
Lipidomics Provides New Insight into Pathogenesis and Therapeutic Targets of the Ischemia—Reperfusion Injury

 ,
,  ,
,  , ,
, ,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. Lipidomics
2.1. Lipid Transport Across Cell Membranes
2.2. The Role of Fatty Acids in Metabolic Pathways
2.3. Lipids as Signaling Molecules
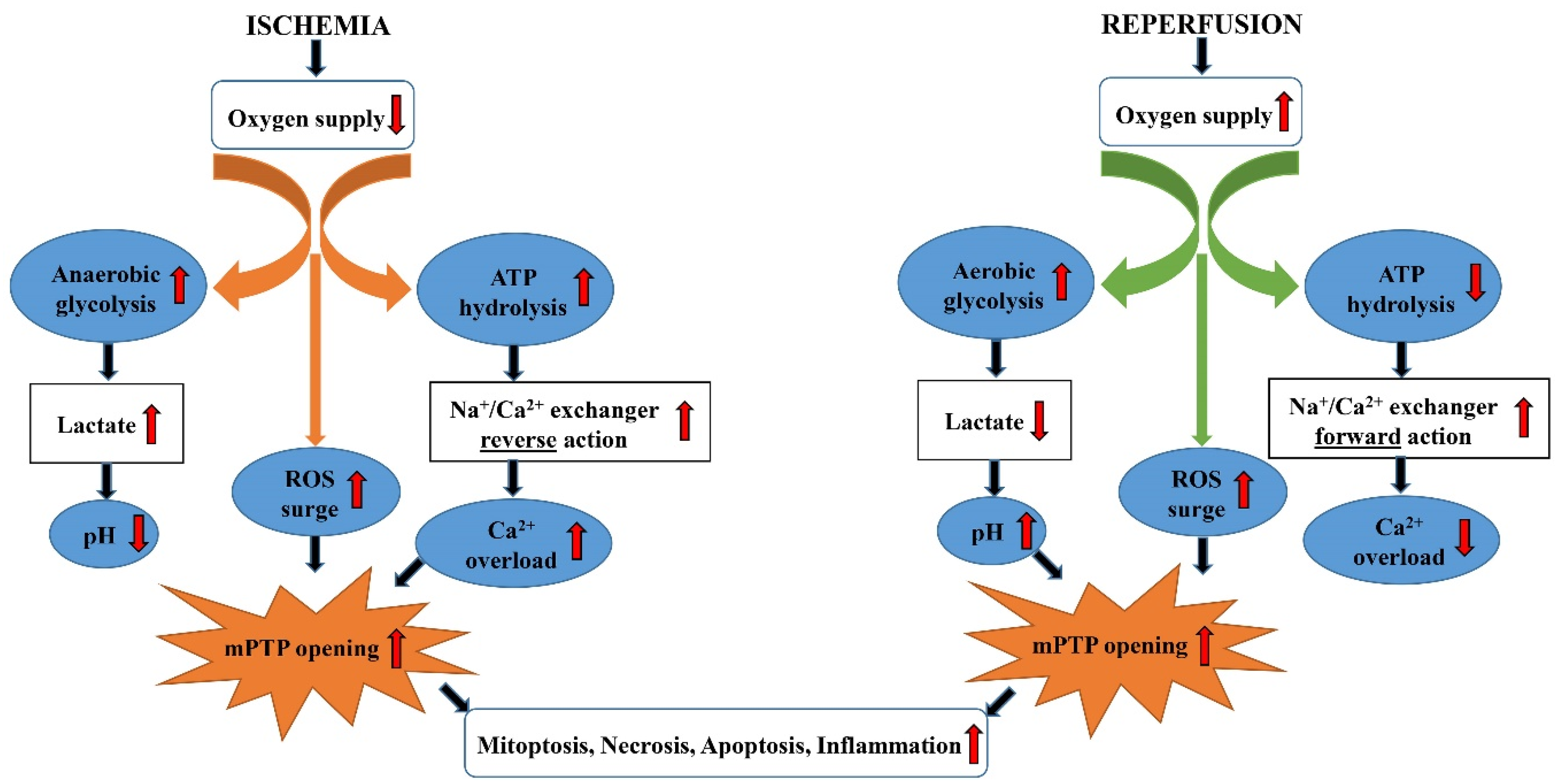
3. Lipid Metabolism in I/R Injury
4. Therapeutic Approach to I/R Injury-Related Lipotoxicity
- Reduction of the lipid overload in I/R injured tissues by increasing their catabolism and/or extraction;
- Transportation of lipids towards the adipose tissue;
- Blockade of the main pathways of FA-induced cell death.
5. Meldonium as the Therapeutic Approach in I/R Injury-Related Lipotoxicity
5.1. Meldonium as the Therapeutic Approach in I/R-Mediated Heart Injury
5.2. Meldonium as the Therapeutic Approach in I/R-Mediated Brain Injury
5.3. Meldonium as the Therapeutic Approach in I/R-Mediated Hepatic Injury
5.4. Meldonium as the Therapeutic Approach in I/R-Mediated Renal Injury
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Konishi, T.; Lentsch, A.B. Hepatic Ischemia/Reperfusion: Mechanisms of Tissue Injury, Repair, and Regeneration. Gene Expr. 2017, 17, 277–287. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Chen, S.; Li, S. The Na+/Ca2+ exchanger in cardiac ischemia/reperfusion injury. Med. Sci. Monit. 2012, 18, RA161–RA165. [Google Scholar] [CrossRef] [Green Version]
- Shoda, W.; Nomura, N.; Ando, F.; Tagashira, H.; Iwamoto, T.; Ohta, A.; Isobe, K.; Mori, T.; Susa, K.; Sohara, E.; et al. Sodium-calcium exchanger 1 is the key molecule for urinary potassium excretion against acute hyperkalemia. PLoS ONE 2020, 15, e0235360. [Google Scholar] [CrossRef]
- Liao, Q.S.; Du, Q.; Lou, J.; Xu, J.Y.; Xie, R. Roles of Na+/Ca2+ exchanger 1 in digestive system physiology and pathophysiology. World J. Gastroenterol. 2019, 25, 287–299. [Google Scholar] [CrossRef]
- Tanaka, H.; Shimada, H.; Namekata, I.; Kawanishi, T.; Iida-Tanaka, N.; Shigenobu, K. Involvement of the Na+/Ca2+ exchanger in ouabain-induced inotropy and arrhythmogenesis in guinea-pig myocardium as revealed by SEA0400. J. Pharmacol. Sci. 2007, 103, 241–246. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.R.; Piacentino, V., 3rd; Houser, S.R.; Bers, D.M. Dynamic regulation of sodium/calcium exchange function in human heart failure. Circulation 2003, 108, 2224–2229. [Google Scholar] [CrossRef] [Green Version]
- Carroll, J.; He, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase. Proc. Natl. Acad. Sci. USA 2019, 116, 12816–12821. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Tang, D.; Kang, R. Oxidative stress-mediated HMGB1 biology. Front. Physiol. 2015, 6, 93. [Google Scholar] [CrossRef] [Green Version]
- Durasevic, S.; Stojkovic, M.; Bogdanovic, L.; Pavlovic, S.; Borkovic-Mitic, S.; Grigorov, I.; Bogojevic, D.; Jasnic, N.; Tosti, T.; Durovic, S.; et al. The Effects of Meldonium on the Renal Acute Ischemia/Reperfusion Injury in Rats. Int. J. Mol. Sci. 2019, 20, 5747. [Google Scholar] [CrossRef] [Green Version]
- Durasevic, S.; Stojkovic, M.; Sopta, J.; Pavlovic, S.; Borkovic-Mitic, S.; Ivanovic, A.; Jasnic, N.; Tosti, T.; Durovic, S.; Dordevic, J.; et al. The effects of meldonium on the acute ischemia/reperfusion liver injury in rats. Sci. Rep. 2021, 11, 1305. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.A.; Kwak, M.K. The Nrf2 system as a potential target for the development of indirect antioxidants. Molecules 2010, 15, 7266–7291. [Google Scholar] [CrossRef] [Green Version]
- Lane, K.; Dixon, J.J.; MacPhee, I.A.; Philips, B.J. Renohepatic crosstalk: Does acute kidney injury cause liver dysfunction? Nephrol. Dial. Transplant. 2013, 28, 1634–1647. [Google Scholar] [CrossRef] [Green Version]
- Bonora, M.; Pinton, P. The mitochondrial permeability transition pore and cancer: Molecular mechanisms involved in cell death. Front. Oncol. 2014, 4, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narita, M.; Shimizu, S.; Ito, T.; Chittenden, T.; Lutz, R.J.; Matsuda, H.; Tsujimoto, Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 14681–14686. [Google Scholar] [CrossRef] [Green Version]
- McCafferty, K.; Forbes, S.; Thiemermann, C.; Yaqoob, M.M. The challenge of translating ischemic conditioning from animal models to humans: The role of comorbidities. Dis. Model. Mech. 2014, 7, 1321–1333. [Google Scholar] [CrossRef] [Green Version]
- Todorović, Z.; Prostran, M.; Nešić, Z.; Stojanović, R.; Stojanov, M. The influence of drugs on biochemical markers of ischemia-reperfusion injury. In The Analysis of Pharmacologically Active Compounds in Biomolecules in Real Samples; Injac, R., Karljiković-Rajić, K., Štrukelj, B., Eds.; Transworld Research Network: Kerala, India, 2009; pp. 217–238. [Google Scholar]
- Chatterjee, P.K. Novel pharmacological approaches to the treatment of renal ischemia-reperfusion injury: A comprehensive review. Naunyn Schmiedebergs Arch. Pharmacol. 2007, 376, 1–43. [Google Scholar] [CrossRef] [PubMed]
- Todorovic, Z.; Medic, B.; Basta-Jovanovic, G.; Radojevic Skodric, S.; Stojanovic, R.; Rovcanin, B.; Prostran, M. Acute pretreatment with chloroquine attenuates renal I/R injury in rats. PLoS ONE 2014, 9, e92673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, F.L.; Patel, N.S.A.; Purvis, G.S.D.; Chiazza, F.; Chen, J.; Sordi, R.; Hache, G.; Merezhko, V.V.; Collino, M.; Yaqoob, M.M.; et al. Inhibition of IkappaB Kinase at 24 Hours After Acute Kidney Injury Improves Recovery of Renal Function and Attenuates Fibrosis. J. Am. Heart Assoc. 2017, 6, e005092. [Google Scholar] [CrossRef]
- Todorovic, Z.; Nesic, Z.; Stojanovic, R.; Basta-Jovanovic, G.; Radojevic-Skodric, S.; Velickovic, R.; Chatterjee, P.K.; Thiemermann, C.; Prostran, M. Acute protective effects of simvastatin in the rat model of renal ischemia-reperfusion injury: It is never too late for the pretreatment. J. Pharmacol. Sci. 2008, 107, 465–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, W.J.; Yutuc, E.; Davies, D.; Dickson, A.; Angelini, R.; El Assad, D.; Frache, G.; Wang, Y. Chapter 1: Lipidomics Basics. In Lipidomics: Current and Emerging Techniques; Griffiths, W., Wang, Y., Eds.; Royal Society of Chemistry: London, UK, 2020; pp. 1–24. [Google Scholar]
- Zullig, T.; Trotzmuller, M.; Kofeler, H.C. Lipidomics from sample preparation to data analysis: A primer. Anal. Bioanal. Chem. 2020, 412, 2191–2209. [Google Scholar] [CrossRef] [Green Version]
- Hyotylainen, T.; Oresic, M. Systems biology strategies to study lipidomes in health and disease. Prog. Lipid Res. 2014, 55, 43–60. [Google Scholar] [CrossRef]
- Simon, N.; Hertig, A. Alteration of Fatty Acid Oxidation in Tubular Epithelial Cells: From Acute Kidney Injury to Renal Fibrogenesis. Front. Med. 2015, 2, 52. [Google Scholar] [CrossRef] [Green Version]
- Erpicum, P.; Rowart, P.; Defraigne, J.O.; Krzesinski, J.M.; Jouret, F. What we need to know about lipid-associated injury in case of renal ischemia-reperfusion. Am. J. Physiol. Renal Physiol. 2018, 315, F1714–F1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.C.; Stahl, A.; Zager, R.A. Triglyceride accumulation in injured renal tubular cells: Alterations in both synthetic and catabolic pathways. Kidney Int. 2005, 67, 2196–2209. [Google Scholar] [CrossRef] [Green Version]
- Cases, S.; Smith, S.J.; Zheng, Y.W.; Myers, H.M.; Lear, S.R.; Sande, E.; Novak, S.; Collins, C.; Welch, C.B.; Lusis, A.J.; et al. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 1998, 95, 13018–13023. [Google Scholar] [CrossRef] [Green Version]
- Shu, H.; Peng, Y.; Hang, W.; Nie, J.; Zhou, N.; Wang, D.W. The role of CD36 in cardiovascular disease. Cardiovasc. Res. 2020, cvaa319. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Stahl, A. SLC27 fatty acid transport proteins. Mol. Aspects Med. 2013, 34, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Black, P.N.; Ahowesso, C.; Montefusco, D.; Saini, N.; DiRusso, C.C. Fatty Acid Transport Proteins: Targeting FATP2 as a Gatekeeper Involved in the Transport of Exogenous Fatty Acids. Medchemcomm 2016, 7, 612–622. [Google Scholar] [CrossRef] [Green Version]
- Furuhashi, M.; Hotamisligil, G.S. Fatty acid-binding proteins: Role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 2008, 7, 489–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smathers, R.L.; Petersen, D.R. The human fatty acid-binding protein family: Evolutionary divergences and functions. Hum. Genom. 2011, 5, 170–191. [Google Scholar] [CrossRef]
- Yang, X.L.; Mi, J.H.; Dong, Q. FABP4 alleviates endoplasmic reticulum stress-mediated ischemia-reperfusion injury in PC12 cells via regulation of PPARgamma. Exp. Ther. Med. 2021, 21, 181. [Google Scholar] [CrossRef]
- Huang, R.; Shi, M.; Guo, F.; Feng, Y.; Feng, Y.; Liu, J.; Li, L.; Liang, Y.; Xiang, J.; Lei, S.; et al. Pharmacological Inhibition of Fatty Acid-Binding Protein 4 (FABP4) Protects Against Rhabdomyolysis-Induced Acute Kidney Injury. Front. Pharmacol. 2018, 9, 917. [Google Scholar] [CrossRef]
- Hu, B.; Guo, Y.; Garbacz, W.G.; Jiang, M.; Xu, M.; Huang, H.; Tsung, A.; Billiar, T.R.; Ramakrishnan, S.K.; Shah, Y.M.; et al. Fatty acid binding protein-4 (FABP4) is a hypoxia inducible gene that sensitizes mice to liver ischemia/reperfusion injury. J. Hepatol. 2015, 63, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Doran, A.C.; Yurdagul, A., Jr.; Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2020, 20, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; Le Bloc’h, J.; Siliart, B.; Dumon, H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.R.; Sobhi, H.F. Advances in Fatty Acid Analysis for Clinical Investigation and Diagnosis using GC/MS Methodology. J. Biochem. Analyt. Stud. 2017, 3. [Google Scholar] [CrossRef]
- Montero Bullón, J.F.; Melo, T.; Martins-Marques, T.; Girão, H.; Domingues, R.; Domingues, P. P 129—Alteration of phospholipidome profile in the heart of an animal model of acute myocardial infarction. Free Radic. Biol. Med. 2017, 108, S61. [Google Scholar] [CrossRef]
- Kirac, E.; Ozcan, F.; Tuzcu, H.; Elpek, G.O.; Aslan, M. Analysis of polyunsaturated fatty acids and the omega-6 inflammatory pathway in hepatic ischemia/re-perfusion injury. Mol. Med. Rep. 2015, 12, 4149–4156. [Google Scholar] [CrossRef] [Green Version]
- Adibhatla, R.M.; Hatcher, J.F.; Dempsey, R.J. Lipids and lipidomics in brain injury and diseases. AAPS J. 2006, 8, E314–E321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Xie, C.; Zheng, J.; Dong, Q.; Si, T.; Zhang, J.; Hou, S.T. An imbalanced ratio between PC(16:0/16:0) and LPC(16:0) revealed by lipidomics supports the role of the Lands’ cycle in ischemic brain injury. J. Biol. Chem. 2020, 296, 100151. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Gallucci, G.M.; Lin, A.; Tong, M.; Chen, X.; Stonestreet, B.S. Critical Shifts in Cerebral White Matter Lipid Profiles After Ischemic–Reperfusion Brain Injury in Fetal Sheep as Demonstrated by the Positive Ion Mode MALDI-Mass Spectrometry. Cell Med. 2020, 12, 2155179019897002. [Google Scholar] [CrossRef]
- Bagchi, A.K.; Surendran, A.; Malik, A.; Jassal, D.S.; Ravandi, A.; Singal, P.K. IL-10 attenuates OxPCs-mediated lipid metabolic responses in ischemia reperfusion injury. Sci. Rep. 2020, 10, 12120. [Google Scholar] [CrossRef]
- Feng, L.; Yang, J.; Liu, W.; Wang, Q.; Wang, H.; Shi, L.; Fu, L.; Xu, Q.; Wang, B.; Li, T. Lipid Biomarkers in Acute Myocardial Infarction Before and After Percutaneous Coronary Intervention by Lipidomics Analysis. Med. Sci. Monit. 2018, 24, 4175–4182. [Google Scholar] [CrossRef]
- Zhang, D.X.; Fryer, R.M.; Hsu, A.K.; Zou, A.P.; Gross, G.J.; Campbell, W.B.; Li, P.L. Production and metabolism of ceramide in normal and ischemic-reperfused myocardium of rats. Basic Res. Cardiol. 2001, 96, 267–274. [Google Scholar] [CrossRef]
- He, X.; Schuchman, E.H. Ceramide and Ischemia/Reperfusion Injury. J. Lipids 2018, 2018, 3646725. [Google Scholar] [CrossRef] [Green Version]
- Ueda, N. Ceramide-induced apoptosis in renal tubular cells: A role of mitochondria and sphingosine-1-phoshate. Int. J. Mol. Sci. 2015, 16, 5076–5124. [Google Scholar] [CrossRef] [Green Version]
- Skotland, T.; Sandvig, K.; Llorente, A. Lipids in exosomes: Current knowledge and the way forward. Prog. Lipid Res. 2017, 66, 30–41. [Google Scholar] [CrossRef]
- Djurasevic, S.; Todorovic, Z.; Pavlovic, S.; Pejic, S. Chapter 27—Cadmium and Fullerenes in Liver Diseases. In Dietary Interventions in Liver Disease; Watson, R.R., Preedy, V.R., Eds.; Elsevier Academic Press: Cambridge, MA, USA, 2019; pp. 333–344. [Google Scholar] [CrossRef]
- Zager, R.A.; Johnson, A.C.; Becker, K. Acute unilateral ischemic renal injury induces progressive renal inflammation, lipid accumulation, histone modification, and “end-stage” kidney disease. Am. J. Physiol. Renal Physiol. 2011, 301, F1334–F1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zager, R.A.; Johnson, A.C.; Hanson, S.Y. Renal tubular triglyercide accumulation following endotoxic, toxic, and ischemic injury. Kidney Int. 2005, 67, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Hauck, A.K.; Bernlohr, D.A. Oxidative stress and lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Kim, K.M.; Cho, S.S.; Ki, S.H. Emerging roles of ferroptosis in liver pathophysiology. Arch. Pharm. Res. 2020, 43, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Nesic, Z.; Todorovic, Z.; Stojanovic, R.; Basta-Jovanovic, G.; Radojevic-Skodric, S.; Velickovic, R.; Chatterjee, P.K.; Thiemermann, C.; Prostran, M. Single-dose intravenous simvastatin treatment attenuates renal injury in an experimental model of ischemia-reperfusion in the rat. J. Pharmacol. Sci. 2006, 102, 413–417. [Google Scholar] [CrossRef] [Green Version]
- Grygiel-Gorniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivarajah, A.; Chatterjee, P.K.; Hattori, Y.; Brown, P.A.; Stewart, K.N.; Todorovic, Z.; Mota-Filipe, H.; Thiemermann, C. Agonists of peroxisome-proliferator activated receptor-alpha (clofibrate and WY14643) reduce renal ischemia/reperfusion injury in the rat. Med. Sci. Monit. 2002, 8, BR532–BR539. [Google Scholar] [PubMed]
- Zhou, Y.; Du, D.; Liu, S.; Zhao, M.; Yuan, Y.; Li, L.; Chen, Y.; Lu, Y.; Cheng, J.; Liu, J. Polyacetylene glycoside attenuates ischemic kidney injury by co-inhibiting inflammation, mitochondria dysfunction and lipotoxicity. Life Sci. 2018, 204, 55–64. [Google Scholar] [CrossRef]
- Alvarez-Guardia, D.; Palomer, X.; Coll, T.; Serrano, L.; Rodriguez-Calvo, R.; Davidson, M.M.; Merlos, M.; El Kochairi, I.; Michalik, L.; Wahli, W.; et al. PPARbeta/delta activation blocks lipid-induced inflammatory pathways in mouse heart and human cardiac cells. Biochim. Biophys. Acta 2011, 1811, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Wepler, M.; Hafner, S.; Scheuerle, A.; Reize, M.; Groger, M.; Wagner, F.; Simon, F.; Matallo, J.; Gottschalch, F.; Seifritz, A.; et al. Effects of the PPAR-beta/delta agonist GW0742 during resuscitated porcine septic shock. Intensive Care Med. Exp. 2013, 1, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collino, M.; Benetti, E.; Miglio, G.; Castiglia, S.; Rosa, A.C.; Aragno, M.; Thiemermann, C.; Fantozzi, R. Peroxisome proliferator-activated receptor beta/delta agonism protects the kidney against ischemia/reperfusion injury in diabetic rats. Free Radic. Biol. Med. 2011, 50, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Shintani, Y.; Collino, M.; Osuchowski, M.F.; Busch, D.; Patel, N.S.; Sepodes, B.; Castiglia, S.; Fantozzi, R.; Bishop-Bailey, D.; et al. Protective role of peroxisome proliferator-activated receptor-beta/delta in septic shock. Am. J. Respir. Crit. Care Med. 2010, 182, 1506–1515. [Google Scholar] [CrossRef]
- Sivarajah, A.; Chatterjee, P.K.; Patel, N.S.; Todorovic, Z.; Hattori, Y.; Brown, P.A.; Stewart, K.N.; Mota-Filipe, H.; Cuzzocrea, S.; Thiemermann, C. Agonists of peroxisome-proliferator activated receptor-gamma reduce renal ischemia/reperfusion injury. Am. J. Nephrol. 2003, 23, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Mister, M.; Noris, M.; Szymczuk, J.; Azzollini, N.; Aiello, S.; Abbate, M.; Trochimowicz, L.; Gagliardini, E.; Arduini, A.; Perico, N.; et al. Propionyl-L-carnitine prevents renal function deterioration due to ischemia/reperfusion. Kidney Int. 2002, 61, 1064–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjakste, N.; Gutcaits, A.; Kalvinsh, I. Mildronate: An antiischemic drug for neurological indications. CNS Drug Rev. 2005, 11, 151–168. [Google Scholar] [CrossRef]
- Porter, C.; Constantin-Teodosiu, D.; Constantin, D.; Leighton, B.; Poucher, S.M.; Greenhaff, P.L. Muscle carnitine availability plays a central role in regulating fuel metabolism in the rodent. J. Physiol. 2017, 595, 5765–5780. [Google Scholar] [CrossRef]
- Dambrova, M.; Makrecka-Kuka, M.; Vilskersts, R.; Makarova, E.; Kuka, J.; Liepinsh, E. Pharmacological effects of meldonium: Biochemical mechanisms and biomarkers of cardiometabolic activity. Pharmacol. Res. 2016, 113, 771–780. [Google Scholar] [CrossRef]
- Speijer, D.; Manjeri, G.R.; Szklarczyk, R. How to deal with oxygen radicals stemming from mitochondrial fatty acid oxidation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonfeld, P.; Wojtczak, L. Fatty acids as modulators of the cellular production of reactive oxygen species. Free Radic. Biol. Med. 2008, 45, 231–241. [Google Scholar] [CrossRef]
- Schonfeld, P.; Wojtczak, L. Short- and medium-chain fatty acids in energy metabolism: The cellular perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef] [Green Version]
- Seifert, E.L.; Estey, C.; Xuan, J.Y.; Harper, M.E. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J. Biol. Chem. 2010, 285, 5748–5758. [Google Scholar] [CrossRef] [Green Version]
- Berlato, D.G.; Baiross, A.V. Meldonium: Pharmacological, toxicological, and analytical aspects. Toxicol. Res. Appl. 2020, 4, 2397847320915143. [Google Scholar] [CrossRef]
- Dambrova, M.; Liepinsh, E.; Kalvinsh, I. Mildronate: Cardioprotective Action through Carnitine-Lowering Effect. Trends Cardiovasc. Med. 2002, 12, 275–279. [Google Scholar] [CrossRef]
- Sokolovska, J.; Isajevs, S.; Sugoka, O.; Sharipova, J.; Lauberte, L.; Svirina, D.; Rostoka, E.; Sjakste, T.; Kalvinsh, I.; Sjakste, N. Correction of glycaemia and GLUT1 level by mildronate in rat streptozotocin diabetes mellitus model. Cell Biochem. Funct. 2011, 29, 55–63. [Google Scholar] [CrossRef]
- Kuwajima, M.; Harashima, H.; Hayashi, M.; Ise, S.; Sei, M.; Lu, K.; Kiwada, H.; Sugiyama, Y.; Shima, K. Pharmacokinetic analysis of the cardioprotective effect of 3-(2,2, 2-trimethylhydrazinium) propionate in mice: Inhibition of carnitine transport in kidney. J. Pharmacol. Exp. Ther. 1999, 289, 93–102. [Google Scholar] [PubMed]
- Nechaeva, G.I.; Zheltikova, E.N. Effects of Meldonium in Early Postmyocardial Infarction Period. Kardiologiia 2015, 55, 35–42. [Google Scholar] [CrossRef]
- Statsenko, M.E.; Belenkova, S.V.; Sporova, O.E.; Shilina, N.N. The use of mildronate in combined therapy of postinfarction chronic heart failure in patients with type 2 diabetes mellitus. Klin. Med. 2007, 85, 39–42. [Google Scholar]
- Nedoshivin, A.O.; Petrova, N.N.; Kutuzova, A.E.; Perepech, N.B. Effect of mildronate on life quality of patients with chronic heart failure. Ter. Arkh. 1999, 71, 10–12. [Google Scholar] [PubMed]
- Kuka, J. The Regulation of Carnitine System for Cardioprotection; Rīga Stradiņš University: Riga, Latvia, 2011. [Google Scholar]
- Liepinsh, E.; Vilskersts, R.; Skapare, E.; Svalbe, B.; Kuka, J.; Cirule, H.; Pugovics, O.; Kalvinsh, I.; Dambrova, M. Mildronate decreases carnitine availability and up-regulates glucose uptake and related gene expression in the mouse heart. Life Sci. 2008, 83, 613–619. [Google Scholar] [CrossRef]
- Kuka, J.; Vilskersts, R.; Cirule, H.; Makrecka, M.; Pugovics, O.; Kalvinsh, I.; Dambrova, M.; Liepinsh, E. The cardioprotective effect of mildronate is diminished after co-treatment with L-carnitine. J. Cardiovasc. Pharmacol. Ther. 2012, 17, 215–222. [Google Scholar] [CrossRef]
- Liepinsh, E.; Vilskersts, R.; Loca, D.; Kirjanova, O.; Pugovichs, O.; Kalvinsh, I.; Dambrova, M. Mildronate, an inhibitor of carnitine biosynthesis, induces an increase in gamma-butyrobetaine contents and cardioprotection in isolated rat heart infarction. J. Cardiovasc. Pharmacol. 2006, 48, 314–319. [Google Scholar] [CrossRef]
- Hayashi, Y.; Muranaka, Y.; Kirimoto, T.; Asaka, N.; Miyake, H.; Matsuura, N. Effects of MET-88, a gamma-butyrobetaine hydroxylase inhibitor, on tissue carnitine and lipid levels in rats. Biol. Pharm. Bull. 2000, 23, 770–773. [Google Scholar] [CrossRef] [Green Version]
- Liepinsh, E.; Vilskersts, R.; Zvejniece, L.; Svalbe, B.; Skapare, E.; Kuka, J.; Cirule, H.; Grinberga, S.; Kalvinsh, I.; Dambrova, M. Protective effects of mildronate in an experimental model of type 2 diabetes in Goto-Kakizaki rats. Br. J. Pharmacol. 2009, 157, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Klusa, V.; Beitnere, U.; Pupure, J.; Isajevs, S.; Rumaks, J.; Svirskis, S.; Dzirkale, Z.; Kalvinsh, I. Mildronate and its neuroregulatory mechanisms: Targeting the mitochondria, neuroinflammation, and protein expression. Medicina 2013, 49, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Isajevs, S.; Isajeva, D.; Beitnere, U.; Jansone, B.; Kalvinsh, I.; Klusa, V. Mildronate as a regulator of protein expression in a rat model of Parkinson’s disease. Medicina 2011, 47, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Klusa, V.Z.; Isajevs, S.; Svirina, D.; Pupure, J.; Beitnere, U.; Rumaks, J.; Svirskis, S.; Jansone, B.; Dzirkale, Z.; Muceniece, R.; et al. Neuroprotective properties of mildronate, a small molecule, in a rat model of Parkinson’s disease. Int. J. Mol. Sci. 2010, 11, 4465–4487. [Google Scholar] [CrossRef] [PubMed]
- Pupure, J.; Isajevs, S.; Skapare, E.; Rumaks, J.; Svirskis, S.; Svirina, D.; Kalvinsh, I.; Klusa, V. Neuroprotective properties of mildronate, a mitochondria-targeted small molecule. Neurosci. Lett. 2010, 470, 100–105. [Google Scholar] [CrossRef]
- Rumaks, J.; Pupure, J.; Svirskis, S.; Isajevs, S.; Duburs, G.; Kalvinsh, I.; Klusa, V. Search for stroke-protecting agents in endothelin-1-induced ischemic stroke model in rats. Medicina 2012, 48, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Sokolovska, J.; Rumaks, J.; Karajeva, N.; Grinvalde, D.; Shapirova, J.; Klusa, V.; Kalvinsh, I.; Sjakste, N. The influence of mildronate on peripheral neuropathy and some characteristics of glucose and lipid metabolism in rat streptozotocin-induced diabetes mellitus model. Biomed. Khim. 2011, 57, 490–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klusa, V.; Muceniece, R.; Isajevs, S.; Isajeva, D.; Beitnere, U.; Mandrika, I.; Pupure, J.; Rumaks, J.; Jansone, B.; Kalvinsh, I.; et al. Mildronate enhances learning/memory and changes hippocampal protein expression in trained rats. Pharmacol. Biochem. Behav. 2013, 106, 68–76. [Google Scholar] [CrossRef]
- Schobersberger, W.; Dunnwald, T.; Gmeiner, G.; Blank, C. Story behind meldonium-from pharmacology to performance enhancement: A narrative review. Br. J. Sports Med. 2017, 51, 22–25. [Google Scholar] [CrossRef]
- Svalbe, B.; Zvejniece, L.; Vavers, E.; Pugovics, O.; Muceniece, R.; Liepinsh, E.; Dambrova, M. Mildronate treatment improves functional recovery following middle cerebral artery occlusion in rats. Behav. Brain Res. 2011, 222, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Makrecka, M.; Svalbe, B.; Volska, K.; Sevostjanovs, E.; Liepins, J.; Grinberga, S.; Pugovics, O.; Liepinsh, E.; Dambrova, M. Mildronate, the inhibitor of L-carnitine transport, induces brain mitochondrial uncoupling and protects against anoxia-reoxygenation. Eur. J. Pharmacol. 2014, 723, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Ribas, G.S.; Vargas, C.R.; Wajner, M. L-carnitine supplementation as a potential antioxidant therapy for inherited neurometabolic disorders. Gene 2014, 533, 469–476. [Google Scholar] [CrossRef]
- Suslina, Z.A.; Fedorova, T.N.; Maksimova, M.; Kim, E.K. Antioxidant activity of mildronate and L-carnitine in the treatment of patients with cerebrovascular diseases. Eksp. Klin. Farmakol. 2003, 66, 32–35. [Google Scholar] [PubMed]
- Vinichuk, S.M. The efficacy of the mildronate treatment of patients with ischemic stroke. Vrach. Delo 1991, 7, 77–79. [Google Scholar]
- Dziak, L.A.; Golik, V.A. Use of mildronate for the treatment of patients with circulatory encephalopathy against a background of stenosis of major arteries of the head. Lik. Sprava 2003, 5–6, 98–101. [Google Scholar]
- Abeuov, B.A.; Raimkulov, B.N.; Mitrokhin, D.A.; Nurzhanova, R.B.; Esenbekov, K.A.; Esmuratov, M.E.; Orsarieva, K.A.; Kudaibergenova, A.S. Condition of the higher brain functions in patients with dyscirculatory encephalopathy treated with mildronate. Meditsina 2004, 2, 78–81. [Google Scholar]
- Vetra, A.; Shefere, M.; Skarda, I.; Matveja, L.; Kalvinsh, I.S. Significance of mildronate for improvement of results of early rehabilitation results of neurological patients. Latv. Arstu Zurnals 1999, 12, 33–37. [Google Scholar]
- Muslin, V.P.; Pohorielov, O.V. Meldonium and neuroprotection. Theory, experiment and clinical practice. Med. Perspekt. 2018, 23, 131–137. [Google Scholar] [CrossRef]
- Soares, R.O.S.; Losada, D.M.; Jordani, M.C.; Evora, P.; Castro, E.S.O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. Int. J. Mol. Sci. 2019, 20, 5034. [Google Scholar] [CrossRef] [Green Version]
- op den Dries, S.; Karimian, N.; Sutton, M.E.; Westerkamp, A.C.; Nijsten, M.W.; Gouw, A.S.; Wiersema-Buist, J.; Lisman, T.; Leuvenink, H.G.; Porte, R.J. Ex vivo normothermic machine perfusion and viability testing of discarded human donor livers. Am. J. Transplant. 2013, 13, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Fingas, C.D.; Beste, M.; Penndorf, V.; Sydor, S.; Nadalin, S.; Bechmann, L.; Paul, A.; Gerken, G.; Canbay, A.; Jochum, C. Liver Regeneration-Related Cytokine Profiles in Donors and Recipients Before and After Living-Donor Liver Transplant. Exp. Clin. Transplant. 2018, 16, 554–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orci, L.A.; Toso, C.; Mentha, G.; Morel, P.; Majno, P.E. Systematic review and meta-analysis of the effect of perioperative steroids on ischaemia-reperfusion injury and surgical stress response in patients undergoing liver resection. Br. J. Surg. 2013, 100, 600–609. [Google Scholar] [CrossRef]
- Aliakbarian, M.; Nikeghbalian, S.; Ghaffaripour, S.; Bahreini, A.; Shafiee, M.; Rashidi, M.; Rajabnejad, Y. Effects of N-Acetylcysteine Addition to University of Wisconsin Solution on the Rate of Ischemia-Reperfusion Injury in Adult Orthotopic Liver Transplant. Exp. Clin. Transplant. 2017, 15, 432–436. [Google Scholar] [CrossRef]
- Klune, J.R.; Dhupar, R.; Cardinal, J.; Billiar, T.R.; Tsung, A. HMGB1: Endogenous danger signaling. Mol. Med. 2008, 14, 476–484. [Google Scholar] [CrossRef]
- Chien, C.T.; Chang, T.C.; Tsai, C.Y.; Shyue, S.K.; Lai, M.K. Adenovirus-mediated bcl-2 gene transfer inhibits renal ischemia/reperfusion induced tubular oxidative stress and apoptosis. Am. J. Transplant. 2005, 5, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yenari, M.A.; Cheng, D.; Sapolsky, R.M.; Steinberg, G.K. Bcl-2 overexpression protects against neuron loss within the ischemic margin following experimental stroke and inhibits cytochrome c translocation and caspase-3 activity. J. Neurochem. 2003, 85, 1026–1036. [Google Scholar] [CrossRef]
- Zahedi, K.; Barone, S.; Soleimani, M. Polyamine Catabolism in Acute Kidney. Int. J. Mol. Sci. 2019, 19, 4790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Liepinsh, E.; Kuka, J.; Svalbe, B.; Vilskersts, R.; Skapare, E.; Cirule, H.; Pugovics, O.; Kalvinsh, I.; Dambrova, M. Effects of long-term mildronate treatment on cardiac and liver functions in rats. Basic Clin. Pharmacol. Toxicol. 2009, 105, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Lipid Categories | |
|---|---|
| 01. Fatty Acyls [FA] [FA01] Fatty Acids and Conjugates [FA02] Octadecanoids [FA03] Eicosanoids [FA04] Docosanoids [FA05] Fatty alcohols [FA06] Fatty aldehydes [FA07] Fatty esters [FA08] Fatty amides [FA09] Fatty nitriles [FA10] Fatty ethers [FA11] Hydrocarbons [FA12] Oxygenated hydrocarbons [FA13] Fatty acyl glycosides [FA00] Other Fatty Acyls 02. Glycerolipids [GL] [GL01] Monoradylglycerols [GL02] Diradylglycerols [GL03] Triradylglycerols [GL04] Glycosylmonoradylglycerols [GL05] Glycosyldiradylglycerols [GL00] Other Glycerolipids 03. Glycerophospholipids [GP] [GP01] Glycerophosphocholines [GP02] Glycerophosphoethanolamines [GP03] Glycerophosphoserines [GP04] Glycerophosphoglycerols [GP05] Glycerophosphoglycerophosphates [GP06] Glycerophosphoinositols [GP07] Glycerophosphoinositol monophosphates [GP08] Glycerophosphoinositol bisphosphates [GP09] Glycerophosphoinositol trisphosphates [GP10] Glycerophosphates [GP11] Glyceropyrophosphates [GP12] Glycerophosphoglycerophosphoglycerols [GP13] CDP-Glycerols [GP14] Glycosylglycerophospholipids [GP15] Glycerophosphoinositolglycans [GP16] Glycerophosphonocholines [GP17] Glycerophosphonoethanolamines [GP18] Di-glycerol tetraether phospholipids [GP19] Glycerol-nonitol tetraether phospholipids [GP20] Oxidized glycerophospholipids [GP00] Other Glycerophospholipids | 04. Sphingolipids [SP] [SP01] Sphingoid bases [SP02] Ceramides [SP03] Phosphosphingolipids [SP04] Phosphonosphingolipids [SP05] Neutral glycosphingolipids [SP06] Acidic glycosphingolipids [SP07] Basic glycosphingolipids [SP08] Amphoteric glycosphingolipids [SP09] Arsenosphingolipids [SP00] Other Sphingolipids 05. Sterol Lipids [ST] [ST01] Sterols [ST02] Steroids [ST03] Secosteroids [ST04] Bile acids and derivatives [ST05] Steroid conjugates [ST00] Other Sterol lipids 06. Prenol Lipids [PR] [PR01] Isoprenoids [PR02] Quinones and hydroquinones [PR03] Polyprenols [PR04] Hopanoids [PR00] Other Prenol lipids 07. Saccharolipids [SL] [SL01] Acylaminosugars [SL02] Acylaminosugar glycans [SL03] Acyltrehaloses [SL04] Acyltrehalose glycans [SL05] Other acyl sugars [SL00] Other Saccharolipids 08. Polyketides [PK] [PK01] Linear polyketides [PK02] Halogenated acetogenins [PK03] Annonaceae acetogenins [PK04] Macrolides and lactone polyketides [PK05] Ansamycins and related polyketides [PK06] Polyenes [PK07] Linear tetracyclines [PK08] Angucyclines [PK09] Polyether antibiotics [PK10] Aflatoxins and related substances [PK11] Cytochalasins [PK12] Flavonoids [PK13] Aromatic polyketides [PK14] Non-ribosomal peptide/polyketide hybrids [PK15] Phenolic lipids [PK00] Other Polyketides |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todorović, Z.; Đurašević, S.; Stojković, M.; Grigorov, I.; Pavlović, S.; Jasnić, N.; Tosti, T.; Macut, J.B.; Thiemermann, C.; Đorđević, J. Lipidomics Provides New Insight into Pathogenesis and Therapeutic Targets of the Ischemia—Reperfusion Injury. Int. J. Mol. Sci. 2021, 22, 2798. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062798
Todorović Z, Đurašević S, Stojković M, Grigorov I, Pavlović S, Jasnić N, Tosti T, Macut JB, Thiemermann C, Đorđević J. Lipidomics Provides New Insight into Pathogenesis and Therapeutic Targets of the Ischemia—Reperfusion Injury. International Journal of Molecular Sciences. 2021; 22(6):2798. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062798
Chicago/Turabian StyleTodorović, Zoran, Siniša Đurašević, Maja Stojković, Ilijana Grigorov, Slađan Pavlović, Nebojša Jasnić, Tomislav Tosti, Jelica Bjekić Macut, Christoph Thiemermann, and Jelena Đorđević. 2021. "Lipidomics Provides New Insight into Pathogenesis and Therapeutic Targets of the Ischemia—Reperfusion Injury" International Journal of Molecular Sciences 22, no. 6: 2798. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062798