
Additive Role of Immune System Infiltration and Angiogenesis in Uveal Melanoma Progression

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Clinical Description
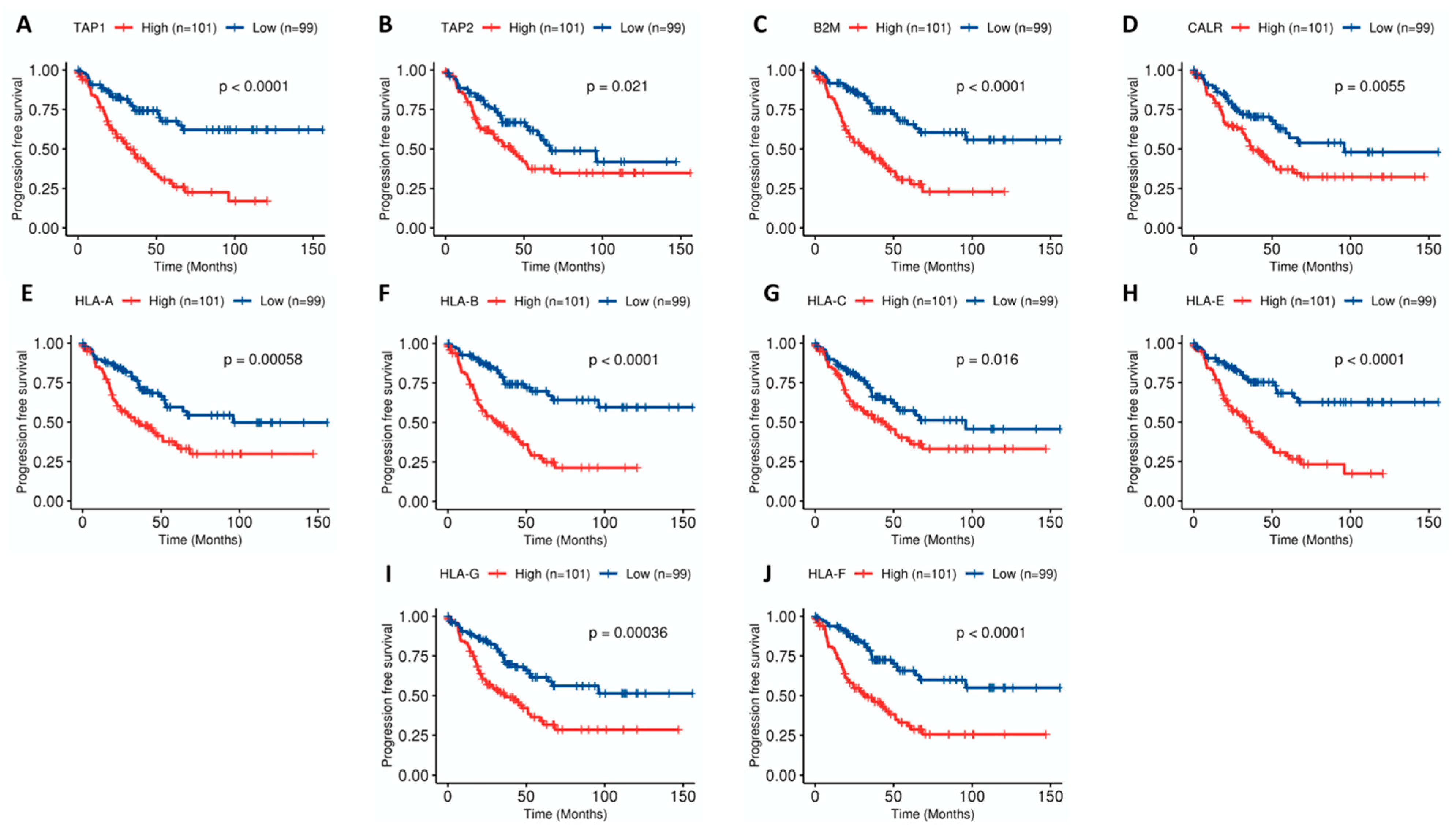
2.2. Stromal and Immune Cell Infiltration Is Associated with Poor Prognosis in Uveal Melanoma (UM)
2.3. Combination of Angiogenesis and Antigen Presentation Confers Poor Prognosis
2.4. Metabolic and Tyrosine Kinase Pathways Are Activated in Poor-Prognosis Tumors
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Microenvironment Characterization
4.3. TMB (Tumor Mutational Burden)
4.4. Angiogenesis and Antigen Presentation Enrichment Analysis
4.5. Hierarchical Clustering
4.6. Functional Analysis
4.7. Statistical Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| aDC | activated dendritic cell |
| Angio | Angiogenesis |
| AP | Antigen Presentation |
| B2M | Beta-2-Microglobulin |
| BAP1 | BRCA1 Associated Protein 1 |
| CALR | Calreticulin |
| CI | Confidence Interval |
| CM | Cutaneous Melanoma |
| CTLA-4 | Cytotoxic T-Lymphocyte Antigen-4 |
| D3 | Disomy of Chromosome 3 |
| DC | dendritic cell |
| DEG | Differentially Expressed Genes |
| DFS | Disease Free Survival |
| FPKM | Fragments per Kilobase per Million |
| GEO | Gene Expression Omnibus |
| GSEA | Gene Set Enrichment Analysis |
| GSVA | Gene Set Variation Analysis |
| GZMA | Granzyme A |
| HLA | Human Leukocyte Antigen |
| HR | Hazard Ratio |
| iDC | immature dendritic cell |
| IL2 | Interleukin-2 |
| IPS | Immunophenoscore |
| KM | Kaplan-Meier |
| M1 | liver metastasis |
| M1a | small lesion liver metastasis |
| M3 | Monosomy of Chromosome 3 |
| MSigDB | Molecular Signatures Database |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| MUM | Metastatic Uveal Melanoma |
| NES | Normalized Enrichment Score |
| NK | Natural Killer |
| OS | Overall Survival |
| PD-1 | Programmed Cell death protein 1 |
| PDL1 | Programmed Death Ligand 1 |
| PET-TC | Positron emission tomography |
| PFS | Progression Free Survival |
| PI3K | Phosphoinositide 3-kinase |
| PRF | Perforin |
| TAP | Tapasin |
| TIL | Tumor Infiltrating Lymphocyte |
| TIS | T-cell Inflammatory Signature |
| TMB | Tumor Mutational Burden |
| TME | Tumor microenvironment |
| TNF-α | Tumor Necrosis Factor alpha |
| UM | Uveal Melanoma |
| VEGF | Vascular Endothelial Growth Factor |
References
- Carvajal, R.D.; Schwartz, G.K.; Tezel, T.; Marr, B.; Francis, J.H.; Nathan, P.D. Metastatic Disease from Uveal Melanoma: Treatment Options and Future Prospects. Br. J. Ophthalmol. 2017, 101, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallone, S.; De Vries, E.; Guzzo, M.; Midena, E.; Verne, J.; Coebergh, J.W.; Marcos-Gragera, R.; Ardanaz, E.; Martinez, R.; Chirlaque, M.D.; et al. Descriptive Epidemiology of Malignant Mucosal and Uveal Melanomas and Adnexal Skin Carcinomas in Europe. Eur. J. Cancer 2012, 48, 1167–1175. [Google Scholar] [CrossRef]
- Yang, J.; Manson, D.K.; Marr, B.P.; Carvajal, R.D. Treatment of Uveal Melanoma: Where Are We Now? Ther. Adv. Med. Oncol. 2018, 10, 175883401875717. [Google Scholar] [CrossRef] [PubMed]
- Caminal, J.M.; Ribes, J.; Clèries, R.; Ibáñez, N.; Arias, L.; Piulats, J.M.; Pera, J.; Gutierrez, C.; Arruga, J. Relative Survival of Patients with Uveal Melanoma Managed in a Single Center. Melanoma Res. 2012, 22, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Pons, F.; Plana, M.; Caminal, J.M.; Pera, J.; Fernandes, I.; Perez, J.; Garcia-Del-Muro, X.; Marcoval, J.; Penin, R.; Fabra, A.; et al. Metastatic Uveal Melanoma: Is There a Role for Conventional Chemotherapy?—A Single Center Study Based on 58 Patients. Melanoma Res. 2011, 21, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Piulats, J.M.; Espinosa, E.; de la Cruz Merino, L.; Varela, M.; Alonso Carrión, L.; Martín-Algarra, S.; López Castro, R.; Curiel, T.; Rodríguez-Abreu, D.; Redrado, M.; et al. Nivolumab Plus Ipilimumab for Treatment-Naïve Metastatic Uveal Melanoma: An Open-Label, Multicenter, Phase II Trial by the Spanish Multidisciplinary Melanoma Group (GEM-1402). J. Clin. Oncol. 2021, JCO2000550. [Google Scholar] [CrossRef]
- Algazi, A.P.; Tsai, K.K.; Shoushtari, A.N.; Munhoz, R.R.; Eroglu, Z.; Piulats, J.M.; Ott, P.A.; Johnson, D.B.; Hwang, J.; Daud, A.I.; et al. Clinical Outcomes in Metastatic Uveal Melanoma Treated with PD-1 and PD-L1 Antibodies: PD-1 Blockade in Uveal Melanoma. Cancer 2016, 122, 3344–3353. [Google Scholar] [CrossRef]
- Rossi, E.; Schinzari, G.; Zizzari, I.G.; Maiorano, B.A.; Pagliara, M.M.; Sammarco, M.G.; Fiorentino, V.; Petrone, G.; Cassano, A.; Rindi, G.; et al. Immunological Backbone of Uveal Melanoma: Is There a Rationale for Immunotherapy? Cancers 2019, 11, 1055. [Google Scholar] [CrossRef] [Green Version]
- Jager, M.J.; Shields, C.L.; Cebulla, C.M.; Abdel-Rahman, M.H.; Grossniklaus, H.E.; Stern, M.-H.; Carvajal, R.D.; Belfort, R.N.; Jia, R.; Shields, J.A.; et al. Uveal Melanoma. Nat. Rev. Dis. Primers 2020, 6, 24. [Google Scholar] [CrossRef]
- Toomey, C.B.; Protopsaltis, N.J.; Phou, S.; Bakhoum, M.F.; Thorson, J.A.; Ediriwickrema, L.S.; Korn, B.S.; Kikkawa, D.O.; Goldbaum, M.H.; Lin, J.H. Prevalence of mismatch repair gene mutations in uveal melanoma. Retina 2020, 40, 2216–2220. [Google Scholar] [CrossRef]
- Cross, N.A.; Murray, A.K.; Rennie, I.G.; Ganesh, A.; Sisley, K. Instability of Microsatellites Is an Infrequent Event in Uveal Melanoma. Melanoma Res. 2003, 13, 435–440. [Google Scholar] [CrossRef]
- Johansson, P.A.; Brooks, K.; Newell, F.; Palmer, J.M.; Wilmott, J.S.; Pritchard, A.L.; Broit, N.; Wood, S.; Carlino, M.S.; Leonard, C.; et al. Whole Genome Landscapes of Uveal Melanoma Show an Ultraviolet Radiation Signature in Iris Tumours. Nat. Commun. 2020, 11, 2408. [Google Scholar] [CrossRef]
- Niederkorn, J.Y. Immune Escape Mechanisms of Intraocular Tumors. Prog. Retin. Eye Res. 2009, 28, 329–347. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; Lichtenberg, M.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, M.; Rullan, A.J.; Piulats, J.M. Uveal Melanoma as a Target for Immune-Therapy. Ann. Transl. Med. 2016, 4, 172. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, C.R.; Kalirai, H.; Sacco, J.J.; Azevedo, R.A.; Duckworth, A.; Slupsky, J.R.; Coulson, J.M.; Coupland, S.E. Loss of BAP1 Expression Is Associated with an Immunosuppressive Microenvironment in Uveal Melanoma, with Implications for Immunotherapy Development. J. Pathol. 2020, 250, 420–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castet, F.; Garcia-Mulero, S.; Sanz-Pamplona, R.; Cuellar, A.; Casanovas, O.; Caminal, J.; Piulats, J. Uveal Melanoma, Angiogenesis and Immunotherapy, Is There Any Hope? Cancers 2019, 11, 834. [Google Scholar] [CrossRef] [Green Version]
- Gangemi, R.; Mirisola, V.; Barisione, G.; Fabbi, M.; Brizzolara, A.; Lanza, F.; Mosci, C.; Salvi, S.; Gualco, M.; Truini, M.; et al. Mda-9/Syntenin Is Expressed in Uveal Melanoma and Correlates with Metastatic Progression. PLoS ONE 2012, 7, e29989. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Valet, F.; Planque, N.; Silveri, L.; Maacha, S.; Anezo, O.; Hupe, P.; Plancher, C.; Reyes, C.; Albaud, B.; et al. High PTP4A3 Phosphatase Expression Correlates with Metastatic Risk in Uveal Melanoma Patients. Cancer Res. 2011, 71, 666–674. [Google Scholar] [CrossRef] [Green Version]
- Field, M.G.; Decatur, C.L.; Kurtenbach, S.; Gezgin, G.; van der Velden, P.A.; Jager, M.J.; Kozak, K.N.; Harbour, J.W. PRAME as an Independent Biomarker for Metastasis in Uveal Melanoma. Clin. Cancer Res. 2016, 22, 1234–1242. [Google Scholar] [CrossRef] [Green Version]
- Fagone, P.; Caltabiano, R.; Russo, A.; Lupo, G.; Anfuso, C.D.; Basile, M.S.; Longo, A.; Nicoletti, F.; De Pasquale, R.; Libra, M.; et al. Identification of Novel Chemotherapeutic Strategies for Metastatic Uveal Melanoma. Sci. Rep. 2017, 7, 44564. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Ma, C. Identification of Prognostic Genes in Uveal Melanoma Microenvironment. PLoS ONE 2020, 15, e0242263. [Google Scholar] [CrossRef]
- Bronkhorst, I.H.G.; Vu, T.H.K.; Jordanova, E.S.; Luyten, G.P.M.; van der Burg, S.H.; Jager, M.J. Different Subsets of Tumor-Infiltrating Lymphocytes Correlate with Macrophage Influx and Monosomy 3 in Uveal Melanoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Cruz, P.O.; Specht, C.S.; McLean, I.W. Lymphocytic Infiltration in Uveal Malignant Melanoma. Cancer 1990, 65, 112–115. [Google Scholar] [CrossRef]
- Whelchel, J.C.; Farah, S.E.; McLean, I.W.; Burnier, M.N. Immunohistochemistry of Infiltrating Lymphocytes in Uveal Malignant Melanoma. Investig. Ophthalmol. Vis. Sci. 1993, 34, 2603–2606. [Google Scholar] [PubMed]
- Buder, K.; Gesierich, A.; Gelbrich, G.; Goebeler, M. Systemic Treatment of Metastatic Uveal Melanoma: Review of Literature and Future Perspectives. Cancer Med. 2013, 2, 674–686. [Google Scholar] [CrossRef]
- Ellerhorst, J.A.; Cooksley, C.D.; Grimm, E.A. Autoimmunity and Hypothyroidism in Patients with Uveal Melanoma. Melanoma Res. 2001, 11, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.-W.; Sun, C.-M.; Calderaro, J.; Jeng, Y.-M.; Hsiao, L.-P.; Lacroix, L.; Bougoüin, A.; et al. B Cells Are Associated with Survival and Immunotherapy Response in Sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary Lymphoid Structures Improve Immunotherapy and Survival in Melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B Cells and Tertiary Lymphoid Structures Promote Immunotherapy Response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing Cancer Immunotherapy Using Antiangiogenics: Opportunities and Challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Khan, K.A.; Kerbel, R.S. Improving Immunotherapy Outcomes with Anti-Angiogenic Treatments and Vice Versa. Nat. Rev. Clin. Oncol. 2018, 15, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamborero, D.; Rubio-Perez, C.; Muiños, F.; Sabarinathan, R.; Piulats, J.M.; Muntasell, A.; Dienstmann, R.; Lopez-Bigas, N.; Gonzalez-Perez, A. A Pan-Cancer Landscape of Interactions between Solid Tumors and Infiltrating Immune Cell Populations. Clin. Cancer Res. 2018, 24, 3717–3728. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-Tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, C.; Oba, J.; Roszik, J.; Marszalek, J.R.; Chen, K.; Qi, Y.; Eterovic, K.; Robertson, A.G.; Burks, J.K.; McCannel, T.A.; et al. Elevated Endogenous SDHA Drives Pathological Metabolism in Highly Metastatic Uveal Melanoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4187–4195. [Google Scholar] [CrossRef] [Green Version]
- Del Carpio Huerta, L.P.; Algarra, M.; Llobera, A.S.; Rodríguez-Vida, A.; Ruiz, S.; Leiva, D.; Lorenzo, D.; Navarro, V.; Gutiérrez, C.; Caminal, J.M.; et al. Metabolic Activity of Liver Metastases May Predict Survival in Patients with Metastatic Uveal Melanoma. Ann. Oncol. 2020, 31, S672–S710. [Google Scholar]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-Cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring Tumour Purity and Stromal and Immune Cell Admixture from Expression Data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Sturm, G.; Finotello, F.; List, M. Immunedeconv: An R Package for Unified Access to Computational Methods for Estimating Immune Cell Fractions from Bulk RNA-Sequencing Data. Methods Mol. Biol. 2020, 2120, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Sánchez, A.; Cast, O.; Miller, M.L. Comprehensive Benchmarking and Integration of Tumor Microenvironment Cell Estimation Methods. Cancer Res. 2019, 79, 6238–6246. [Google Scholar] [CrossRef] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Schilling, B.; Liu, D.; Sucker, A.; Livingstone, E.; Jerby-Arnon, L.; Zimmer, L.; Gutzmer, R.; Satzger, I.; Loquai, C.; et al. Integrative Molecular and Clinical Modeling of Clinical Outcomes to PD1 Blockade in Patients with Metastatic Melanoma. Nat. Med. 2019, 25, 1916–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Entire Cohort n = 213 | GSE22138 n = 63 | GSE27831 n = 29 | GSE73652 n = 13 | GSE84976 n = 28 | TCGA n = 80 | p-Value |
|---|---|---|---|---|---|---|---|
| Age | 62.3 | 61.0 | 66.0 | 61.6 | 62.2 | 0.411 | |
| Sex | 0.801 | ||||||
| Female | 71 (41.3%) | 24 (38.1%) | 12 (41.4%) | 0 (0.0%) | 0 (0.0%) | 35 (43.8%) | |
| Male | 101 (58.7%) | 39 (61.9%) | 17 (58.6%) | 0 (0.0%) | 0 (0.00%) | 45 (56.2%) | |
| NA | 41 | 0 (0.0%) | 0 (0.0%) | 13 (100%) | 28 (100%) | 0 (0.0%) | |
| Chr 3 status | <0.001 | ||||||
| Disomy | 64 (32.2%) | 18 (28.6%) | 11 (37.9%) | 0 (0.0%) | 14 (50.0%) | 21 (26.2%) | |
| Partial monosomy | 5 (2.5%) | 5 (7.94%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | |
| Monosomy | 94 (47.2%) | 32 (50.8%) | 17 (58.6%) | 0 (0.0%) | 14 (50.0%) | 31 (38.8%) | |
| NA | 36 (18.1%) | 8 (12.7%) | 1 (3.5%) | 13 (100%) | 0 (0.0%) | 28 (35.0%) | |
| Cell type | <0.001 | ||||||
| Epithelioid | 40 (18.7%) | 21 (33.3%) | 6 (20.7%) | 0 (0.0%) | 0 (0.0%) | 13 (16.2%) | |
| Mixed | 72 (33.8%) | 23 (36.5%) | 12 (41.4%) | 0 (0.0%) | 0 (0.0%) | 37 (46.2%) | |
| Spindle | 39 (18.3%) | 0 (0.0%) | 9 (31%) | 0 (0.0%) | 0 (0.0%) | 30 (37.5%) | |
| NA | 60 (28.2%) | 19 (30.2%) | 0 (0.0%) | 13 (100%) | 28 (100%) | 0 (0.0%) | |
| Recurrence | 0.245 | ||||||
| Non-recurrent | 119 (55.9%) | 28 (44.4%) | 18 (62.1%) | 8 (61.5%) | 15 (53.6%) | 50 (62.5%) | |
| Recurrent | 94 (44.1%) | 35 (55.6%) | 11 (37.9%) | 5 (38.5%) | 13 (46.4%) | 30 (37.5%) | |
| DFS Months | 38.6 | 41.1 | 37.2 | 77.8 | 23.2 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Mulero, S.; Alonso, M.H.; del Carpio, L.P.; Sanz-Pamplona, R.; Piulats, J.M. Additive Role of Immune System Infiltration and Angiogenesis in Uveal Melanoma Progression. Int. J. Mol. Sci. 2021, 22, 2669. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052669
García-Mulero S, Alonso MH, del Carpio LP, Sanz-Pamplona R, Piulats JM. Additive Role of Immune System Infiltration and Angiogenesis in Uveal Melanoma Progression. International Journal of Molecular Sciences. 2021; 22(5):2669. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052669
Chicago/Turabian StyleGarcía-Mulero, Sandra, Maria Henar Alonso, Luis P. del Carpio, Rebeca Sanz-Pamplona, and Josep M. Piulats. 2021. "Additive Role of Immune System Infiltration and Angiogenesis in Uveal Melanoma Progression" International Journal of Molecular Sciences 22, no. 5: 2669. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052669