino, A.; Carrì, M.T.; et al. UsnRNP Trafficking Is Regulated by Stress Granules and Compromised by Mutant ALS Proteins. Neurobiol. Dis. 2020, 138, 104792. [Google Scholar] [CrossRef] [PubMed]
- Boeynaems, S.; Bogaert, E.; Michiels, E.; Gijselinck, I.; Sieben, A.; Jovičić, A.; De Baets, G.; Scheveneels, W.; Steyaert, J.; Cuijt, I.; et al. Drosophila Screen Connects Nuclear Transport Genes to DPR Pathology in c9ALS/FTD. Sci. Rep. 2016, 6, 20877. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, B.; Robberecht, W.; Bosch, L.V.D. RNA Toxicity in Non-Coding Repeat Expansion Disorders. EMBO J. 2020, 39, e101112. [Google Scholar] [CrossRef]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 Repeat Expansion Disrupts Nucleocytoplasmic Transport. Nat. Cell Biol. 2015, 525, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, N.; Daley, E.L.; Gleixner, A.M.; Mann, J.R.; Kour, S.; Mawrie, D.; Anderson, E.N.; Kofler, J.; Donnelly, C.J.; Kiskinis, E.; et al. RNA Dependent Suppression of C9orf72 ALS/FTD Associated Neurodegeneration by Matrin-3. Acta Neuropathol. Commun. 2020, 8, 1–20. [Google Scholar] [CrossRef]
- Yin, S.; Lopez-Gonzalez, R.; Kunz, R.C.; Gangopadhyay, J.; Borufka, C.; Gygi, S.P.; Gao, F.-B.; Reed, R. Evidence that C9ORF72 Dipeptide Repeat Proteins Associate with U2 snRNP to Cause Mis-Splicing in ALS/FTD Patients. Cell Rep. 2017, 19, 2244–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nihei, Y.; German Consortium for Frontotemporal Lobar Degeneration; Mori, K.; Werner, G.; Arzberger, T.; Zhou, Q.; Khosravi, B.; Japtok, J.; Hermann, A.; Sommacal, A.; et al. Poly-Glycine–Alanine Exacerbates C9orf72 Repeat Expansion-Mediated DNA Damage via Sequestration of Phosphorylated ATM and Loss of Nuclear hnRNPA3. Acta Neuropathol. 2020, 139, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, Y.S.; Flood, L.; Robinson, A.C.; Nihei, Y.; Mori, K.; Rollinson, S.; Richardson, A.; Benson, B.C.; Jones, M.; Snowden, J.S.; et al. Heterogeneous Ribonuclear Protein a3 (hnRNP A3) Is Present in Dipeptide Repeat Protein Containing Inclusions in Frontotem-Poral Lobar Degeneration and Motor Neurone Disease Associated with Expansions in C9orf72 Gene. Acta Neuropathol. Commun. 2017, 5, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Shu, S.; Wang, R.R.; Liu, F.; Cui, B.; Guo, X.N.; Lu, C.X.; Li, X.G.; Liu, M.S.; Peng, B.; et al. Whole-Exome Sequencing Identifies a Missense Mutation in hnRNPA1 in a Family with Flail Arm ALS. Neurology 2016, 87, 1763–1769. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Lowe, P.; Koppers, M.; McKenna-Yasek, D.; Baron, D.M.; et al. Mutations in the Profilin 1 Gene Cause Familial Amyotrophic Lateral Sclerosis. Nat. Cell Biol. 2012, 488, 499–503. [Google Scholar] [CrossRef]
- Smith, B.N.; Vance, C.; Scotter, E.L.; Troakes, C.; Wong, C.H.; Topp, S.; Maekawa, S.; King, A.; Mitchell, J.C.; Lund, K.; et al. Novel Mutations Support a Role for Profilin 1 in the Pathogenesis of ALS. Neurobiol. Aging 2015, 36, 1602.e17–1602.e27. [Google Scholar] [CrossRef] [Green Version]
- Giampetruzzi, A.; Danielson, E.W.; Gumina, V.; Jeon, M.; Boopathy, S.; Brown, R.H.; Ratti, A.; Landers, J.E.; Fallini, C. Modulation of Actin Polymerization Affects Nucleocytoplasmic Transport in Multiple Forms of Amyotrophic Lateral Sclerosis. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wolozin, B.; Ivanov, P. Stress Granules and Neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef]
- Samir, P.; Kesavardhana, S.; Patmore, D.M.; Gingras, S.; Malireddi, R.K.S.; Karki, R.; Guy, C.S.; Briard, B.; Place, D.E.; Bhattacharya, A.; et al. DDX3X Acts as a Live-or-Die Checkpoint in Stressed Cells by Regulating NLRP3 Inflammasome. Nat. Cell Biol. 2019, 573, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Aulas, A.; Velde, C.V. Alterations in Stress Granule Dynamics Driven by TDP-43 and FUS: A Link to Pathological Inclusions in ALS? Front. Cell. Neurosci. 2015, 9, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress Granules as Crucibles of ALS Pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Liu-Yesucevitz, L.; Lin, A.Y.; Ebata, A.; Boon, J.Y.; Reid, W.; Xu, Y.-F.; Kobrin, K.; Murphy, G.J.; Petrucelli, L.; Wolozin, B. ALS-Linked Mutations Enlarge TDP-43-Enriched Neuronal RNA Granules in the Dendritic Arbor. J. Neurosci. 2014, 34, 4167–4174. [Google Scholar] [CrossRef] [Green Version]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress Granule Assembly Is Mediated by Prion-like Aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef] [Green Version]
- Nonhoff, U.; Ralser, M.; Welzel, F.; Piccini, I.; Balzereit, D.; Yaspo, M.-L.; Lehrach, H.; Krobitsch, S. Ataxin-2 Interacts with the DEAD/H-Box RNA Helicase DDX6 and Interferes with P-Bodies and Stress Granules. Mol. Biol. Cell 2007, 18, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Farg, M.A.; Soo, K.Y.; Warraich, S.T.; Sundaramoorthy, V.; Blair, I.P.; Atkin, J.D. Ataxin-2 Interacts with FUS and Intermediate-Length Polyglutamine Expansions Enhance FUS-Related Pathology in Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2012, 22, 717–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Z.; Jiao, B.; Hou, L.; Xiao, T.; Liu, X.; Wang, J.; Xu, J.; Zhou, L.; Yan, X.; Tang, B.; et al. Mutation Analysis of the TIA1 Gene in Chinese Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Neurobiol. Aging 2018, 64, 160.e9–160.e12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liu, Q.; Shen, D.; Tai, H.; Fu, H.; Liu, S.; Wang, Z.; Shi, J.; Ding, Q.; Li, X.; et al. Genetic Analysis of TIA1 Gene in Chinese Patients with Amyotrophic Lateral Sclerosis. Neurobiol. Aging 2018, 67, 201.e9–201.e10. [Google Scholar] [CrossRef]
- Zhao, M.; Kim, J.R.; Van Bruggen, R.; Park, J. RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. Mol. Cells 2018, 41, 818–829. [Google Scholar] [PubMed]
- Fernandes, N.; Eshleman, N.; Buchan, J.R. Stress Granules and ALS: A Case of Causation or Correlation? In RNA Metabolism in Neurodegenerative Diseases; Springer: Cham, Switzerland, 2018; pp. 173–212. [Google Scholar] [CrossRef]
- Baron, D.M.; Kaushansky, L.J.; Ward, C.L.; Sama, R.R.K.; Chian, R.-J.; Boggio, K.J.; Quaresma, A.J.; Nickerson, J.A.; Bosco, D.A. Amyotrophic Lateral Sclerosis-Linked FUS/TLS Alters Stress Granule Assembly and Dynamics. Mol. Neurodegener. 2013, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase Separation by Low Complexity Domains Promotes Stress Granule Assembly and Drives Pathological Fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.L.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Demen. Stem Cells 2016, 34, 2063–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, J.; Cook, C.; Gendron, T.F.; Jansen-West, K.; Del Rosso, G.; Daughrity, L.M.; Castanedes-Casey, M.; Kurti, A.; Stankowski, J.N.; Disney, M.D.; et al. Aberrant Deposition of Stress Granule-Resident Proteins Linked to C9orf72-Associated TDP-43 Proteinopathy. Mol. Neurodegener. 2019, 14, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Boeynaems, S.; Bogaert, E.; Kovacs, D.; Konijnenberg, A.; Timmerman, E.; Volkov, A.; Guharoy, M.; De Decker, M.; Jaspers, T.; Ryan, V.H.; et al. Phase Separation of C9orf72 Dipeptide Repeats Perturbs Stress Granule Dynamics. Mol. Cell 2017, 65, 1044–1055.e5. [Google Scholar] [CrossRef] [Green Version]
- Marmor-Kollet, H.; Siany, A.; Kedersha, N.; Knafo, N.; Rivkin, N.; Danino, Y.M.; Moens, T.G.; Olender, T.; Sheban, D.; Cohen, N.; et al. Spatiotemporal Proteomic Analysis of Stress Granule Disassembly Using APEX Reveals Regulation by SUMOylation and Links to ALS Pathogenesis. Mol. Cell 2020, 80, 876–891. [Google Scholar] [CrossRef]
- Yang, P.; Mathieu, C.; Kolaitis, R.-M.; Zhang, P.; Messing, J.; Yurtsever, U.; Yang, Z.; Wu, J.; Li, Y.; Pan, Q.; et al. G3BP1 Is a Tunable Switch that Triggers Phase Separation to Assemble Stress Granules. Cell 2020, 181, 325–345.e28. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef] [Green Version]
- Markmiller, S.; Soltanieh, S.; Server, K.L.; Mak, R.; Jin, W.; Fang, M.Y.; Luo, E.-C.; Krach, F.; Yang, D.; Sen, A.; et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018, 172, 590–604.e13. [Google Scholar] [CrossRef] [Green Version]
- Naruse, H.; Ishiura, H.; Mitsui, J.; Date, H.; Takahashi, Y.; Matsukawa, T.; Tanaka, M.; Ishii, A.; Tamaoka, A.; Hokkoku, K.; et al. Molecular Epidemiological Study of Familial Amyotrophic Lateral Sclerosis in Japanese Population by Whole-Exome Sequencing and Identification of Novel HNRNPA1 Mutation. Neurobiol. Aging 2018, 61, 255.e9–255.e16. [Google Scholar] [CrossRef]
- Youn, J.-Y.; Dunham, W.H.; Hong, S.J.; Knight, J.D.; Bashkurov, M.; Chen, G.I.; Bagci, H.; Rathod, B.; MacLeod, G.; Eng, S.W.; et al. High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol. Cell 2018, 69, 517–532.e11. [Google Scholar] [CrossRef] [PubMed]
- Figley, M.D.; Bieri, G.; Kolaitis, R.-M.; Taylor, J.P.; Gitler, A.D. Profilin 1 Associates with Stress Granules and ALS-Linked Mutations Alter Stress Granule Dynamics. J. Neurosci. 2014, 34, 8083–8097. [Google Scholar] [CrossRef] [Green Version]
- Gal, J.; Kuang, L.; Barnett, K.R.; Zhu, B.Z.; Shissler, S.C.; Korotkov, K.V.; Hayward, L.J.; Kasarskis, E.J.; Zhu, H. ALS Mutant SOD1 Interacts with G3BP1 and Affects Stress Granule Dynamics. Acta Neuropathol. 2016, 132, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; Boczek, E.E.; Maharana, S.; Lee, H.O.; Carra, S.; Hyman, A.A.; Alberti, S. An Aberrant Phase Transition of Stress Granules Triggered by Misfolded Protein and Prevented by Chaperone Function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef]
- Da Ros, M.; Deol, H.K.; Savard, A.; Guo, H.; Meiering, E.M.; Gibbings, D. Wild-Type and Mutant SOD1 Localizes to RNA-Rich Structures in Cells and Mice but Does Not Bind RNA. J. Neurochem. 2021, 156, 524–538. [Google Scholar] [CrossRef]
- Luisier, R.; Tyzack, G.E.; Hall, C.E.; Mitchell, J.S.; Devine, H.; Taha, D.M.; Malik, B.; Meyer, I.; Greensmith, L.; Newcombe, J.; et al. Intron Retention and Nuclear Loss of SFPQ Are Molecular Hallmarks of ALS. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Herrmann, D.; Parlato, R. C9orf72-Associated Neurodegeneration in ALS-FTD: Breaking New Ground in Ribosomal RNA and Nucleolar Dysfunction. Cell Tissue Res. 2018, 373, 351–360. [Google Scholar] [CrossRef]
- Latonen, L. Phase-to-Phase with Nucleoli—Stress Responses, Protein Aggregation and Novel Roles of RNA. Front. Cell. Neurosci. 2019, 13, 151. [Google Scholar] [CrossRef]
- Mangan, H.; Gailín, M.Ó.; McStay, B. Integrating the Genomic Architecture of Human Nucleolar Organizer Regions with the Biophysical Properties of Nucleoli. FEBS J. 2017, 284, 3977–3985. [Google Scholar] [CrossRef]
- Lee, J.; Hwang, Y.J.; Ryu, H.; Kowall, N.W.; Ryu, H. Nucleolar Dysfunction in Huntington’s Disease. Biochim. Biophys. Acta (BBA) Bioenerg. 2014, 1842, 785–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evsyukov, V.; Domanskyi, A.; Bierhoff, H.; Gispert, S.; Mustafa, R.; Schlaudraff, F.; Liss, B.; Parlato, R. Genetic Mutations Linked to Parkinson’s Disease Differentially Control Nucleolar Activity in Pre-symptomatic Mouse Models. Dis. Model. Mech. 2017, 10, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Jesse, S.; Bayer, H.; Alupei, M.C.; Zügel, M.; Mulaw, M.; Tuorto, F.; Malmsheimer, S.; Singh, K.; Steinacker, J.; Schumann, U.; et al. Ribosomal Transcription is Regulated by PGC-1alpha and Disturbed in Huntington’s Disease. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.R.; Mitrea, D.M.; Zhang, P.; Stanley, C.B.; Cassidy, D.E.; Nourse, A.; Phillips, A.H.; Tolbert, M.; Taylor, J.P.; Kriwacki, R.W. C9orf72 Poly(PR) Dipeptide Repeats Disturb Biomolecular Phase Separation and Disrupt Nucleolar Function. Mol. Cell 2019, 74, 713–728.e6. [Google Scholar] [CrossRef] [PubMed]
- Kanekura, K.; Harada, Y.; Fujimoto, M.; Yagi, T.; Hayamizu, Y.; Nagaoka, K.; Kuroda, M. Characterization of Membrane Penetration and Cytotoxicity of C9orf72-Encoding Arginine-Rich Dipeptides. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Suzuki, H.; Shibagaki, Y.; Hattori, S.; Matsuoka, M. The Proline–Arginine Repeat Protein Linked to C9-ALS/FTD Causes Neuronal Toxicity by Inhibiting the DEAD-Box RNA Helicase-Mediated Ribosome Biogenesis. Cell Death Dis. 2018, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Tao, Z.; Wang, H.; Xia, Q.; Li, K.; Li, K.; Jiang, X.; Xu, G.; Wang, G.; Ying, Z. Nucleolar Stress and Impaired Stress Granule Formation Contribute to C9orf72 RAN Translation-Induced Cytotoxicity. Hum. Mol. Genet. 2015, 24, 2426–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense Proline-Arginine RAN Dipeptides Linked to C9ORF72-ALS/FTD Form Toxic Nuclear Aggregates that Initiate In Vitro and In Vivo Neuronal Death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeusler, A.R.; Donnelly, C.J.; Rothstein, J.D. The Expanding Biology of the C9orf72 Nucleotide Repeat Expansion in Neurodegenerative Disease. Nat. Rev. Neurosci. 2016, 17, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, A.R.; Donnelly, C.J.; Periz, G.; Simko, E.A.J.; Shaw, P.G.; Kim, M.-S.; Maragakis, N.J.; Troncoso, J.C.; Pandey, A.; Sattler, R.; et al. C9orf72 Nucleotide Repeat Structures Initiate Molecular Cascades of Disease. Nat. Cell Biol. 2014, 507, 195–200. [Google Scholar] [CrossRef]
- Zhang, Q.; An, Y.; Chen, Z.S.; Koon, A.C.; Lau, K.-F.; Ngo, J.C.K.; Chan, H.Y.E. A Peptidylic Inhibitor for Neutralizing (GGGGCC)-Associated Neurodegeneration in C9ALS-FTD. Mol. Ther. Nucleic Acids 2019, 16, 172–185. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, J.G.; Bogdanik, L.; Muhammad, A.; Gendron, T.F.; Kim, K.J.; Austin, A.; Cady, J.; Liu, E.Y.; Zarrow, J.; Grant, S.; et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron 2015, 88, 892–901. [Google Scholar] [CrossRef] [Green Version]
- Trinkle-Mulcahy, L.; Sleeman, J.E. The Cajal Body and the Nucleolus: “In a Relationship” or “It’s Complicated”? RNA Biol. 2016, 14, 739–751. [Google Scholar] [CrossRef] [Green Version]
- Jády, B.E.; Richard, P.; Bertrand, E.; Kiss, T. Cell Cycle-Dependent Recruitment of Telomerase RNA and Cajal Bodies to Human Telomeres. Mol. Biol. Cell 2006, 17, 944–954. [Google Scholar] [CrossRef] [Green Version]
- Spector, D.L.; Lamond, A.I. Nuclear Speckles. Cold Spring Harb. Perspect. Biol. 2010, 3, a000646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galganski, L.; Urbanek, M.O.; Krzyzosiak, W.J. Nuclear Speckles: Molecular Organization, Biological Function and Role in Disease. Nucleic Acids Res. 2017, 45, 10350–10368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobbins, A.M.; Reichenbach, L.F.; Lucas, C.M.; Hudson, A.J.; Burley, G.A.; Eperon, I.C. The Mechanisms of a Mammalian Splicing Enhancer. Nucleic Acids Res. 2018, 46, 2145–2158. [Google Scholar] [CrossRef] [Green Version]
- Liao, E.S.; Regev, O. Splicing at the Phase-Separated Nuclear Speckle Interface: A Model. Nucleic Acids Res. 2021, 49, 636–645. [Google Scholar] [CrossRef]
- Ha, M. Transcription Boosting by Nuclear Speckles. Nat. Rev. Mol. Cell Biol. 2019, 21, 64–65. [Google Scholar] [CrossRef]
- Wang, K.; Wang, L.; Wang, J.; Chen, S.; Shi, M.; Cheng, H. Intronless mRNAs Transit through Nuclear Speckles to Gain Export Competence. J. Cell Biol. 2018, 217, 3912–3929. [Google Scholar] [CrossRef] [Green Version]
- Kanekura, K.; Yagi, T.; Cammack, A.J.; Mahadevan, J.; Kuroda, M.; Harms, M.B.; Miller, T.M.; Urano, F. Poly-Dipeptides Encoded by the C9ORF72 Repeats Block Global Protein Translation. Hum. Mol. Genet. 2016, 25, 1803–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakkar, N.; Kovalik, T.; Lorenzini, I.; Spangler, S.; Lacoste, A.; Sponaugle, K.; Ferrante, P.; Argentinis, E.; Sattler, R.; Bowser, R. Artificial Intelligence in Neurodegenerative Disease Research: Use of IBM Watson to Identify Additional RNA-Binding Proteins Altered in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2018, 135, 227–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, I.-F.; Reddy, N.M.; Shen, C.-K.J. Higher Order Arrangement of the Eukaryotic Nuclear Bodies. Proc. Natl. Acad. Sci. USA 2002, 99, 13583–13588. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, L.; Lin, W.-L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS Disease-Associated Mutant TDP-43 Impairs Mitochondrial Dynamics and Function in Motor Neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [Green Version]
- Ling, J.P.; Pletnikova, O.; Troncoso, J.C.; Wong, P.C. TDP-43 Repression of Nonconserved Cryptic Exons Is Compromised in ALS-FTD. Sciences 2015, 349, 650–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.-C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long Pre-mRNA Depletion and RNA Missplicing Contribute to Neuronal Vulnerability from Loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Shibagaki, Y.; Hattori, S.; Matsuoka, M. C9-ALS/FTD-Linked Proline–Arginine Dipeptide Repeat Protein Associates with Paraspeckle Components and Increases Paraspeckle Formation. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Fox, A.H.; Nakagawa, S.; Hirose, T.; Bond, C.S. Paraspeckles: Where Long Noncoding RNA Meets Phase Separation. Trends Biochem. Sci. 2018, 43, 124–135. [Google Scholar] [CrossRef] [Green Version]
- Pisani, G.; Baron, B. Nuclear Paraspeckles Function in Mediating Gene Regulatory and Apoptotic Pathways. Non-Coding RNA Res. 2019, 4, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Yamazaki, T.; Hirose, T. Molecular Dissection of Nuclear Paraspeckles: Towards Understanding the Emerging World of the RNP Milieu. Open Biol. 2018, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesi, A.; Staahl, B.T.; Jovičić, A.; Couthouis, J.; Fasolino, M.; Raphael, A.R.; Yamazaki, T.; Elias, L.; Polak, M.; Kelly, C.; et al. Exome Sequencing to Identify de Novo Mutations in Sporadic ALS Trios. Nat. Neurosci. 2013, 16, 851–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapeli, K.; Martinez, F.J.; Yeo, G.W. Genetic Mutations in RNA-Binding Proteins and Their Roles in ALS. Qual. Life Res. 2017, 136, 1193–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas-Jinu, S.; Gordon, P.M.; Fielding, T.; Taylor, R.; Smith, B.N.; Snowden, V.; Blanc, E.; Vance, C.; Topp, S.; Wong, C.-H.; et al. Non-nuclear Pool of Splicing Factor SFPQ Regulates Axonal Transcripts Required for Normal Motor Development. Neuron 2017, 94, 322–336.e5. [Google Scholar] [CrossRef] [Green Version]
- Nishimoto, Y.; Nakagawa, S.; Hirose, T.; Okano, H.J.; Takao, M.; Shibata, S.; Suyama, S.; Kuwako, K.-I.; Imai, T.; Murayama, S.; et al. The Long Non-coding RNA Nuclear-Enriched Abundant Transcript 1_2 Induces Paraspeckle Formation in the Motor Neuron during the Early Phase of Amyotrophic Lateral Sclerosis. Mol. Brain 2013, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, P.; Bandopadhyay, R. Pathophysiological Implications of RNP Granules in Frontotemporal Dementia and ALS. Neurochem. Int. 2020, 140, 104819. [Google Scholar] [CrossRef]
- Wang, C.; Duan, Y.; Duan, G.; Wang, Q.; Zhang, K.; Deng, X.; Qian, B.; Gu, J.; Ma, Z.; Zhang, S.; et al. Stress Induces Dynamic, Cytotoxicity-Antagonizing TDP-43 Nuclear Bodies via Paraspeckle LncRNA NEAT1-Mediated Liquid-Liquid Phase Separation. Mol. Cell 2020, 79, 443–458.e7. [Google Scholar] [CrossRef]
- Yamazaki, T.; Nakagawa, S.; Hirose, T. Architectural RNAs for Membraneless Nuclear Body Formation. Cold Spring Harb. Symp. Quant. Biol. 2019, 84, 227–237. [Google Scholar] [CrossRef]
- An, H.; Skelt, L.; Notaro, A.; Highley, J.R.; Fox, A.H.; La Bella, V.; Buchman, V.L.; Shelkovnikova, T.A. ALS-Linked FUS Mutations Confer Loss and Gain of Function in the Nucleus by Promoting Excessive Formation of Dysfunctional Paraspeckles. Acta Neuropathol. Commun. 2019, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Česnik, A.B.; Darovic, S.; Mihevc, S.P.; Štalekar, M.; Malnar, M.; Motaln, H.; Lee, Y.-B.; Mazej, J.; Pohleven, J.; Grosch, M.; et al. Nuclear RNA Foci from C9ORF72 Expansion Mutation Form Paraspeckle-like Bodies. J. Cell Sci. 2019, 132, jcs224303. [Google Scholar] [CrossRef] [Green Version]
- Bampton, A.; Gittings, L.M.; Fratta, P.; Lashley, T.; Gatt, A. The Role of hnRNPs in Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2020, 140, 599–623. [Google Scholar] [CrossRef] [PubMed]
- Gittings, L.M.; Foti, S.C.; Benson, B.C.; Gami-Patel, P.; Isaacs, A.M.; Lashley, T. Heterogeneous Nuclear Ribonucleoproteins R and Q Accumulate in Pathological Inclusions in FTLD-FUS. Acta Neuropathol. Commun. 2019, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Conlon, E.G.; Lu, L.; Sharma, A.; Yamazaki, T.; Tang, T.; Shneider, N.A.; Manley, J.L. The C9ORF72 GGGGCC Expansion Forms RNA G-Quadruplex Inclusions and Sequesters hnRNP H to Disrupt Splicing in ALS Brains. eLife 2016, 5, e17820. [Google Scholar] [CrossRef]
- Deshaies, J.-E.; Shkreta, L.; Moszczynski, A.J.; Sidibé, H.; Semmler, S.; Fouillen, A.; Bennett, E.R.; Bekenstein, U.; Destroismaisons, L.; Toutant, J.; et al. TDP-43 Regulates the Alternative Splicing of hnRNP A1 to Yield an Aggregation-Prone Variant in Amyotrophic Lateral Sclerosis. Brain 2018, 141, 1320–1333. [Google Scholar] [CrossRef] [PubMed]
- Frottin, F.; Schueder, F.; Tiwary, S.; Gupta, R.; Körner, R.; Schlichthaerle, T.; Cox, J.; Jungmann, R.; Hartl, F.U.; Hipp, M.S. The Nucleolus Functions as a Phase-Separated Protein Quality Control Compartment. Science 2019, 365, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Woo, K.-C.; Kim, D.-Y.; Kim, T.-D.; Shin, J.; Park, S.M.; Jang, S.K.; Kim, K.-T. Rhythmic Interaction between Period1 mRNA and hnRNP Q Leads to Circadian Time-Dependent Translation. Mol. Cell. Biol. 2011, 32, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.; Allada, R. Emerging Roles for Post-transcriptional Regulation in Circadian Clocks. Nat. Neurosci. 2013, 16, 1544–1550. [Google Scholar] [CrossRef]
- Patton, J.G.; Mayer, S.A.; Tempst, P.; Nadal-Ginard, B. Characterization and Molecular Cloning of Polypyrimidine Tract-Binding Protein: A Component of a Complex Necessary for Pre-mRNA Splicing. Genes Dev. 1991, 5, 1237–1251. [Google Scholar] [CrossRef] [Green Version]
- Galbán, S.; Kuwano, Y.; Pullmann, R.; Martindale, J.L.; Kim, H.H.; Lal, A.; Abdelmohsen, K.; Yang, X.; Dang, Y.; Liu, J.O.; et al. RNA-Binding Proteins HuR and PTB Promote the Translation of Hypoxia-Inducible Factor 1α. Mol. Cell. Biol. 2007, 28, 93–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, K.-C.; Kim, T.-D.; Lee, K.-Y.; Kim, D.-Y.; Kim, W.; Kim, K.-T. Mouse Period 2 mRNA cIrcadian Oscillation Is Modulated by PTB–Mediated Rhythmic mRNA Degradation. Nucleic Acids Res. 2008, 37, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Couthouis, J.; Hart, M.P.; Shorter, J.; DeJesus-Hernandez, M.; Erion, R.; Oristano, R.; Liu, A.X.; Ramos, D.; Jethava, N.; Hosangadi, D.; et al. A Yeast Functional Screen Predicts New Candidate ALS Disease Genes. Proc. Natl. Acad. Sci. USA 2011, 108, 20881–20890. [Google Scholar] [CrossRef] [Green Version]
- Couthouis, J.; Raphael, A.R.; Daneshjou, R.; Gitler, A.D. Targeted Exon Capture and Sequencing in Sporadic Amyotrophic Lateral Sclerosis. PLoS Genet. 2014, 10, e1004704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emde, A.; Eitan, C.; Liou, L.; Libby, R.T.; Rivkin, N.; Magen, I.; Reichenstein, I.; Oppenheim, H.; Eilam, R.; Silvestroni, A.; et al. Dysregulated mi RNA Biogenesis Downstream of Cellular Stress and ALS-Causing Mutations: A New Mechanism for ALS. EMBO J. 2015, 34, 2633–2651. [Google Scholar] [CrossRef] [Green Version]
- Butti, Z.; Patten, S.A. RNA Dysregulation in Amyotrophic Lateral Sclerosis. Front. Genet. 2019, 9, 712. [Google Scholar] [CrossRef] [Green Version]
- Aakalu, G.; Smith, W.; Nguyen, N.; Jiang, C.; Schuman, E.M. Dynamic Visualization of Local Protein Synthesis in Hippocampal Neurons. Neuron 2001, 30, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Seo, J.-Y.; Ryu, H.G.; Kim, D.-Y.; Lee, K.-H.; Kim, K.-T. BDNF-Induced Local Translation of GluA1 is Regulated by HNRNP A2/B1. Sci. Adv. 2020, 6, eabd2163. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.; Holste, D.; Kreiman, G.; Burge, C.B. Variation in Alternative Splicing across Human Tissues. Genome Biol. 2004, 5, R74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Membrane-less organelles that are affected by amyotrophic lateral sclerosis (ALS)-related RNA-binding proteins. Membrane-less organelles described in this review are illustrated, including the nucleolus, nuclear speckle, Cajal body, stress granule, along with RNA foci and nuclear pore complex. Dysregulation of RNA-binding proteins (RBPs) such as ALS-causing mutations leads to abnormal dynamics and function of the nucleolus, nuclear speckles, Cajal bodies, and stress granules. This dysfunction of membrane-less organelles is also interconnected to nucleocytoplasmic shuttling and RNA foci that may sequestrate RNA-binding proteins. These ALS-related cellular abnormalities influence each other. Created with BioRender.com.
Figure 1.
Membrane-less organelles that are affected by amyotrophic lateral sclerosis (ALS)-related RNA-binding proteins. Membrane-less organelles described in this review are illustrated, including the nucleolus, nuclear speckle, Cajal body, stress granule, along with RNA foci and nuclear pore complex. Dysregulation of RNA-binding proteins (RBPs) such as ALS-causing mutations leads to abnormal dynamics and function of the nucleolus, nuclear speckles, Cajal bodies, and stress granules. This dysfunction of membrane-less organelles is also interconnected to nucleocytoplasmic shuttling and RNA foci that may sequestrate RNA-binding proteins. These ALS-related cellular abnormalities influence each other. Created with BioRender.com.


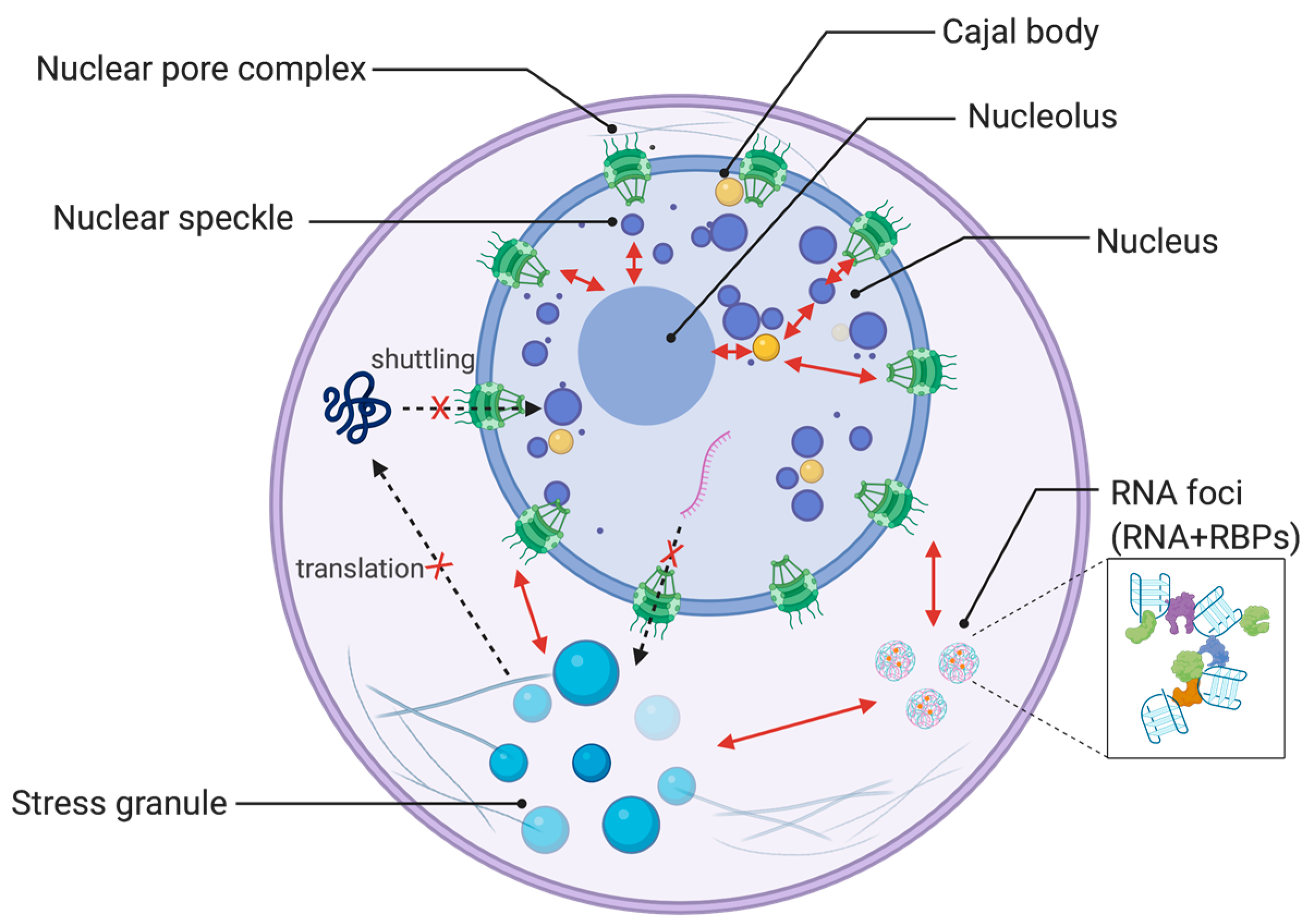{kind=link}
Table 1.
Membrane-less organelles and the corresponding RNA-binding proteins.
| Location | RNA-Binding Proteins | Function |
|---|---|---|
| Stress granules | FUS | Abnormal stress granule formation [32,93] |
| TARDBP | Abnormal stress granule formation [33,85]; GR or PR DPR interactor [47,96] | |
| ATXN2 | Stress granule components; cause ALS or increase its risk [16,25] | |
| TIA-1 | Stress granule components; abnormal stress granule dynamics [28,91]; GR or PR DPR interactor [47] | |
| HNRNPA2B1 | Formation of aberrant stress granules [26]; GR or PR DPR interactor [47,101] | |
| HNRNPA1 | Increased incorporation into stress granules [94]; GR or PR DPR interactor [47,102] | |
| G3BP1 | GR or PR DPR interactor [47,99] | |
| PABPC1 | GR or PR DPR interactor [47,101] | |
| PTBP1 | GR or PR DPR interactor [47,101] | |
| SFPQ | GR or PR DPR interactor [47] | |
| YBX1 | GR or PR DPR interactor [47,101] | |
| HNRNPUL1 | GR or PR DPR interactor [47] | |
| FXR | GR or PR DPR interactor [47] | |
| LBR | GR or PR DPR interactor [47] | |
| SYNCRIP | GR or PR DPR interactor [47,101] | |
| HNRNPH | GR or PR DPR interactor [47,101] | |
| PFN1 | Impairments in stress granule dynamics [80,104] | |
| SOD1 | Incorporation into stress granule; delaying stress granule assembly [105,106] | |
| Nucleolus | NPM1 | Colocalization and interaction with GR or PR DPR [47,109]; mislocalization with impaired nucleolus dynamics [118,120] |
| NCL | Colocalization and interaction with GR or PR DPR also with repeat RNA [38,47,118,119], altered distribution, and nucleolar stress [122,123] | |
| FBL | Colocalization DPR [109,155] | |
| ADARB2 | Interaction with repeat RNA of C9orf72 [38] | |
| HNRNPA3 | Interaction with repeat RNA of C9orf72 [38] | |
| HNRNPH | Interaction with repeat RNA of C9orf72 [38] | |
| Cajal bodies | SRRM2 | GR and PR DPR interactor [47]; DPR altered dynamics [69] |
| FUS | Decreasing number of Cajal bodies [69]; altering distribution of snRNP into Cajal bodies [68,69] | |
| Nuclear speckles | SRSF7 | Altered dynamics by interaction with GR or PR DPR [47] |
| SRSF2 | Colocalize with C9orf72 RNA foci [38,133] | |
| TARDBP | Disruption of nuclear speckles [127,135]; disturbing splicing event [136] and expression of MALAT1 [137] | |
| FUS | Disruption of nuclear speckles [127] | |
| Paraspeckles | SFPQ | Paraspeckle-enriched and mutation in ALS [141,144], associated with C9orf72 [138,150,151] |
| FUS | Paraspeckle-enriched and mutation in ALS [141,143,145], dysfunction of paraspeckle [149], associated with C9orf72 RNA foci [150] | |
| EWSR1 | Paraspeckle-enriched and mutation in ALS [24,143] | |
| TAF15 | Paraspeckle-enriched and mutation in ALS [141,143] | |
| TARDBP | Paraspeckle-enriched and mutation in ALS [141,143,145]; defect in paraspeckle [146,147] | |
| SS18L1 | Paraspeckle-enriched and mutation in ALS [141,143] | |
| HNRNPA1 | Paraspeckle-enriched and mutation in ALS [141,143] | |
| NONO | Associated with C9orf72 RNA foci [150] and DPR [138,151] | |
| RBM14 | Associated with C9orf72 RNA foci [150] and DPR [138,151] | |
| HNRNPH | Associated with C9orf72 RNA foci [150] and regulated by poly PR [138] | |
| HNRNPF | GR or PR DPR interactor [138,151] and regulated by poly PR [138] | |
| HNRNPM | GR or PR DPR interactor [138,151] | |
| FAM98 | GR or PR DPR interactor [138,151] | |
| MATR3 | GR or PR DPR interactor [138,151] | |
| Nucleocytoplasmic shuttling | FUS | Dysregulation of nucleocytoplasmic shuttling [32,63] |
| TARDBP | Dysregulation of nucleocytoplasmic shuttling [33,61] | |
| HNRNPA3 | Nucleocytoplasmic transport capability [71]; mislocalization by DPR [75] | |
| MATR3 | Sequestrated by RNA foci [73] | |
| PARP1 | GR or PR DPR interactor; nucleocytoplasmic shuttling modulator [47] | |
| YBX1 | GR or PR DPR interactor; nucleocytoplasmic shuttling modulator [47] | |
| LBR | GR or PR DPR interactor; nucleocytoplasmic shuttling modulator [47] | |
| U2 snRNP | Mislocalization by DPR [74] | |
| HNRNPA1 | Cytoplasmic inclusion formation [26] | |
| HNRNPA2B1 | Cytoplasmic inclusion formation [26] | |
| PFN1 | Altered distribution of RNA-binding proteins [80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, W.; Kim, D.-Y.; Lee, K.-H. RNA-Binding Proteins and the Complex Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 2598. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052598
AMA Style
Kim W, Kim D-Y, Lee K-H. RNA-Binding Proteins and the Complex Pathophysiology of ALS. International Journal of Molecular Sciences. 2021; 22(5):2598. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052598
Chicago/Turabian StyleKim, Wanil, Do-Yeon Kim, and Kyung-Ha Lee. 2021. "RNA-Binding Proteins and the Complex Pathophysiology of ALS" International Journal of Molecular Sciences 22, no. 5: 2598. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052598
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.