
JNK and p38 Inhibitors Prevent Transforming Growth Factor-β1-Induced Myofibroblast Transdifferentiation in Human Graves’ Orbital Fibroblasts


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
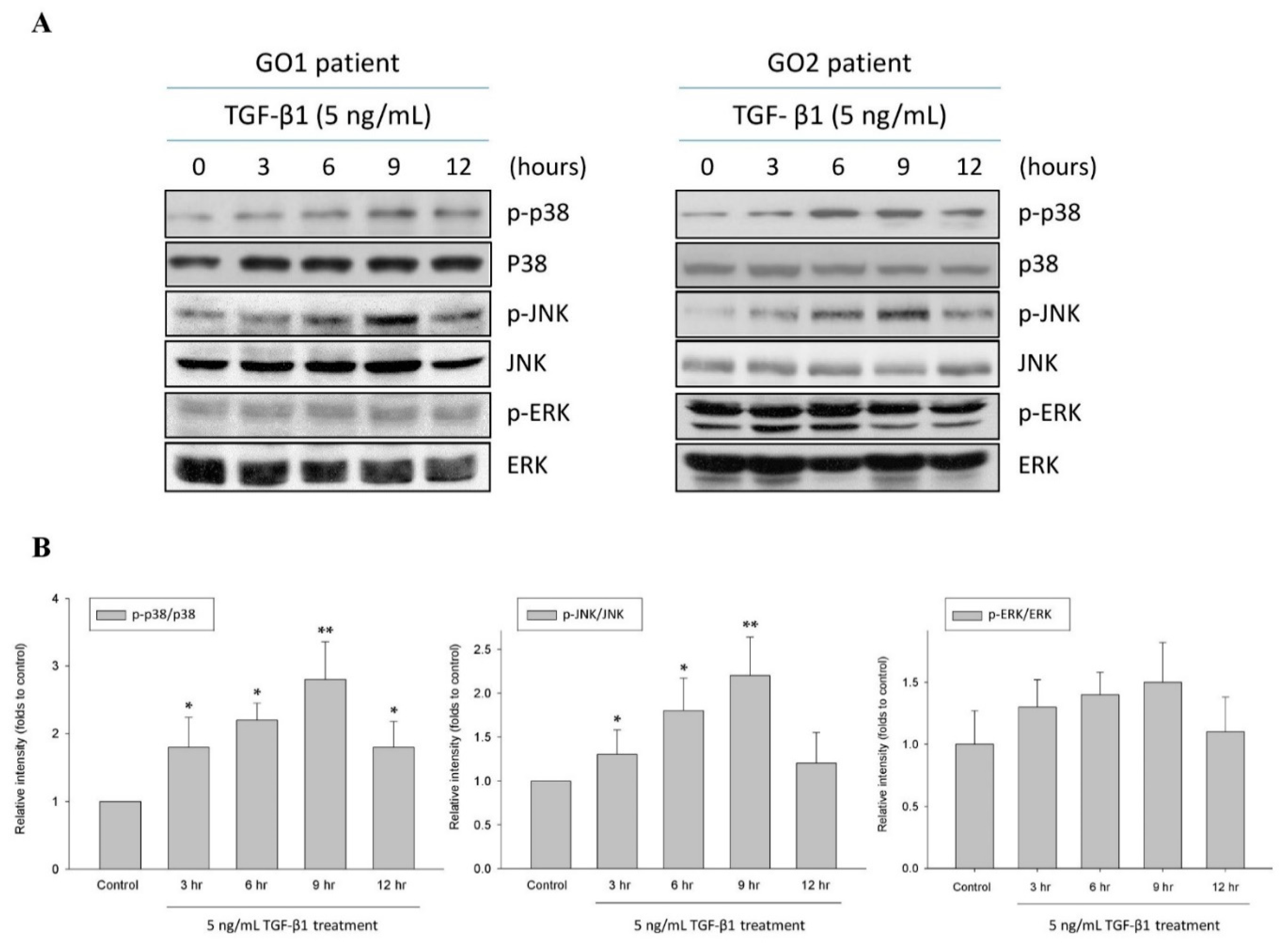
2.1. TGF-β1 Upregulated p38 and JNK Phosphorylation in Human Graves’ Orbital Fibroblasts
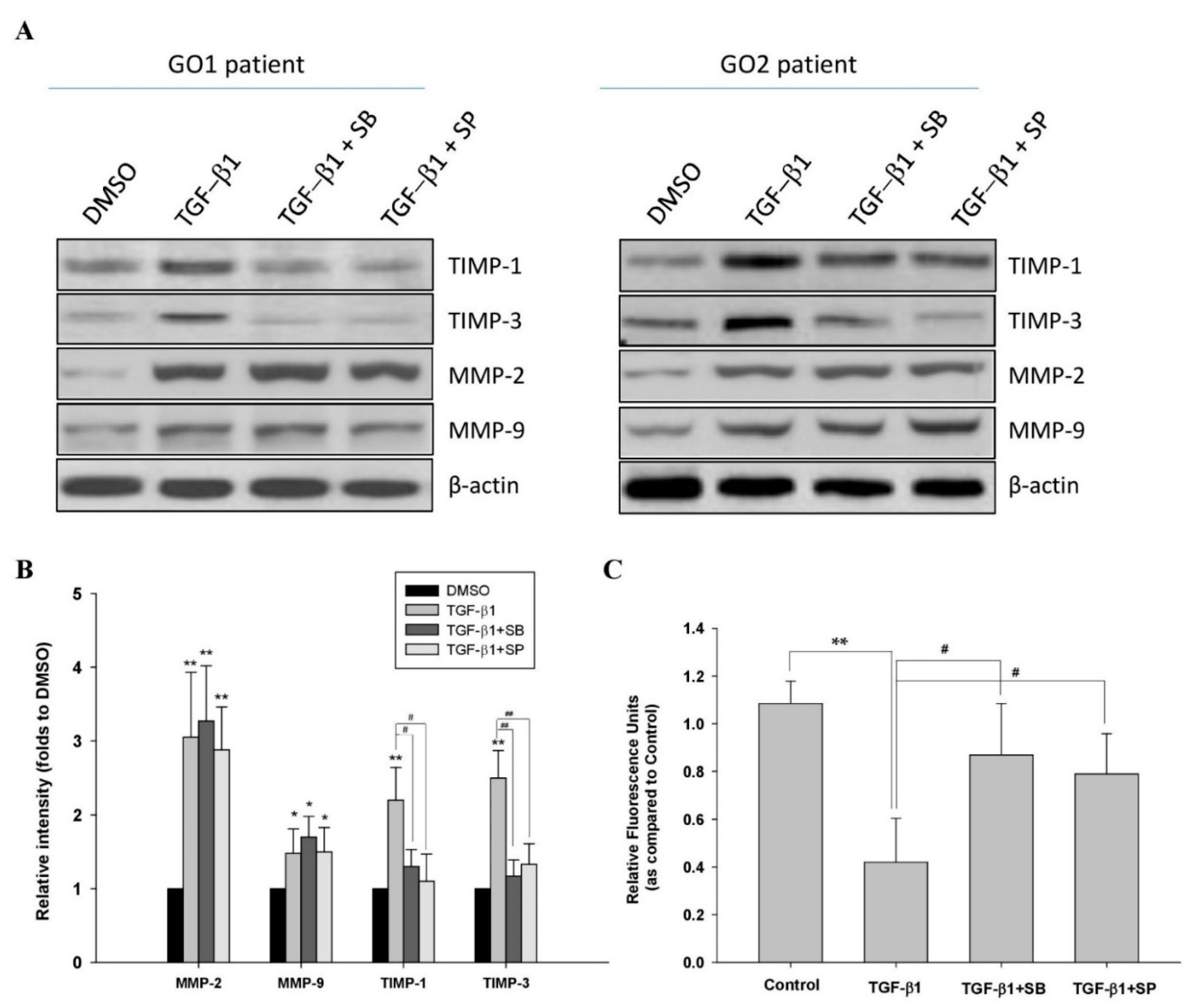
2.2. p38 and JNK Inhibitors Suppressed TGF-β1-Enhanced Fibrogenesis in GO
2.3. TGF-β1 Affected ECM Metabolism through p38 and JNK Mediators in GO
3. Discussion
4. Materials and Methods
4.1. Tissue Acquisition and Cell Culture
4.2. Chemicals and Antibodies
4.3. Western Blot Analysis
4.4. MMP-2 and MMP-9 Enzyme Activity Assay
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AP-1 | Activating protein-1 |
| ATF | Activating transcription factor |
| CTGF | Connective tissue growth factor |
| ECM | Extracellular matrix |
| ERK | Extracellular-signal-regulated kinase |
| FBS | Fetal bovine serum |
| GO | Graves’ ophthalmopathy |
| HRP | Horseradish peroxidase |
| JNK | c-Jun N-terminal kinase |
| MAPK | Mitogen-activated protein kinase |
| MMP | Matrix metalloproteinase |
| mRNA | Messenger ribonucleic acid |
| PBS | Phosphate-buffered saline |
| S1P | Sphingosine-1-phosphate |
| TGF-β1 | Transforming growth factor-β1 |
| TIMP | Tissue inhibitors of metalloproteinase |
| TSHR | Thyroid-stimulating hormone receptor |
| α-SMA | α-smooth muscle actin |
References
- Łacheta, D.; Miśkiewicz, P.; Głuszko, A.; Nowicka, G.; Struga, M.; Kantor, I.; Poślednik, K.B.; Mirza, S.; Szczepański, M.J. Immunological aspects of Graves’ ophthalmopathy. BioMed Res. Int. 2019, 2019, 7453260. [Google Scholar] [CrossRef]
- Wang, Y.; Smith, T.J. Current concepts in the molecular pathogenesis of thyroid-associated ophthalmopathy. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1735–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.J. Insights into the role of fibroblasts in human autoimmune diseases. Clin. Exp. Immunol. 2005, 141, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Longo, C.M.; Higgins, P.J. Molecular biomarkers of Graves’ ophthalmopathy. Exp. Mol. Pathol. 2019, 106, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.M.; Chang, P.C.; Wu, S.B.; Kau, H.C.; Tsai, C.C.; Liu, C.J.; Wei, Y.H. Expression and clinical significance of connective tissue growth factor (CTGF) in Graves’ ophthalmopathy. Br. J. Ophthalmol. 2017, 101, 676–680. [Google Scholar] [CrossRef]
- Tsai, C.C.; Wu, S.B.; Kau, H.C.; Wei, Y.H. Essential role of connective tissue growth factor (CTGF) in transforming growth factor-β1 (TGF-β1)-induced myofibroblast transdifferentiation from Graves’ orbital fibroblasts. Sci. Rep. 2018, 8, 7276. [Google Scholar] [CrossRef] [PubMed]
- Koumas, L.; Smith, T.J.; Feldon, S.; Blumberg, N.; Phipps, R.P. Thy-1 expression in human fibroblast subsets defines myofibroblastic or lipofibroblastic phenotypes. Am. J. Pathol. 2003, 163, 1291–1300. [Google Scholar] [CrossRef] [Green Version]
- Van Steensel, L.; Paridaens, D.; Schrijver, B.; Dingjan, G.M.; van Daele, P.L.; van Hagen, P.M.; van den Bosch, W.A.; Drexhage, H.A.; Hooijkaas, H.; Dik, W.A. Imatinib mesylate and AMN107 inhibit PDGF-signaling in orbital fibroblasts: A potential treatment for Graves’ ophthalmopathy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3091–3098. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.C.; Wu, S.B.; Kao, S.C.; Kau, H.C.; Lee, F.L.; Wei, Y.H. The protective effect of antioxidants on orbital fibroblasts from patients with Graves’ ophthalmopathy in response to oxidative stress. Mol. Vis. 2013, 19, 927–934. [Google Scholar]
- Pawlowski, P.; Reszec, J.; Eckstein, A.; Johnson, K.; Grzybowski, A.; Chyczewski, L.; Mysliwiec, J. Markers of inflammation and fibrosis in the orbital fat/connective tissue of patients with Graves’ orbitopathy: Clinical implications. Mediat. Inflamm. 2014, 2014, 412158. [Google Scholar] [CrossRef]
- Shu, D.Y.; Lovicu, F.J. Myofibroblast transdifferentiation: The dark force in ocular wound healing and fibrosis. Prog. Retin. Eye Res. 2017, 60, 44–65. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Leivonen, S.K.; Lazaridis, K.; Decock, J.; Chantry, A.; Edwards, D.R.; Kähäri, V.M. TGF-β-elicited induction of tissue inhibitor of metalloproteinases (TIMP)-3 expression in fibroblasts involves complex interplay between Smad3, p38α, and ERK1/2. PLoS ONE 2013, 8, e57474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Xu, C.; Cao, P.; Tian, Y.; Zhang, Y.; Shi, C.; Xu, J.; Yuan, W.; Chen, H. TGF-β stimulates expression of chondroitin polymerizing factor in nucleus pulposus cells through the Smad3, RhoA/ROCK1, and MAPK signaling pathways. J. Cell Biochem. 2018, 119, 566–579. [Google Scholar] [CrossRef]
- Kim, Y.I.; Kim, K.S.; Ahn, H.J.; Kang, I.H.; Shin, M.K. Reduced matrix metalloproteinase and collagen transcription mediated by the TGF-β/Smad pathway in passaged normal human dermal fibroblasts. J. Cosmet. Dermatol. 2020, 19, 1211–1218. [Google Scholar] [CrossRef]
- Kapelko-Słowik, K.; Słowik, M.; Szaliński, M.; Dybko, J.; Wołowiec, D.; Prajs, I.; Bohdanowicz-Pawlak, A.; Biernat, M.; Urbaniak-Kujda, D. Elevated serum concentrations of metalloproteinases (MMP-2, MMP-9) and their inhibitors (TIMP-1, TIMP-2) in patients with Graves’ orbitopathy. Adv. Clin. Exp. Med. 2018, 27, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.S.; Chae, M.K.; Jang, S.Y.; Lee, S.Y.; Lee, E.J. Antifibrotic effects of quercetin in primary orbital fibroblasts and orbital fat tissue cultures of Graves’ orbitopathy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5921–5929. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. The role of connective tissue growth factor, a multifunctional matricellular protein, in fibroblast biology. Biochem. Cell Biol. 2003, 81, 355–363. [Google Scholar] [CrossRef]
- Grotendorst, G.R.; Duncan, M.R. Individual domains of connective tissue growth factor regulate fibroblast proliferation and myofibroblast differentiation. FASEB J. 2005, 19, 729–738. [Google Scholar] [CrossRef]
- Khong, J.J.; McNab, A.A.; Ebeling, P.R.; Craig, J.E.; Selva, D. Pathogenesis of thyroid eye disease: Review and update on molecular mechanisms. Br. J. Ophthalmol. 2016, 100, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Cano, M.; Guerrero-Castilla, A.; Nabavi, S.M.; Ayala, A.; Argüelles, S. Targeting pro-senescence mitogen activated protein kinase (Mapk) enzymes with bioactive natural compounds. Food Chem. Toxicol. 2019, 131, 110544. [Google Scholar] [CrossRef]
- Cui, J.; Jin, S.; Jin, C.; Jin, Z. Syndecan-1 regulates extracellular matrix expression in keloid fibroblasts via TGF-β1/Smad and MAPK signaling pathways. Life Sci. 2020, 254, 117326. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, Y.; Gao, Y.; Li, Q.; Zeng, Z.; Li, Y.; Chen, H. Kindlin-2 regulates hepatic stellate cells activation and liver fibrogenesis. Cell. Death Discov. 2018, 4, 34. [Google Scholar] [CrossRef]
- Tian, M.; Chang, X.; Zhang, Q.; Li, C.; Li, S.; Sun, Y. TGF-β1 mediated MAPK signaling pathway promotes collagen formation induced by Nano NiO in A549 cells. Environ. Toxicol. 2019, 34, 719–727. [Google Scholar] [CrossRef]
- Lee, J.; An, J.N.; Hwang, J.H.; Lee, H.; Lee, J.P.; Kim, S.G. p38 MAPK activity is associated with the histological degree of interstitial fibrosis in IgA nephropathy patients. PLoS ONE 2019, 14, e0213981. [Google Scholar] [CrossRef] [PubMed]
- Grynberg, K.; Ma, F.Y.; Nikolic-Paterson, D.J. The JNK signaling pathway in renal fibrosis. Front. Physiol. 2017, 8, 829. [Google Scholar] [CrossRef]
- Luo, K. Signaling cross talk between TGF-β/Smad and other signaling pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammone, T.; Chidlow, G.; Casson, R.J.; Wood, J.P.M. Expression and activation of mitogen-activated protein kinases in the optic nerve head in a rat model of ocular hypertension. Mol. Cell Neurosci. 2018, 88, 270–291. [Google Scholar] [CrossRef]
- Frangogiannis, N. Transforming growth factor-β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Hu, Y.; Peng, J.; Feng, D.; Chu, L.; Li, X.; Jin, Z.; Lin, Z.; Zeng, Q. Role of extracellular signal-regulated kinase, p38 kinase, and activator protein-1 in transforming growth factor-beta1-induced alpha smooth muscle actin expression in human fetal lung fibroblasts in vitro. Lung 2006, 184, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Sonbol, H.S. Extracellular matrix remodeling in human disease. J. Microsci. Ultrastruct. 2018, 6, 123–128. [Google Scholar] [CrossRef]
- Hu, H.H.; Chen, D.Q.; Wang, Y.N.; Feng, Y.L.; Cao, G.; Vaziri, N.D.; Zhao, Y.Y. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 2018, 292, 76–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.T.; Meng, X.M. TGF-β/Smad and renal fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 347–364. [Google Scholar] [PubMed]
- Takahara, T.; Furui, K.; Funaki, J.; Nakayama, Y.; Itoh, H.; Miyabayashi, C.; Sato, H.; Seiki, M.; Ooshima, A.; Watanabe, A. Increased expression of matrix metalloproteinase-II in experimental liver fibrosis in rats. Hepatology 1995, 21, 787–795. [Google Scholar] [CrossRef]
- Piek, E.; Ju, W.J.; Heyer, J.; Escalante–Alcalde, D.; Stewart, C.L.; Weinstein, M.; Deng, C.; Kucherlapati, R.; Bottinger, E.P.; Roberts, A.B. Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J. Biol. Chem. 2001, 276, 19945–19953. [Google Scholar] [CrossRef] [Green Version]
- Kellenberger, T.; Krag, S.; Danielsen, C.C.; Wang, X.F.; Nyengaard, J.R.; Pedersen, L.; Yang, C.; Gao, S.; Wogensen, L. Differential effects of Smad3 targeting in a murine model of chronic kidney disease. Physiol. Rep. 2013, 1, e00181. [Google Scholar] [CrossRef]
- Edwards, D.R.; Leco, K.J.; Beaudry, P.P.; Atadja, P.W.; Veillette, C.; Riabowol, K.T. Differential effects of transforming growth factor-β1 on the expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in young and old human fibroblasts. Exp. Gerontol. 1996, 31, 207–223. [Google Scholar] [CrossRef]
- Dik, W.A.; Virakul, S.; Steensel, L.V. Current perspectives on the role of orbital fibroblasts in the pathogenesis of Graves’ ophthalmopathy. Exp. Eye Res. 2016, 142, 83–91. [Google Scholar] [CrossRef]
- Valyasevi, R.W.; Jyonouchi, S.C.; Dutton, C.M.; Munsakul, N.; Bahn, R.S. Effect of tumor necrosis factor-alpha, interferon-gamma, and transforming growth factor-beta on adipogenesis and expression of thyrotropin receptor in human orbital preadipocyte fibroblasts. J. Clin. Endocrinol. Metab. 2001, 86, 903–908. [Google Scholar] [PubMed] [Green Version]
- Ko, J.; Chae, M.K.; Lee, J.H.; Lee, E.J.; Yoon, J.S. Sphingosine-1-phosphate mediates fibrosis in orbital fibroblasts in Graves’ orbitopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2544–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heufelder, A.E.; Bahn, R.S. Modulation of Graves’ orbital fibroblast proliferation by cytokines and glucocorticoid receptor agonists. Investig. Ophthalmol. Vis. Sci. 1994, 35, 120–127. [Google Scholar] [PubMed]
- Tsai, C.C.; Wu, S.B.; Chang, P.C.; Wei, Y.H. Alteration of connective tissue growth factor (CTGF) expression in orbital fibroblasts from patients with Graves’ ophthalmopathy. PLoS ONE 2015, 10, e0143514. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.Y.; Sun, C.Y.; Young, G.H.; Hsieh, Y.T.; Chen, Y.H.; Wu, M.S.; Wu, V.C.; Lee, J.H.; Lee, C.C. Protein-bound uremic toxins induce tissue remodeling by targeting the EGF receptor. J. Am. Soc. Nephrol. 2015, 26, 281–290. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, T.-Y.; Wu, S.-B.; Kau, H.-C.; Tsai, C.-C. JNK and p38 Inhibitors Prevent Transforming Growth Factor-β1-Induced Myofibroblast Transdifferentiation in Human Graves’ Orbital Fibroblasts. Int. J. Mol. Sci. 2021, 22, 2952. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062952
Hou T-Y, Wu S-B, Kau H-C, Tsai C-C. JNK and p38 Inhibitors Prevent Transforming Growth Factor-β1-Induced Myofibroblast Transdifferentiation in Human Graves’ Orbital Fibroblasts. International Journal of Molecular Sciences. 2021; 22(6):2952. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062952
Chicago/Turabian StyleHou, Tzu-Yu, Shi-Bei Wu, Hui-Chuan Kau, and Chieh-Chih Tsai. 2021. "JNK and p38 Inhibitors Prevent Transforming Growth Factor-β1-Induced Myofibroblast Transdifferentiation in Human Graves’ Orbital Fibroblasts" International Journal of Molecular Sciences 22, no. 6: 2952. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062952