
Methamphetamine Blocks Adenosine A2A Receptor Activation via Sigma 1 and Cannabinoid CB1 Receptors

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
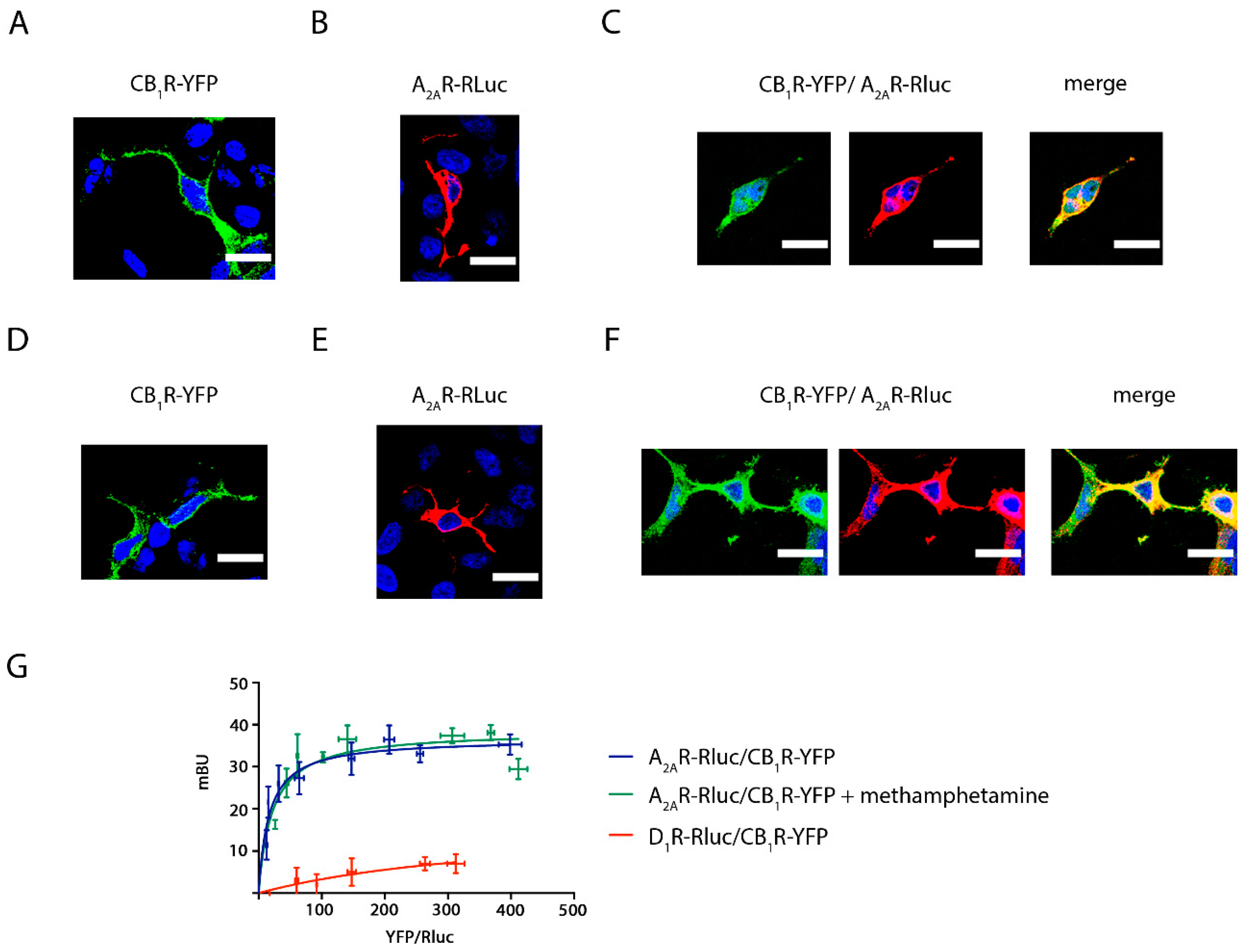
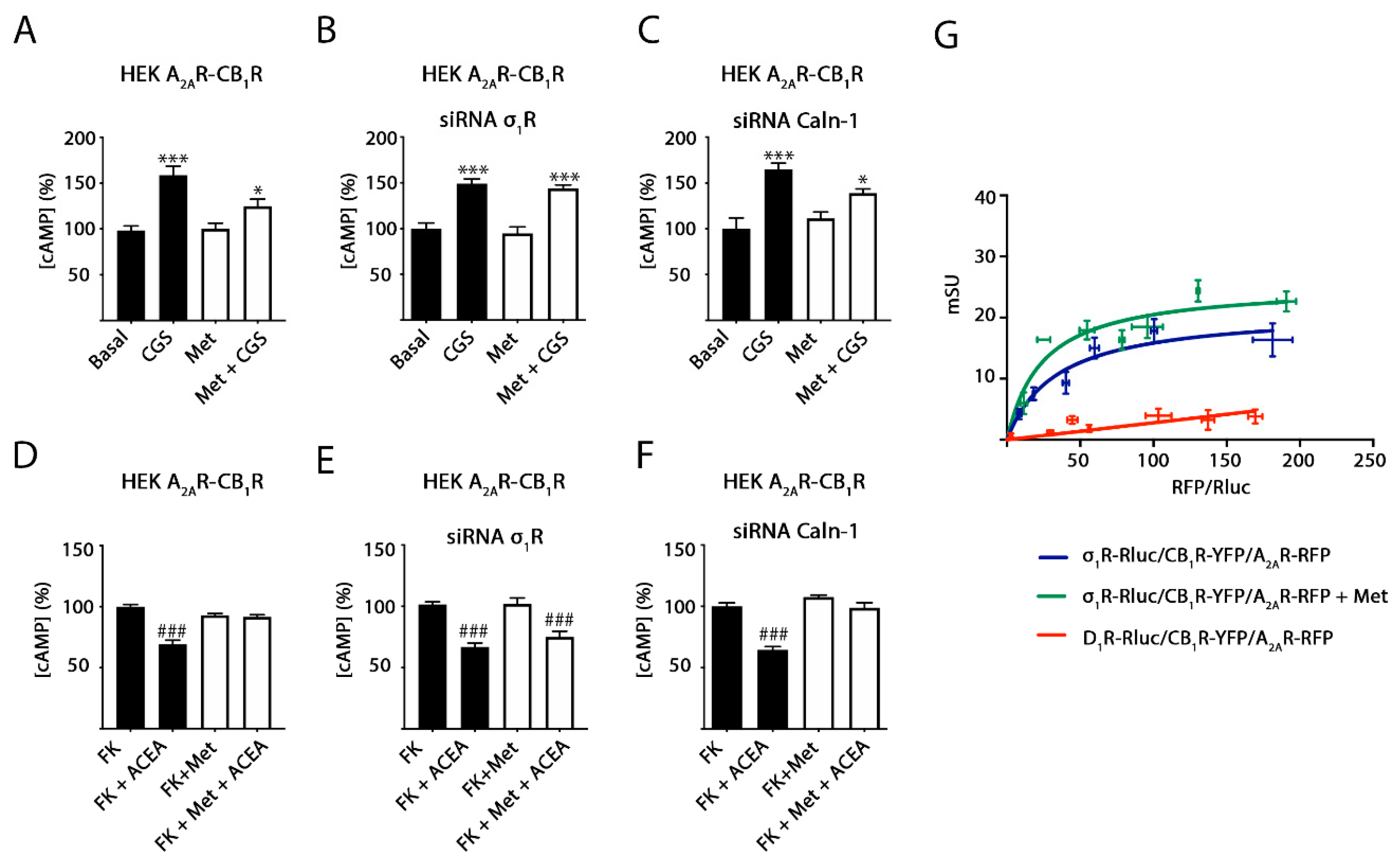
2.1. Methamphetamine Does Not Disrupt the Formation of A2A and CB1 Receptor Complexes in a Heterologous Expression System
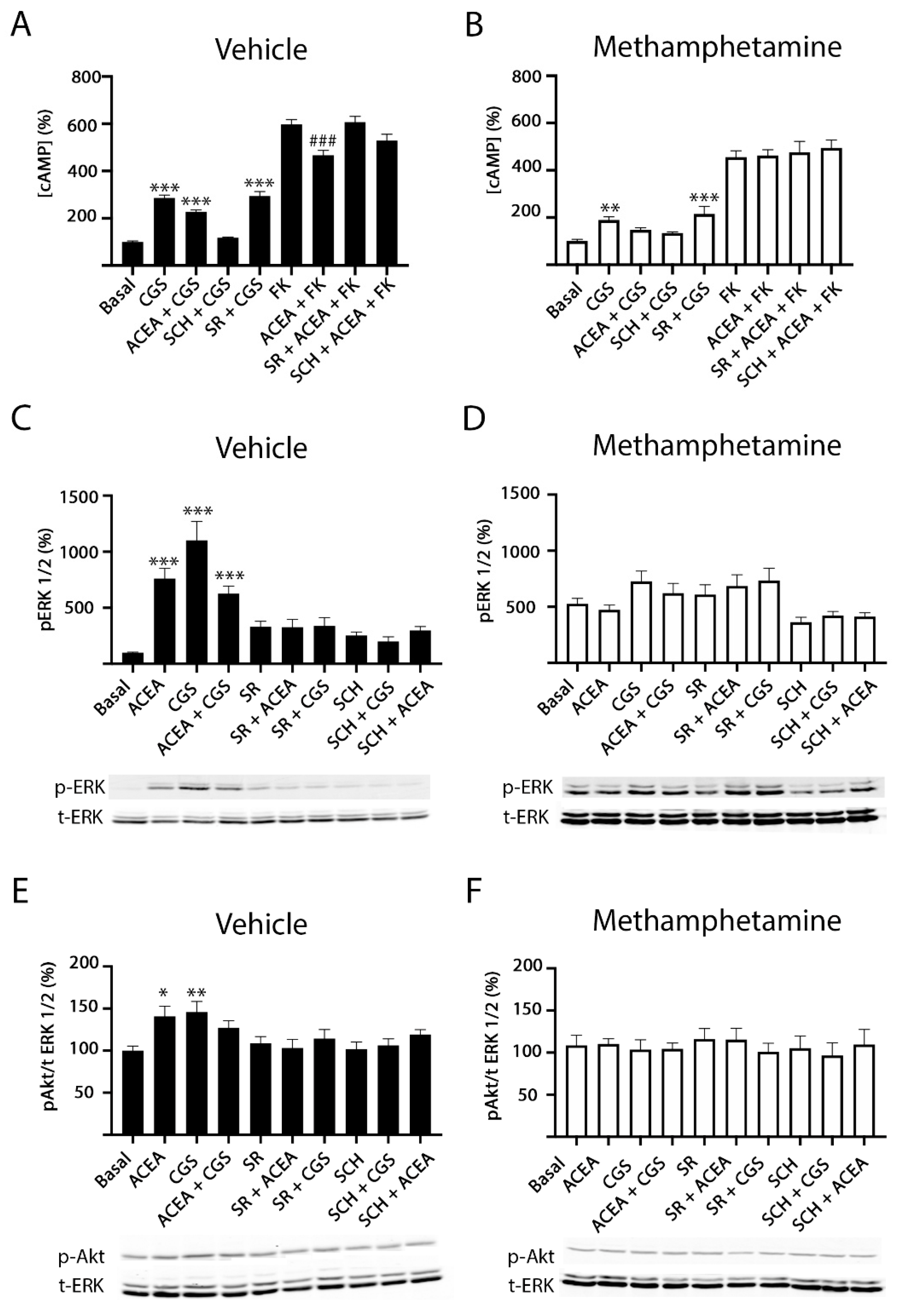
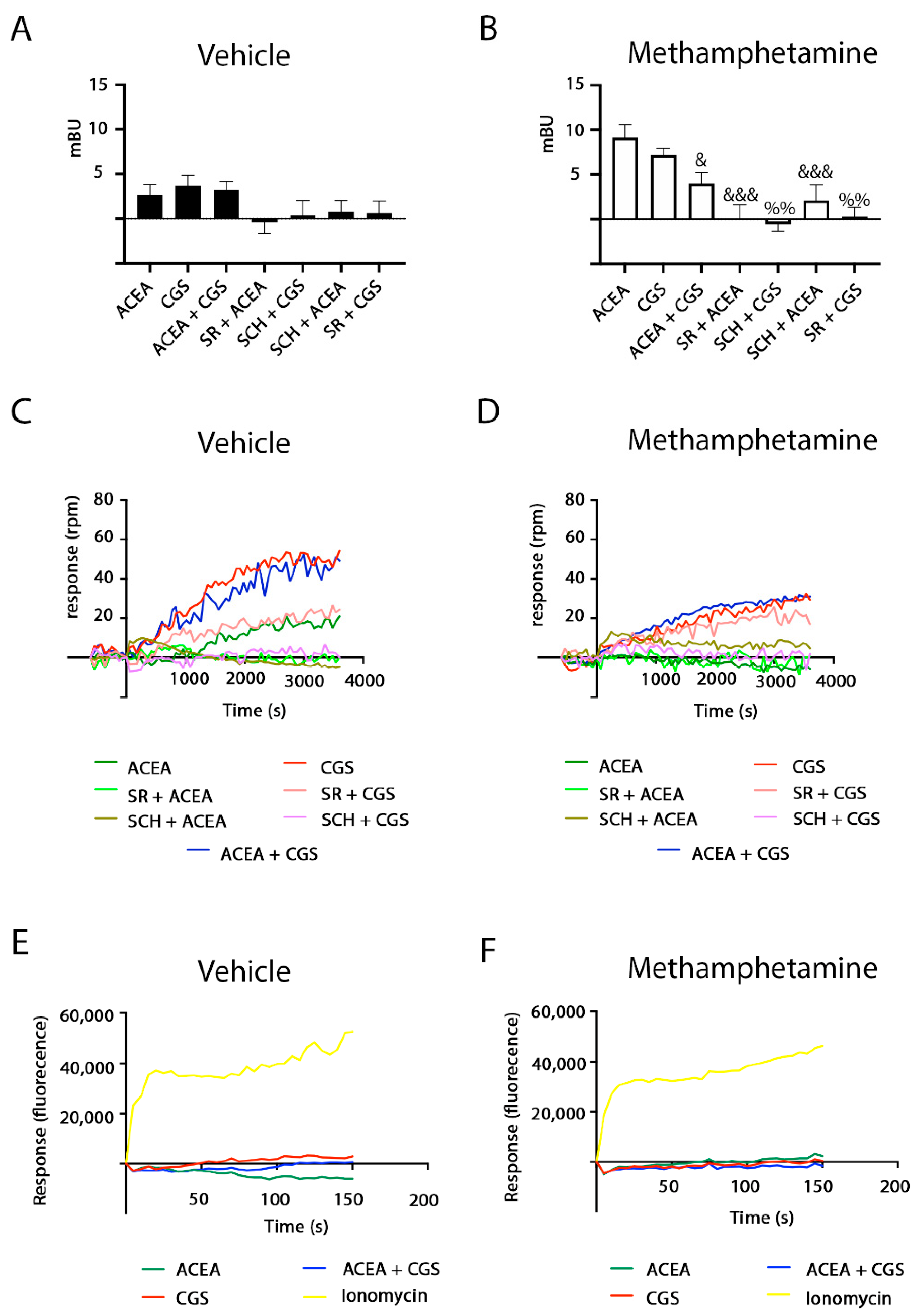
2.2. Methamphetamine Blocks CB1R Function in HEK-293T Cells Expressing the A2A–CB1Het
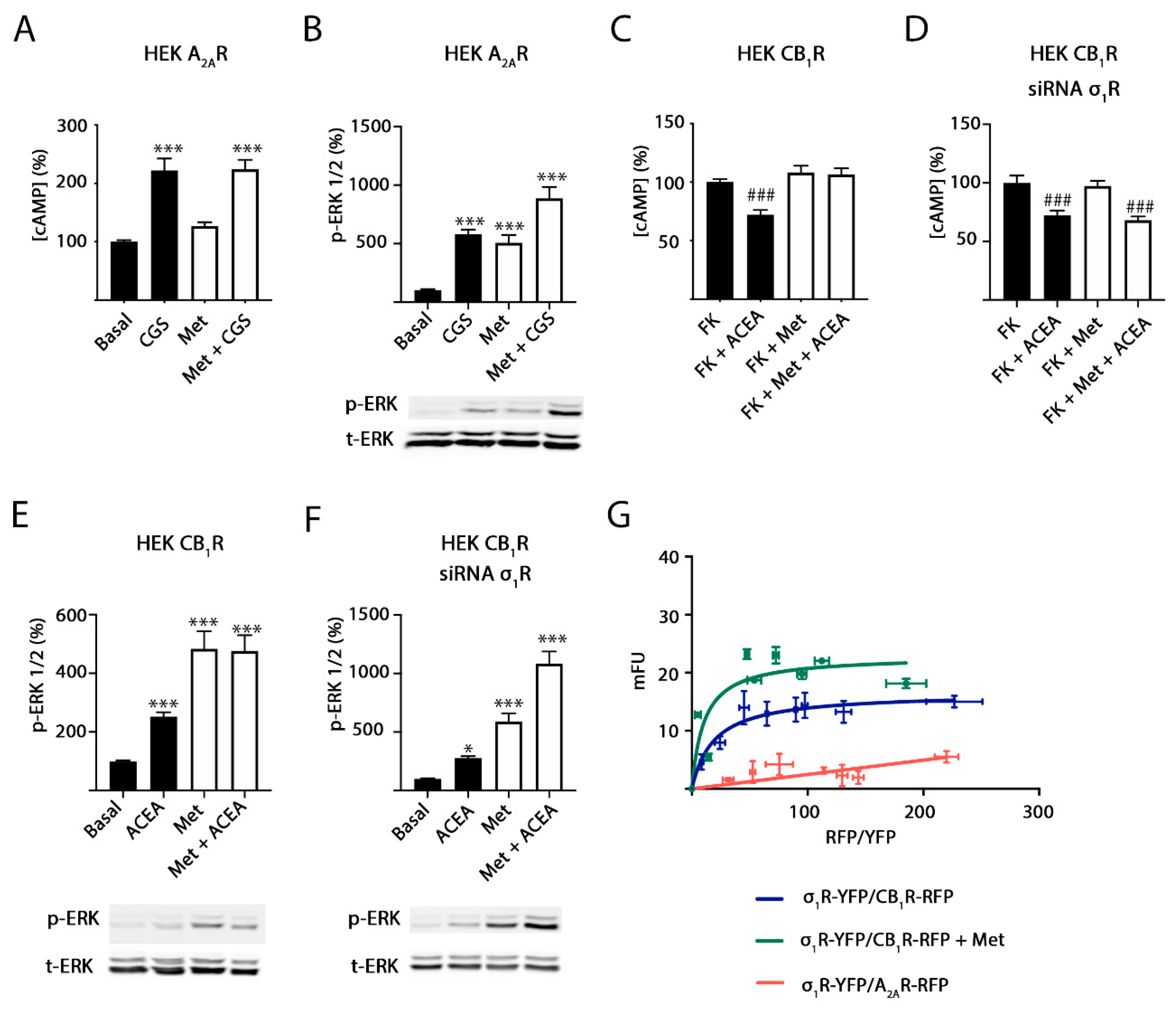
2.3. Methamphetamine Action in Cells Expressing A2A or CB1 Receptors
2.4. σ1 Receptor Involvement in Methamphetamine Action in Cells Expressing A2A–CB1Hets
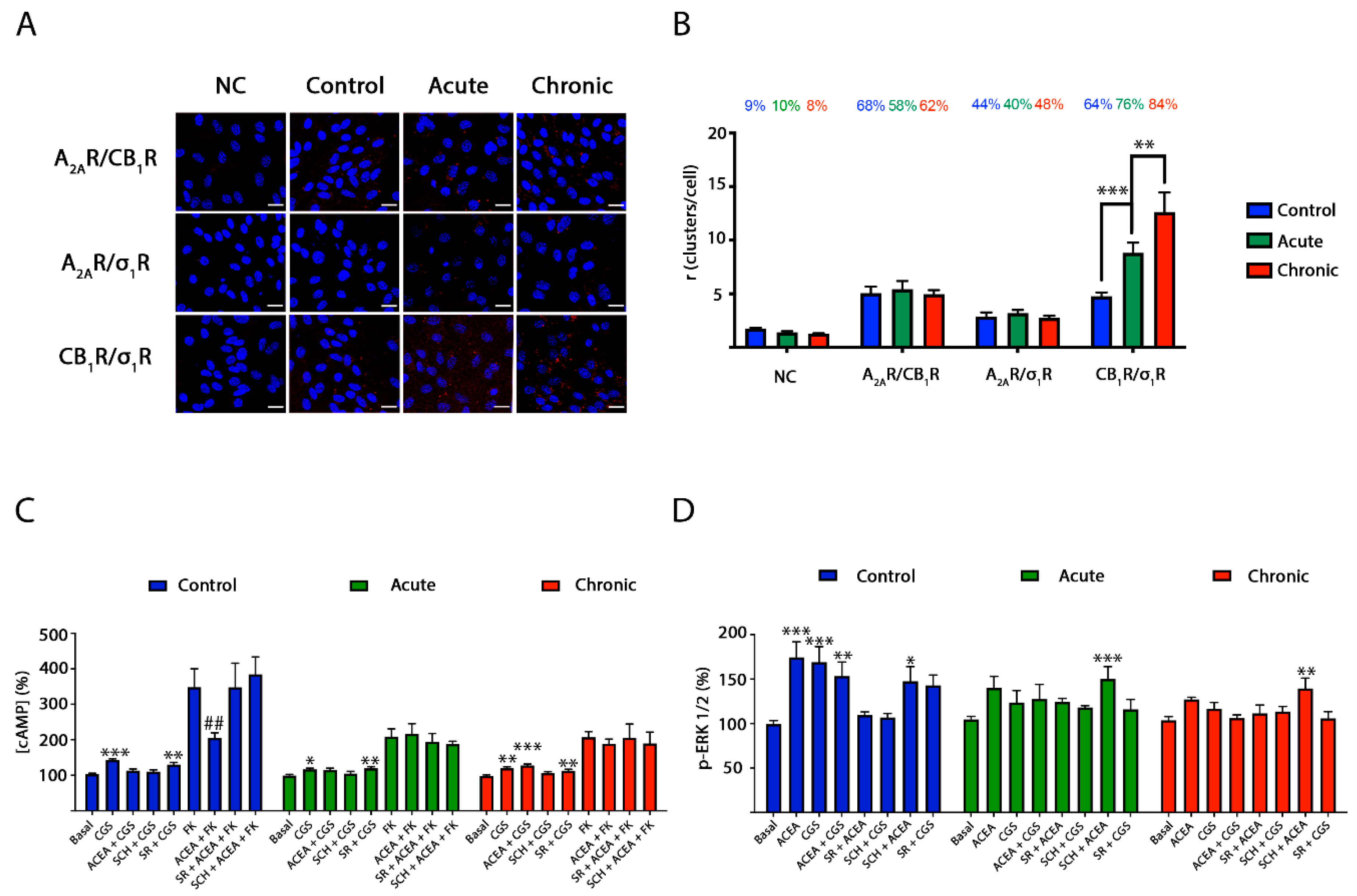
2.5. CB1R-σ1R Expression Is Altered When Striatal Primary Cultures of Neurons Are Treated with Methamphetamine
2.6. Blockade by Methamphetamine of CB1R–A2AR Complex Signaling in Striatal Neurons
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Expression Vectors
4.3. Cell Culture and Transfection
4.4. Immunocytochemistry
4.5. Resonance Energy Transfer Assays
4.6. Cytosolic cAMP Determination
4.7. Extracellular Signal-Regulated Kinase (ERK) and Protein Kinase B (Akt) Phosphorylation Assays
4.8. Dynamic Mass Redistribution (DMR) Assays
4.9. Determination of Cytoplasmic Calcium Ion Levels
4.10. ß-Arrestin 2 Recruitment
4.11. Proximity Ligation Assays (PLA)
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| A2AR | Adenosine A2A receptor |
| BRET | Bioluminescence Resonance Energy Transfer |
| BSA | Bovine Serum Albumin |
| CB1R | Cannabinoid CB1 receptor |
| CB2R | Cannabinoid CB2 receptor |
| CGS | CGS 21680 |
| CNS | Central nervous system |
| D1R | Dopamine D1 receptor |
| DMEM | Dulbeco’s Modified Eagle’s Medium |
| DMR | Dynamic Mass Redistribution |
| GPCR | G Protein-Coupled Receptor |
| FBS | Fetal Bovine Serum |
| FK | Forskolin |
| FRET | Fluorescence Resonance Energy Transfer |
| HTRF | Homogeneous time-resolved fluorescence energy transfer |
| Met | Methamphetamine |
| MAPK | Mitogen activated protein kinase |
| PBS | Phosphate-buffered saline |
| PLA | Proximity Ligation Assay |
| SCH | SCH 58261 |
| SR | SR 141716A |
| SRET | Sequential Resonance Energy Transfer |
| ∆9-THC | ∆9-Tetrahydrocannabinol |
| σ1R | Sigma 1 receptor |
References
- Oñatibia-Astibia, A.; Franco, R.; Martínez-Pinilla, E. Health benefits of methylxanthines in neurodegenerative diseases. Mol. Nutr. Food Res. 2017, 61, 1600670. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Oñatibia-Astibia, A.; Martínez-Pinilla, E. Health benefits of methylxanthines in cacao and chocolate. Nutrients 2013, 5, 4159–4173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskelinen, M.H.; Ngandu, T.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Midlife healthy-diet index and late-life dementia and Alzheimer’s disease. Dement. Geriatr. Cogn. Dis. Extra 2011, 1, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Sindi, S.; Kåreholt, I.; Eskelinen, M.; Hooshmand, B.; Lehtisalo, J.; Soininen, H.; Ngandu, T.; Kivipelto, M. Healthy dietary changes in midlife are associated with reduced dementia risk later in life. Nutrients 2018, 10, 1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskelinen, M.H.; Ngandu, T.; Tuomilehto, J.; Soininen, H.; Kivipelto, M. Midlife coffee and tea drinking and the risk of late-life dementia: A population-based CAIDE study. J. Alzheimer’s Dis. 2009, 16, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskelinen, M.H.; Kivipelto, M. Caffeine as a protective factor in dementia and Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, S167–S174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canas, P.M.; Porciuncula, L.O.; Cunha, G.M.A.; Silva, C.G.; Machado, N.J.; Oliveira, J.M.A.; Oliveira, C.R.; Cunha, R.A. Adenosine A2A Receptor Blockade Prevents Synaptotoxicity and Memory Dysfunction Caused by -Amyloid Peptides via p38 Mitogen-Activated Protein Kinase Pathway. J. Neurosci. 2009, 29, 14741–14751. [Google Scholar] [CrossRef]
- Nobre, H.V.; Cunha, G.M.D.A.; De Vasconcelos, L.M.; Magalhães, H.I.F.; Neto, R.N.O.; Maia, F.D.; De Moraes, M.O.; Leal, L.K.A.M.; Viana, G.S.D.B. Caffeine and CSC, adenosine A2A antagonists, offer neuroprotection against 6-OHDA-induced neurotoxicity in rat mesencephalic cells. Neurochem. Int. 2010, 56, 51–58. [Google Scholar] [CrossRef]
- Espinosa, J.; Rocha, A.; Nunes, F.; Costa, M.S.; Schein, V.; Kazlauckas, V.; Kalinine, E.; Souza, D.O.; Cunha, R.A.; Porciúncula, L.O. Caffeine consumption prevents memory impairment, neuronal damage, and adenosine A2A receptors upregulation in the hippocampus of a rat model of sporadic dementia. J. Alzheimer’s Dis. JAD 2013, 34, 509–518. [Google Scholar] [CrossRef]
- Carriba, P.; Ortiz, O.; Patkar, K.; Justinova, Z.; Stroik, J.; Themann, A.; Müller, C.; Woods, A.S.; Hope, B.T.; Ciruela, F.; et al. Striatal Adenosine A2A and Cannabinoid CB1 Receptors Form Functional Heteromeric Complexes that Mediate the Motor Effects of Cannabinoids. Neuropsychopharmacology 2007, 32, 2249–2259. [Google Scholar] [CrossRef] [Green Version]
- Navarro, G.; Carriba, P.; Gandía, J.; Ciruela, F.; Casadó, V.; Cortés, A.; Mallol, J.; Canela, E.I.; Lluis, C.; Franco, R.; et al. Detection of heteromers formed by cannabinoid CB1, dopamine D2, and adenosine A2A G-protein-coupled receptors by combining bimolecular fluorescence complementation and bioluminescence energy transfer. Sci. World J. 2008, 8, 1088–1097. [Google Scholar] [CrossRef] [Green Version]
- Carriba, P.; Navarro, G.; Ciruela, F.; Ferré, S.; Casadó, V.; Agnati, L.; Cortés, A.; Mallol, J.; Fuxe, K.; Canela, E.I.E.I.; et al. Detection of heteromerization of more than two proteins by sequential BRET-FRET. Nat. Methods 2008, 5, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Castelli, M.P.; Madeddu, C.; Casti, A.; Casu, A.; Casti, P.; Scherma, M.; Fattore, L.; Fadda, P.; Ennas, M.G. Δ9-Tetrahydrocannabinol prevents methamphetamine-induced neurotoxicity. PLoS ONE 2014, 9, e98079. [Google Scholar] [CrossRef]
- Chipana, C.; García-Ratés, S.; Camarasa, J.; Pubill, D.; Escubedo, E. Different oxidative profile and nicotinic receptor interaction of amphetamine and 3,4-methylenedioxy-methamphetamine. Neurochem. Int. 2008, 52, 401–410. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aso, E.; Fernández-Dueñas, V.; López-Cano, M.; Taura, J.; Watanabe, M.; Ferrer, I.; Luján, R.; Ciruela, F. Adenosine A2A-Cannabinoid CB1 Receptor Heteromers in the Hippocampus: Cannabidiol Blunts Δ9-Tetrahydrocannabinol-Induced Cognitive Impairment. Mol. Neurobiol. 2019, 56, 5382–5391. [Google Scholar] [CrossRef] [Green Version]
- Lever, J.R.; Fergason-Cantrell, E.A.; Watkinson, L.D.; Carmack, T.L.; Lord, S.A.; Xu, R.; Miller, D.K.; Lever, S.Z. Cocaine occupancy of sigma 1 receptors and dopamine transporters in mice. Synapse 2016, 70, 98–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romieu, P.; Martin-Fardon, R.; Maurice, T. Involvement of the sigma1 receptor in the cocaine-induced conditioned place preference. Neuroreport 2000, 11, 2885–2888. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T.; Romieu, P. Involvement of the sigma1 receptor in the appetitive effects of cocaine. Pharmacopsychiatry 2004, 37, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Aguinaga, D.; Medrano, M.; Vega-Quiroga, I.; Gysling, K.; Canela, E.I.; Navarro, G.; Franco, R. Cocaine effects on dopaminergic transmission depend on a balance between sigma-1 and sigma-2 receptor expression. Front. Mol. Neurosci. 2018, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Moreno, E.; Bonaventura, J.; Brugarolas, M.; Farré, D.; Aguinaga, D.; Mallol, J.; Cortés, A.; Casadó, V.; Lluís, C.; et al. Cocaine Inhibits Dopamine D2 Receptor Signaling via Sigma-1-D2 Receptor Heteromers. PLoS ONE 2013, 8, e61245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, E.C.; McCracken, K.A.; Liu, Y.; Pouw, B.; Matsumoto, R.R. Involvement of sigma (σ) receptors in the acute actions of methamphetamine: Receptor binding and behavioral studies. Neuropharmacology 2005, 49, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Ujike, H.; Kuroda, S.; Otsuki, S. sigma Receptor antagonists block the development of sensitization to cocaine. Eur. J. Pharmacol. 1996, 296, 123–128. [Google Scholar] [CrossRef]
- Katz, J.L.; Hiranita, T.; Hong, W.C.; Job, M.O.; McCurdy, C.R. A role for sigma receptors in stimulant self-administration and addiction. Handb. Exp. Pharmacol. 2017, 244, 177–218. [Google Scholar] [PubMed] [Green Version]
- Zhang, Y.; Lv, X.; Bai, Y.; Zhu, X.; Wu, X.; Chao, J.; Duan, M.; Buch, S.; Chen, L.; Yao, H. Involvement of sigma-1 receptor in astrocyte activation induced by methamphetamine via up-regulation of its own expression. J. Neuroinflamm. 2015, 12, 29. [Google Scholar] [CrossRef] [Green Version]
- Yasui, Y.; Su, T.P. Potential molecular mechanisms on the role of the sigma-1 receptor in the action of cocaine and methamphetamine. J. Drug Alcohol Res. 2016, 5, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundmann, M.; Kostenis, E. (Eds.) Holistic Methods for the Analysis of cNMP Effects. In Non-Canonical Cyclic Nucleotides; Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2015; Volume 238, pp. 339–357. [Google Scholar] [CrossRef]
- Hinz, S.; Navarro, G.; Borroto-Escuela, D.; Seibt, B.F.; Ammon, C.; De Filippo, E.; Danish, A.; Lacher, S.K.; Červinková, B.; Rafehi, M.; et al. Adenosine A2A receptor ligand recognition and signaling is blocked by A2B receptors. Oncotarget 2018, 9, 13593–13611. [Google Scholar] [CrossRef] [Green Version]
- Kenakin, T. A holistic view of GPCR signaling. Nat. Biotechnol. 2010, 28, 928–929. [Google Scholar] [CrossRef]
- Grundmann, M. Label-free dynamic mass redistribution and bio-impedance methods for drug discovery. Curr. Protoc. Pharmacol. 2017, 2017, 9.24.1–9.24.21. [Google Scholar] [CrossRef] [PubMed]
- Aguinaga, D.; Medrano, M.; Cordomí, A.; Jiménez-Rosés, M.; Angelats, E.; Casanovas, M.; Vega-Quiroga, I.; Canela, E.I.; Petrovic, M.; Gysling, K.; et al. Cocaine Blocks Effects of Hunger Hormone, Ghrelin, Via Interaction with Neuronal Sigma-1 Receptors. Mol. Neurobiol. 2019, 56, 1196–1210. [Google Scholar] [CrossRef] [Green Version]
- Aguinaga, D.; Casanovas, M.; Rivas-Santisteban, R.; Reyes-Resina, I.; Navarro, G.; Franco, R. The sigma-1 receptor as key common factor in cocaine and food seeking behaviors. J. Mol. Endocrinol. 2019, 63, R81–R92. [Google Scholar] [CrossRef] [PubMed]
- Itzhak, Y. Repeated methamphetamine-treatment alters brain δ receptors. Eur. J. Pharmacol. 1993, 230, 243–244. [Google Scholar] [CrossRef]
- Borroto-escuela, D.O.; Hagman, B.; Woolfenden, M.; Pinton, L.; Jiménez-beristain, A.; Ofl, J.; Narvaez, M.; Di Palma, M.; Feltmann, K.; Sartini, S.; et al. Receptor and Ion Channel Detection in the Brain. Neuromethods 2016, 110, 109–124. [Google Scholar]
- Borroto-Escuela, D.O.; Romero-Fernandez, W.; Pérez-Alea, M.; Narvaez, M.; Tarakanov, A.O.; Mudó, G.; Agnati, L.F.; Ciruela, F.; Belluardo, N.; Fuxe, K. The existence of FGFR1-5-HT1A receptor heterocomplexes in midbrain 5-HT neurons of the rat: Relevance for neuroplasticity. J. Neurosci. 2012, 32, 6295–6303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micale, V.; Mazzola, C.; Drago, F. Endocannabinoids and neurodegenerative diseases. Pharmacol. Res. 2007, 56, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-C.C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef] [Green Version]
- Alexander, S.P.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. The concise guide to pharmacology 2019/20: G protein-coupled receptors. Br. J. Pharmacol. 2019, 176, S21–S141. [Google Scholar] [CrossRef] [Green Version]
- Sam, A.H.; Salem, V.; Ghatei, M.A. Rimonabant: From RIO to Ban. J. Obes. 2011, 2011, 432607. [Google Scholar] [CrossRef]
- Rosin, D.L.; Robeva, A.; Woodard, R.L.; Guyenet, P.G.; Linden, J. Immunohistochemical Localization of Adenosine A 2A Receptors in the Rat Central Nervous System. J. Comp. Neurol. 1998, 401, 163–186. [Google Scholar] [CrossRef]
- Jenner, P.; Mori, A.; Hauser, R.; Morelli, M.; Fredholm, B.B.; Chen, J.F. Adenosine, adenosine A 2A antagonists, and Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Navarro, G.; Borroto-Escuela, D.O.D.O.; Fuxe, K.; Franco, R. Purinergic signaling in Parkinson’s disease. Relevance for treatment. Neuropharmacology 2015, 104, 161–168. [Google Scholar] [CrossRef]
- Kondo, T.; Mizuno, Y. Japanese Istradefylline Study Group A long-term study of istradefylline safety and efficacy in patients with Parkinson disease. Clin. Neuropharmacol. 2015, 38, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Kondo, T. Adenosine A2A receptor antagonist istradefylline reduces daily OFF time in Parkinson’s disease. Mov. Disord. 2013, 28, 1138–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinna, A.; Pontis, S.; Borsini, F.; Morelli, M. Adenosine A2A receptor antagonists improve deficits in initiation of movement and sensory motor integration in the unilateral 6-hydroxydopamine rat model of Parkinson’s disease. Synapse 2007, 61, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Simola, N.; Fenu, S.; Baraldi, P.G.; Tabrizi, M.A.; Morelli, M. Blockade of globus pallidus adenosine A2A receptors displays antiparkinsonian activity in 6-hydroxydopamine-lesioned rats treated with D 1 or D2 dopamine receptor agonists. Synapse 2008, 62, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Chu, U.B.; Ruoho, A.E. Biochemical Pharmacology of the Sigma-1 Receptor. Mol. Pharmacol. 2016, 89, 142–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alon, A.; Schmidt, H.; Zheng, S.; Kruse, A.C. Structural perspectives on sigma-1 receptor function. Adv. Exp. Med. Biol. 2017, 964, 5–13. [Google Scholar] [PubMed]
- Kruse, A. Structural Insights into Sigma1 Function. In Sigma Proteins: Evolution of the Concept of Sigma Receptors; Handbook of Experimental Pharmacology; Kim, F., Pasternak, G., Eds.; Springer: Cham, Switzerland, 2016; Volume 244, pp. 13–25. [Google Scholar] [CrossRef]
- Navarro, G.; Quiroz, C.; Moreno-Delgado, D.; Sierakowiak, A.; McDowell, K.; Moreno, E.; Rea, W.; Cai, N.-S.; Aguinaga, D.; Howell, L.A.; et al. Orexin-corticotropin-releasing factor receptor heteromers in the ventral tegmental area as targets for cocaine. J. Neurosci. 2015, 35, 6639–6653. [Google Scholar] [CrossRef] [Green Version]
- Ferré, S.; Baler, R.; Bouvier, M.; Caron, M.G.; Devi, L.A.; Durroux, T.; Fuxe, K.; George, S.R.; Javitch, J.A.; Lohse, M.J.; et al. Building a new conceptual framework for receptor heteromers. Nat. Chem. Biol. 2009, 5, 131–134. [Google Scholar] [CrossRef] [Green Version]
- Franco, R.; Canals, M.; Marcellino, D.; Ferré, S.; Agnati, L.; Mallol, J.; Casadó, V.; Ciruela, F.; Fuxe, K.; Lluis, C.; et al. Regulation of heptaspanning-membrane-receptor function by dimerization and clustering. Trends Biochem. Sci. 2003, 28, 238–243. [Google Scholar] [CrossRef]
- Ginés, S.; Ciruela, F.; Burgueño, J.; Casadó, V.; Canela, E.I.I.; Mallol, J.; Lluís, C.; Franco, R. Involvement of caveolin in ligand-induced recruitment and internalization of A(1) adenosine receptor and adenosine deaminase in an epithelial cell line. Mol. Pharmacol. 2001, 59, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Aguinaga, D.; Jiménez, J.; Lillo, J.; Martínez-Pinilla, E.; Navarro, G. Biased receptor functionality versus biased agonism in G-protein-coupled receptors. Biomol. Concepts 2018, 9, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Ferré, S.; Lluís, C.; Justinova, Z.; Quiroz, C.; Orru, M.; Navarro, G.; Canela, E.I.; Franco, R.; Goldberg, S.R. Adenosine-cannabinoid receptor interactions. Implications for striatal function. Br. J. Pharmacol. 2010, 160, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Tebano, M.T.; Martire, A.; Chiodi, V.; Pepponi, R.; Ferrante, A.; Domenici, M.R.; Frank, C.; Chen, J.-F.; Ledent, C.; Popoli, P. Adenosine A2A receptors enable the synaptic effects of cannabinoid CB1 receptors in the rodent striatum. J. Neurochem. 2009, 110, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Martire, A.; Tebano, M.T.; Chiodi, V.; Ferreira, S.G.; Cunha, R.A.; Köfalvi, A.; Popoli, P. Pre-synaptic adenosine A2A receptors control cannabinoid CB1 receptor-mediated inhibition of striatal glutamatergic neurotransmission. J. Neurochem. 2011, 116, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Chiodi, V.; Ferrante, A.; Ferraro, L.; Potenza, R.L.; Armida, M.; Beggiato, S.; Pèzzola, A.; Bader, M.; Fuxe, K.; Popoli, P.; et al. Striatal adenosine-cannabinoid receptor interactions in rats over-expressing adenosine A2A receptors. J. Neurochem. 2016, 136, 907–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The Role of PI3K/Akt and ERK in Neurodegenerative Disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef]
- Martínez-Pinilla, E.; Rodríguez-Pérez, A.I.I.; Navarro, G.; Aguinaga, D.; Moreno, E.; Lanciego, J.L.L.; Labandeira-García, J.L.L.; Franco, R. Dopamine D2 and angiotensin II type 1 receptors form functional heteromers in rat striatum. Biochem. Pharmacol. 2015, 96, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Hradsky, J.; Mikhaylova, M.; Karpova, A.; Kreutz, M.R.; Zuschratter, W. Super-resolution microscopy of the neuronal calcium-binding proteins Calneuron-1 and Caldendrin. Methods Mol. Biol. 2013, 963, 147–169. [Google Scholar] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casanovas, M.; Reyes-Resina, I.; Lillo, A.; Lillo, J.; López-Arnau, R.; Camarasa, J.; Escubedo, E.; Navarro, G.; Franco, R. Methamphetamine Blocks Adenosine A2A Receptor Activation via Sigma 1 and Cannabinoid CB1 Receptors. Int. J. Mol. Sci. 2021, 22, 2743. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052743
Casanovas M, Reyes-Resina I, Lillo A, Lillo J, López-Arnau R, Camarasa J, Escubedo E, Navarro G, Franco R. Methamphetamine Blocks Adenosine A2A Receptor Activation via Sigma 1 and Cannabinoid CB1 Receptors. International Journal of Molecular Sciences. 2021; 22(5):2743. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052743
Chicago/Turabian StyleCasanovas, Mireia, Irene Reyes-Resina, Alejandro Lillo, Jaume Lillo, Raul López-Arnau, Jorge Camarasa, Elena Escubedo, Gemma Navarro, and Rafael Franco. 2021. "Methamphetamine Blocks Adenosine A2A Receptor Activation via Sigma 1 and Cannabinoid CB1 Receptors" International Journal of Molecular Sciences 22, no. 5: 2743. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052743