
Homologous Recombination Deficiencies and Hereditary Tumors


Department of Clinical Genomic Medicine, Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama University, 2-5-1 Shikata-cho, Kita-ku, Okayama 700-8558, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(1), 348; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010348
Submission received: 28 November 2021
/
Revised: 23 December 2021
/
Accepted: 25 December 2021
/
Published: 29 December 2021
(This article belongs to the Special Issue Molecular Biology of Hereditary Tumors)
Abstract
:Homologous recombination (HR) is a vital process for repairing DNA double-strand breaks. Germline variants in the HR pathway, comprising at least 10 genes, such as BRCA1, BRCA2, ATM, BARD1, BRIP1, CHEK2, NBS1(NBN), PALB2, RAD51C, and RAD51D, lead to inherited susceptibility to specific types of cancers, including those of the breast, ovaries, prostate, and pancreas. The penetrance of germline pathogenic variants of each gene varies, whereas all their associated protein products are indispensable for maintaining a high-fidelity DNA repair system by HR. The present review summarizes the basic molecular mechanisms and components that collectively play a role in maintaining genomic integrity against DNA double-strand damage and their clinical implications on each type of hereditary tumor.
1. Introduction
Homologous recombination (HR) is a tightly regulated molecular mechanism essential for maintaining genomic integrity against DNA double-strand breaks (DSBs) in a wide range of species, including yeast and humans. Compared to other known mechanisms, such as non-homologous end-joining (NHEJ) and microhomology-mediated end-joining (MMEJ), the most noteworthy characteristics of HR for DSB repair are error-free procedures and outcomes using sister chromatids and highly conserved molecular associates [1,2]. The representative genes involved in HR are BRCA1 and BRCA2, whose deleterious variants are associated with hereditary breast and ovarian cancer (HBOC) syndrome [3]. Hereditary tumors, including HBOC, are characterized by constitutional variants which lead to intrafamily accumulation of specific types of cancer, early age onset, and/or multiple tumor development in a synchronous or metachronous manner. Inherited predispositions to cancer, sarcoma, or even benign tumors are mainly due to germline loss-of function in cell cycle regulation or DNA damage repair [4].
For error-free DSB repairing, HR can be considered the most important, or perhaps even the only mechanism. Recent clinical trials of molecular-based precision medicine on patients and their families (i.e., probands and relatives) suffering from hereditary tumors have yielded encouraging evidence for utilizing molecular-based knowledge of HR not only against tumor development associated with BRCA1 and BRCA2, but also for other HR-related gene variants carrying moderate or low tumor development risks [5,6]. In this work, we provide an overview of the mechanisms and essential molecular components involved in an efficient HR with their impacts on hereditary tumors. We also discuss the clinical utility of multi-gene panel testing (MGPT) for the diagnosis of HR-related hereditary tumors as well as for therapeutic and preventive strategies against them.
2. Molecular Components and Cascade Overview of HR
HR repair is an essential molecular mechanism for maintaining genomic stability by repairing double-strand DNA breaks (DSBs) or lesions that stall DNA replication forks [7]. DSBs are potentially more complex and difficult to repair than other types of DNA damage. DSBs can be caused not only by exogenous sources, such as ionizing radiation and toxic chemicals, but also by endogenous genotoxins, including reactive oxygen species and metabolic byproducts. High-fidelity and timely repair of DSBs is necessary for maintaining genomic integrity and cell survival by minimizing mutagenesis or improper apoptosis, which may otherwise lead to carcinogenesis [8,9]. HR is a highly conserved mechanism in eukaryotes, from yeast to humans. HR in budding yeast Saccharomyces cerevisiae, for example, is catalyzed by proteins encoded by RAD52 epistasis group genes such as RAD50, RAD51, RAD54, RAD55, RAD57, RAD59, XRS2 (NBS1 or NBN in humans), and MRE11, including RFA1–3, the heterotrimeric complex of RPA [10]. There is a conserved appearance in the foci formation with DNA-damage-related products at the site of DNA damage [11].
In mammalian cells, the macro-complex consists of BRCA1 and BARD1, which bind through the RING finger domain at the N-terminus; components such as phosphorylated ABRA1 binding through the BRCT domain at the carboxyl-terminus; and NBA1, RAP80, etc. All these components are recruited to the sites of DNA damage along with BRCA1 [12,13]. These macro-complex structures are observed through DNA damage checkpoint activation, which leads to G2-M checkpoint arrest. HR uses undamaged sister chromatids as templates to enable precise repair and functions during the cell cycle phases of late S to G2. RAD51 recombinase, which is the final effector of the HR cascade, and is recruited to the sites of DNA damage as a consequence of the interaction of BRCA1 and BRCA2 through PALB2, which is a key triplet complex of HR (Figure 1, Part 1). RAD51 is carried by binding to BRCA2 through eight conserved motifs of BRC repeats in the middle of the molecule and at the unrelated carboxyl-terminus region of the TR2 domain [14]. Once RAD51 is loaded and released on the end-resected DNA damage site, RAD51 forms a nucleofilament and leads to strand invasion. Repair proceeds from the invading DNA end to the second end, which is captured by two Holliday junctions and is thereafter resolved as a crossover or dissolved in a non-crossover form [15].
2.1. Key Initial Step for HR: Sensing DSBs
To proceed to HR, a key initial step is the sensing of the damaged sites by the MRE11–RAD50–NBS1 complex (MRN complex) [16,17]. The molecular function of MRE11 was originally identified as a 3′ to 5′ exo- and endonuclease, whereas RAD50 was found to be an ATPase [18,19]. Extensive studies have revealed that the functions of MRN complex are categorized in the following three roles. Firstly, it holds the damaged ends of DNA in DSB lesions structurally by RAD50. Secondly, it creates single-stranded DNA (ssDNA) by nucleolytic processing to expose 3′ of DNA, that is, 3′ overhangs by the end resection of the 5′ to 3′ strand for which MRE11 is functional alongside BRCA1 [8]. Both sides of the DSBs are immediately acted upon by human replication protein A (RPA), an ssDNA-binding protein, to melt DNA secondary structures and to prepare for RAD51 nucleoprotein filament formation. Lastly, NBS1 (alternative name: NBN), a substrate of ataxia telangiectasia-mutated (ATM), is responsible for the recruitment and checkpoint activation of ATM.
ATM is traditionally known as a multifunctional serine/threonine kinase, which stabilizes and activates the tumor suppressor protein p53. Upon DNA damage, ATM is recruited to the damaged site and leads to subsequent check-point activation during the G1/S checkpoint [7,20]. ATM and ATM-Rad3-related (ATR) phosphorylate multiple downstream targets such as p53, H2AX, and BRCA1 either directly or via CHEK2 gene coding checkpoint kinase 2 protein (CHK2) activation [21]. BRCA1 is assisted by BRCA1-associated RING domain 1 (BARD1) and BRCA1 interacting helicase 1 (BRIP1), which acts as a scaffolding protein that organizes the assembly with other repair proteins (Figure 1, Part 2). BRCA1 interacts with BARD1 via the RING-finger domain near the N-terminus of BRCA1, both of which encode a nuclear export signal (NES). BRCA1 forms three different types of complexes exclusively with phosphorylated Abraxas (ABRA1), BRCA1-associated C-terminal helicase 1 (BACH1/FANCJ/BRIP1), or CtBP-interacting protein (CtIP) through the BRCT domain near the C-terminus of BRCA1 [22,23]. As a consequence of interacting with these different proteins, BRCA1 plays pleotropic roles, including DNA damage resistance, ubiquitination, gene transcription, and cell cycle progression, such as G2-M checkpoint control [24] (detailed information is found in Molecular Diagnosis and Targeting for Gynecologic Malignancy, ISBN 978-981-33-6013-6 (eBook)) [25].
Under the mediation of PALB2, a linker protein between BRCA1 and BRCA2, RAD51 is recruited to the DSB sites by binding to BRCA2. Once successfully released from BRCA2, RAD51 forms nucleofilaments and allows DNA to invade the homologous double helix. HR progresses by DNA synthesis using homologous DNA as a template, leading to second-end capture and double Holliday junction formation, ultimately ending in either resolution or dissolution.
2.2. ATM, CHEK2, BRIP1, and BARD1: Activators and Partners of BRCA-1 and Their Roles in Cancer Susceptibility
ATM and CHEK2 are recognized as the most common breast cancer susceptibility genes, and their attenuation is associated with BRCAness, namely phenocopies of BRCA1/2 defects. In a single institutional review of 1185 breast cancer cases using multi-gene panel testing (MGPT), a high occurrence of bilateral tumors was observed in 26.3% of the ATM germline variant carriers and in as much as 41.2% of CHEK2 variant carriers [26]. Histological analyses of breast cancer subtypes showed that there were varying trends between ATM and CHEK2. Over half (64.3%) of CHEK2-associated tumors were luminal A-like subtype (enriched for hormonal receptors, less proliferative, and low-grade), whereas nearly half (56.2%) of ATM-associated tumors were luminal B-like subtype (low expression of hormonal receptors, but more proliferative and high-grade) and/or human epidermal growth factor (HER2)-negative type. Lobular carcinoma was observed in 21.4% of CHEK2-related invasive tumors. A quarter of ATM-related breast cancer and approximately one-third (30%) of CHEK2-related breast cancers were in situ carcinomas [26].
ATM proteins are multi-functional. They are conventionally found as an activating factor of p53 in response to DNA damage and phosphorylate BRCA1 following irradiation injury [27]. Phosphorylation of BRCA1 by ATM is an important procedure for G2/M and S-phase checkpoint activation, in which BRCA1 plays a role [28]. CHK2, protein product of CHEK2, is also categorized as a serine/threonine kinase that is activated by ATM-mediated phosphorylation of SQ/TQ sites (cluster motifs containing seven serine (S) or threonine (T) residues followed by glutamine (Q)) in response to DNA damage [29]. Substrate spectra of CHK2 kinase are wide-ranging from cell cycle controls, such as phosphatases including Cdc25A, Cdc25C, and phosphoinositide 3-kinase (PI3K), to cell death signaling constituted by p53-MDM2 interplay, and DNA damage repair pathway through BRCA1. CHK2-mediated BRCA1 phosphorylation is important for the BRCA1–PALB2–BRCA2 effector complex functions in the HR pathway.
BRIP1, an alternative name for BRCA1-associated C-terminal helicase (BACH1) or Fanconi anemia subtype J (FANCJ), interacts with BRCA1 through the BRCT domains at the carboxyl-terminus (Figure 1, Part 2). BRIP1 was originally identified in the investigation of Fanconi anemia [30]. BRIP1 is physiologically essential for the maintenance of genomic integrity, removing DNA-bound proteins, stabilizing replication forks, and unwinding substitutive DNA structures along with RPA [31]. BRIP1 acts as a tumor suppressor through its interaction with BRCA1 [32]. Several truncating variants of BRIP1 are conventionally known to be associated with breast cancer with low or moderate penetrance [33]. In recent years, it has been recognized that deleterious germline variants of BRIP1 are highly susceptible to ovarian cancer [34]. There is an emerging report showing potential associations between germline pathogenic variants (GPVs) of BRIP1 and colorectal cancer based on next-generation sequencing (NGS) analysis of multiple genes [35].
The BRCA1–BARD1 complex is essential for BRCA1 to function as an E3 ubiquitin ligase on a molecular basis [8,28]. GPVs in BARD1 have been associated with early onset of breast cancer [36]. For the clinical management of GPV carriers of ATM, CHEK2, or BARD1, all of which are autosomal dominant, it is recommended to perform imaging screenings for preventive risk management against breast cancer. This may be done by annual mammograms with or without tomosynthesis and, by performing breast magnetic resonance imaging (MRI) starting at age 40, according to the National Comprehensive Cancer Network® (NCCN) Guidelines® Version 1.2022 as of August 2021 (www.nccn.org, accessed on 19 November 2021). Breast cancer risk in females with GVPs of BRIP1 is stated as “potential”, with insufficient evidence for risk management whereas there are increased risks for ovarian cancer, justifying the consideration of risk-reducing salpingo-oophorectomy (RRSO) at around 45–50 years of age (with a discussion) or earlier, based on specific family history of an earlier onset of ovarian cancer (NCCN Guidelines® Version 1.2022, www.nccn.org, accessed on 19 November 2021).
2.3. PALB2: Bridging Mediator of BRCA1 and BRCA2 and the Roles in Cancer Susceptibility
Partner and localizer of BRCA2 (PALB2) was discovered as an interacting protein with BRCA2 [37]. PALB2 mediates complex formation together with BRCA1 and BRCA2 and promotes BRCA2 recruitment to DSB sites. In addition to these functions, PALB2 was identified as an associated gene for Fanconi amenia (subtype N, FANCN) and pediatric malignancies such as Wilms’ tumor or medulloblastoma. It is also implicated in the susceptibility to breast cancer in adults, conferring a 2.3-fold higher risk of cancer development [38]. However, in later studies, penetrance of variants of PALB2 in breast cancer development was found to be distinctively varied in different populations [39]. Breast cancer risk due to loss-of-function variants of PALB2 overlapped with that of BRCA2 variants, validating the direct interaction of PALB2 with BRCA2 [40]. In recent years, the spectrum of clinical preventive risk management for carriers with GPVs of PALB2 has been extended to epithelial ovarian cancer and pancreatic cancer, with absolute risks at 3–5% and 5–10%, respectively, according to NCCN Guidelines® Version1.2022 (www.nccn.org, accessed on 19 November 2021).
There is also a suggestive case report showing a potential involvement of GPVs of PALB2 and NBS1 (NBN) in the susceptibility to pancreatic cancer [41]. For presumed high-risk individuals with a pronounced history (e.g., at least two affected cases within first-degree relatives) of familial pancreatic cancer, it is suggested to conduct routine follow-ups with endoscopic ultrasound (EUS) and MRI or magnetic resonance cholangiopancreatography (MRCP) as surveillance of pancreatic cancer, in accordance with the International Cancer of Pancreas Screening (CAPS) established in 2013 by Johns Hopkins University [42] and with the latest version of the NCCN Guidelines® [43].
2.4. RAD51, RAD51C, RAD51D: The Final Effector of HR and Paralogs
Recombinase RAD51 is a critically important molecule, as it is the final effector in the HR cascade. RAD51 is recruited to the DSB sites after being carried under the mediation of BRCA2. RAD51 binds to BRCA2 via the eight conserved short motifs of BRC repeats and a carboxyl-terminus region called the TR2 domain (Figure 1, Part 3). Deleterious variants of BRCA2 at BRC repeats can, therefore, compromise the interaction of BRCA2 and RAD51, potentially causing HBOC phenotypes [44]. Binding of RAD51 to BRCA2 at the carboxyl terminus is known to be dependent on the cyclin-dependent kinase 2 (CDK2)-mediated phosphorylation status of BRCA2 [45]. After loading RAD51 successfully on the resected DNA ends, RAD51-bound DNA forms nucleofilaments and invades the homologous double helix.
DNA polymerase synthesizes new DNA structures, using homologous DNA as a template with ssDNA as a primer. RAD51C (RAD51L2) and RAD51D (RAD51L3) constitute RAD51 paralogues, with other members such as RAD51B (RAD51L1), XRCC2, and XRCC3. RAD51C and RAD51D form a subcomplex with RAD51B and XRCC2 (BCDX2 complex) (Figure 1, Part 4). They assist the recruitment of RAD51 under the regulation of BRCA1–PALB2–BRCA2 effector complex [28]. GPVs of RAD51C and RAD51D are known to impart susceptibility to breast cancer at absolute risks of 15–40% (NCCN Guidelines® Version1.2022 at www.nccn.org, accessed on 19 November 2021), while both of them are shown to moderately increase susceptibility to ovarian cancer, such as a six-fold higher risks for carriers of RAD51D variant [46]. The penetrance of RAD51D for ovarian cancer is at moderate risk, which is almost consistent across ethnicities [47]. In the NCCN Guidelines® Version 1.2022, it is stated that there are not sufficient data so far to suggest preventive management of breast cancer in carriers of GPVs of RAD51C or RAD51D (RAD51C/D). Conversely, RRSO is considered at age of 45–50 years for the clinical management of ovarian cancer associated with RAD51C/D variants (www.nccn.org, accessed on 19 November 2021). There is also new evidence regarding the clinical significance of RAD51D splicing variants that can be detected in various types of tumors, including non-epithelial sarcoma [48].
3. HR-Based Precision Oncology for Hereditary Tumor Clinics
A lot of clinical evidence has been accumulated concerning HR, the impairment of which is associated with inherited susceptibility to hereditary tumors. A representative hereditary tumor due to germline deficiency of HR, which is represented by alterations in BRCA1 or BRCA2 (BRCA1/2), is the hereditary breast and ovarian cancer (HBOC) syndrome [49]. Pathogenic attenuations in either BRCA1 or BRCA2 account for 25–40% of familial breast cancers and up to 10% of all breast cancers [50,51]. In several large-scale case-control studies of familial cancer patients and whole exome germline sequencing, alternative candidate genes conferring susceptibility at various intensities have been identified in breast cancer and in other HBOC-related tumors such as ovarian, pancreatic, and prostate cancers (Table 1).
According to a sequencing study conducted by Welsh et al. for 360 cases of primary ovarian, peritoneal, or fallopian tube carcinomas, irrespective of the family history, 24% of the cases carried germline loss-of-function variants of either BRCA1, BRCA2, or other genes, including those involved in HR pathways such as CHEK2, MRE11A, NBS1 (NBN), RAD50, RAD51C, or in the FA pathway such as BRIP1/FANCJ, PALB2/FANCN, and BARD1. The rest of the population carried germline variants in TP53, a tumor suppressive DNA damage response gene, or MSH6, a mismatch-repair gene. The proportion of germline variants in any of the genes other than BRCA1/2 accounted for 6% of the total cases examined, which, in turn, accounted for one-third of the patients with germline variants in their study [82]. In tumors with germline alterations of HR-related genes, there are emerging new concepts termed as BRCAness, phenocopies of BRCA1/2 defects, expressing communal phenotypes such as the organs in which malignancy develops and the potential clinical responses to platinum-based therapies or vulnerability to poly(ADP-ribose) polymerase inhibitors (PARPi) [83,84,85]. With respect to the penetrance in each type of cancer, BRCA1/2 is known to be the strongest factor responsible and therefore, is categorized as a high-risk genetic factor in imparting susceptibility to specific types of cancer.
According to the results of an international BRCA1/2 cohort study, the cumulative risk of developing breast cancer among BRCA1/2 variant carriers reaches approximately 70% by the age of 80 (72% in BRCA1 variant carriers and 69% in BRCA2 variant carriers) [60]. For ovarian cancer, the risk is 17% and 44% for BRCA1 and BRCA2 variant carriers, respectively, by the age of 80. The estimated frequencies of developing malignancies are 0.1% for prostate cancer and 0.5% for pancreatic cancer, while the relative risks for their lifetime incidence were reported to increase significantly (up to 20-fold for prostate cancer and 10-fold for pancreatic cancer) upon association with BRCA2 [28,86]. According to a phase II study conducted by Tug et al., germline variants of PALB2 are presumed to have a favorable response to treatment with olaparib, a PARPi, in metastatic breast cancer patients [87]. The overall survival of pancreatic adenocarcinoma patients with germline HR-related gene variants, such as ATM alterations, is significantly longer compared to that of non-variant carriers [88]. This evidence offers the validity of routine sequencing of not only BRCA1/2 but also of the wide range of HR-related genes as part of biological characterization and for treatment decisions using PARPi, at least in the cases of breast and pancreatic cancer.
3.1. HR Deficiency (HRD) and HRD-Associated Genes
HR deficiency (HRD) is most prevalent in ovarian, breast, prostate, and pancreatic cancers. In a pan-cancer cohort reported by Nguyen et al., the HRD frequency in ovarian and breast cancers is followed by that in pancreatic and prostate cancers. The prevalence of HRD in patients with any of four types of cancer was reported to be up to 85% by an analysis in which HRD was predicted through specific single nucleotide variants (SNVs), insertions/deletions (indels), structural variants (SVs), or loss-of-heterozygosity (LOH) [89]. Assessing HRD status by the region of LOH has been proposed as a useful indicator for defective BRCA1/2, at least in ovarian cancer [90]. In a systematic review and meta-analysis of the prevalence of HRD in both somatic and germline contexts among patients with pancreatic cancer, HRD descriptions based on genomic scarring and surrogate markers such as point mutational and structural variants were well-characterized with HR-related genes such as BRCA1, BRCA2, PALB2, ATM, ATR, CHEK2, RAD51, and the FANC genes [91]. Their study showed that HRD prevalence in pancreatic cancer ranged from 14.5–16.5% through targeted NGS and was as high as 24–44% by whole-genome or whole-exome sequencing.
It has been recognized that HRD is associated with cellular sensitivity of ovarian cancer to crosslinking agents such as platinum salts and topoisomerase inhibitors [92]. In contrast, according to the American Society of Clinical Oncology (ASCO) guidelines published in 2020, current HRD assays do not provide significant differentiation of patient response to PARPi to routinely recommend their use [93]. These controversial discussions regarding the clinical significance of HRD in the responsiveness to PARPi might be due to different pipeline methodologies or criteria for HRD evaluation. MyChoice test (Myriad Genetics, Salt Lake City, UT, USA), for example, determines HRD status solely in tumor tissues through the combined analyses of pathogenic variants in BRCA1 and BRCA2 based on sequencing and large rearrangements, and by assessing genomic instability in three biomarkers: LOH, telomeric allelic imbalance (TAI), and large-scale state transition (LST) (https://myriad.com/, accessed on 23 November 2021). Another weighted model, HRDetect, was developed as a tool to predict BRCA1/2 deficiency in tumors that reflects selective sensitivity to PARPi. HRDetect covers both germline and somatic events by integrating all classes of mutational signatures in the analyses [94].
Among PARPi (e.g., niraparib, olaparib, and rucaparib), olaparib was first approved by the US Food and Drug Administration (FDA) in 2018 for the treatment of patients with germline BRCA-mutated, HER2-negative metastatic breast cancer who were treated with chemotherapy (www.fda.gov, accessed on 17 November 2021) based on the OlympiAD trial [95]. Later in the same year, olaparib was approved for the maintenance of response in the first-line treatment of patients with deleterious or suspected deleterious germline or somatic BRCA mutated advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer who respond completely or partially to platinum-based first-line chemotherapy. According to SOLO1 phase III trial, patients showed a reduced risk of progression or death with maintenance olaparib as compared to placebo by 70% [96]. Another phase III trial, the PAOLA-1 trial, showed that when maintenance olaparib was added to the first-line treatment with bevacizumab (a vascular endothelial growth factor (VEGF) monoclonal antibody), patients with advanced ovarian cancer presented significantly favorable outcomes as compared to bevacizumab plus placebo in the progression-free survival of patients with HRD-positive tumors, including those without a BRCA variant (HRD-positive and BRCA wild-type) [97]. In the VELIA trial, however, hazard ratios (HRs) of veliparib maintenance therapy in patients with newly diagnosed, high-grade serous ovarian carcinoma was similar in HRD-positive with BRCA wild-type (HR, 0.74; 95% CI, 0.52 to 1.06) and non-HRD tumors (HR, 0.81; 95% CI, 0.60 to 1.09) [98]. These somewhat controversial results are perhaps due to the different evaluation criteria for HRD using different cutoff values of genomic instability scores. The FDA has approved the indication criteria of niraparib for patients with advanced ovarian cancer, based on the cancer status determined as HRD-positive by HRD testing of BRCA mutated and/or a genomic instability score of over 42. Among HR effector genes potentially presumed as being associated with HRD status, apart from BRCA1/2, would be ATM, BARD1, BRIP1, CHEK1, CHEK2, FANCA, FANCC, MRE11A, NBS1 (NBN), PALB2, RAD50, RAD51, RAD51B, RAD51C, RAD52, and RAD54L [99]. In a pan-cancer study conducted by Nguyen et al., biallelic activation of BRCA1, BRCA2, RAD51C, and PALB2 were reported as the most common genetic causes of HRD [89].
3.2. Differential Features of BRCA1-Type and BRCA2-Type
Considering the distinct molecular roles of BRCA1 and BRCA2, it is understandable that the pathogenic variants of BRCA1 and BRCA2 are associated with different subtypes in cancers. BRCA1-mutant breast tumors, for example, are typically negative for estrogen receptor alpha (ER alpha) [100]. A disease-specific microarray analysis of BRCA1-mutant breast tumor tissue over sporadic tumors expressing wild-type BRCA1 found that the expression of ESR1, which is a coding gene for ER alpha, was significantly lower in BRCA1-mutant tumor tissue than in sporadic tumor tissues. In vitro analysis using breast cancer cell lines further identified a mechanism by which ESR1 expression is positively regulated by direct binding of BRCA1 to the ESR1 promoter, being recruited under the regulation of the transcription factor Oct-1. Clinically, BRCA1 variant carriers have a higher risk of developing triple-negative (TN) status in breast cancer, which is negative for ER, progesterone receptor (PgR), and HER2 [101].
BRCA2 variants typically cause ER-positive luminal subtypes, showing slow proliferation with low grade clinical aggression of breast cancer [32]. In a cohort study of patients with BRCA germline-mutated breast cancer, immunohistochemical analyses showed that the luminal A-like subtype, which is strongly positive for hormone receptors (ER and/or PgR) and low-proliferative and low-grade subtypes, were more frequently observed in BRCA2-mutated (35%) than in BRCA1-mutated (9%) [102].
According to a large-scale international joint consortium, it was shown that histopathological features of breast cancer refine the likelihood estimation for BRCA1 or BRCA2 variant status, which can be informative for genetic testing and clinical management of the patients involved [103]. Histologically, it is well-established that medullary breast carcinoma, which displays a basal-like molecular subtype, is associated with germline variants in BRCA1 [104]. In prostate cancer, germline BRCA2 variant carriers had worse clinical outcomes than non-variant carriers when treated with surgery or radiotherapy [105]. It has been shown that the BRCA2-mutant in prostate cancer might be associated with the histological characteristics of intraductal carcinoma of the prostate (IDC-P), and a tendency for higher incidence of biochemical relapse after surgery. In a pan-cancer HRD prediction study, it was found that biallelic inactivation of BRCA1, BRCA2, RAD51C, or PALB2 is the most common genetic cause of HRD across cancer types, with the inactivation of the latter two genes, RAD51C and PALB2, resulting in the same mutational footprints of BRCA2, the so-called BRCA2-type HRD [89].
3.3. Reversion of BRCA1/2 Variants and Other Resistance Mechanisms to PARP Inhibitors or Platinum Chemotherapy
As somatic events in BRCA1 or BRCA2, genomic reversion is known as a mechanism for acquiring resistance to PARPi or platinum chemotherapy in cancer cells [106,107]. Revertant BRCA1/2 alleles due to secondary intragenic alteration ameliorates the wild-type BRCA1/2 open reading frame. The reconstitution of functional BRCA1/2 protein restores intact HR and cells subsequently acquire resistance to PARPi or platinum-based chemotherapy. By comparing the analyses of tumor whole-exome sequencing before and after acquired resistance to PARPi or platinum chemotherapy, it has also been proposed that HR restoration is caused by increased DNA end resection due to MRE11A amplification [108]. It was also reported that no RAD51 foci were observed in pre-resistant tumors, while they were detected in post-resistance tumors, suggesting that RAD51 foci formation is an indicator of HR restoration and could be a potentially useful biomarker to predict responses to later DNA-damaging therapy. Biallelic inactivation of TP53BP1 and a histone methyltransferase KKMT2C, or the reduced expression of TP53BP1, were also shown as genetic alterations essential for HR proficiency, and correlate with response and resistance to PARPi/platinum therapy.
4. Rational Strategies for Diagnosis of Hereditary Tumors Using Multi-Gene Panel Testing (MGPT)
The development of NGS technologies has paved the way for simultaneous analyses of multiple candidate cancer susceptibility genes through massive parallel sequencing. This recent trend in the research field is becoming widely common with large-scale non-biased or population-based analyses covering multiple genes. These strategies enable genetic characterization for the somatic and effective detection of the germline predisposition to each type of cancer for both probands and their relatives. Since a US Supreme Court Judgement invalidated the patent of Myriad Genetics on BRCA1/2 genetics tests in 2013 [109], multi-gene panel testings (MGPTs) including BRCA1/2 can be provided by other Clinical Laboratory Improvement Amendments (CLIA) or College of American Pathologists (CAP)-certified clinical laboratories for clinical use. MGPTs generally cover at least 10 common HR-related genes such as ATM, BARD1, BRCA1, BRCA2, BRIP1, CHEK2, NBS1 (NBN), PALB2, RAD51C, and RAD51D, all of whose germline alterations are associated with BRCAness phenotypes for predisposition to breast, ovarian, prostate, or pancreatic cancer.
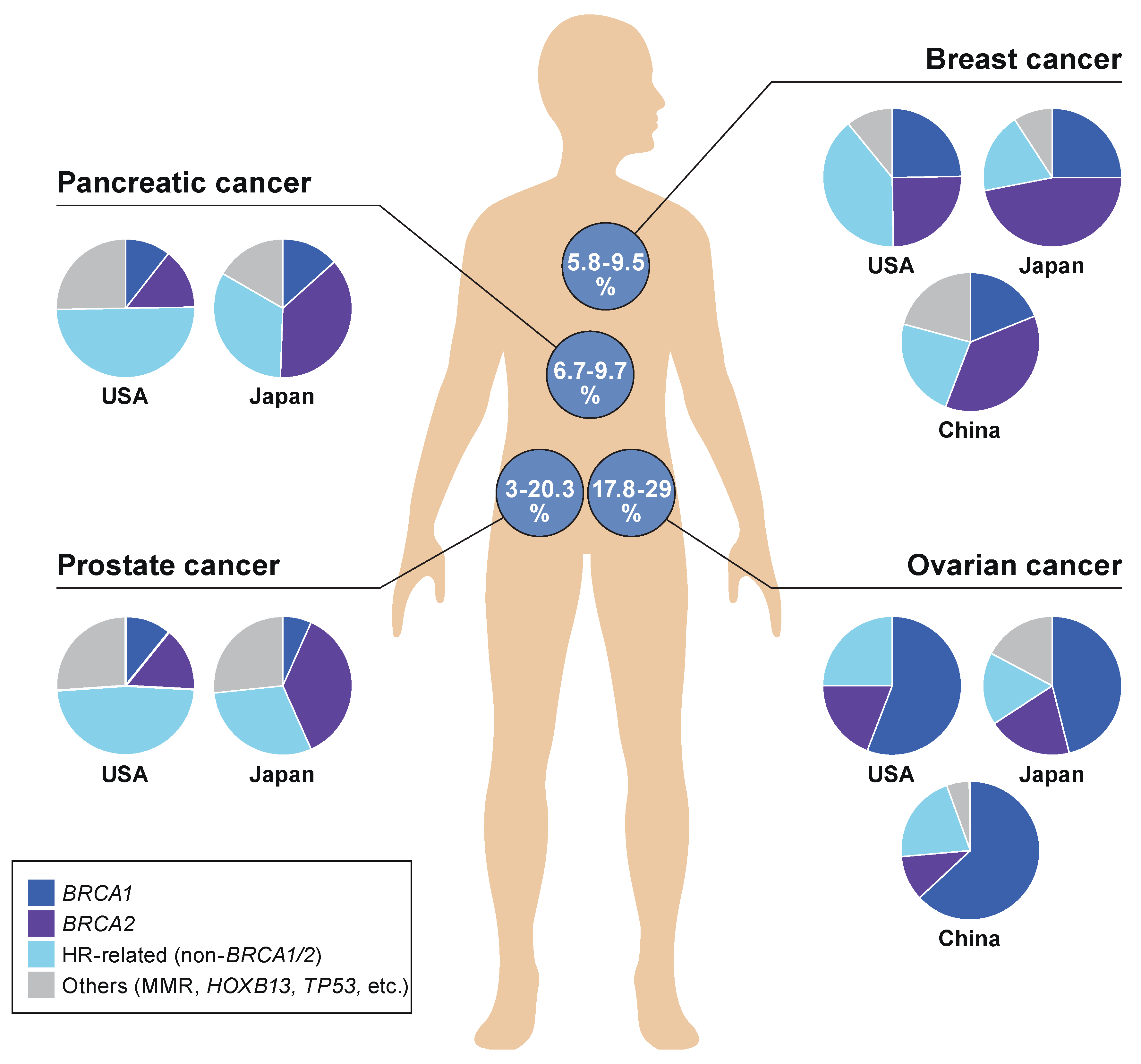
According to literature reviews performed by the authors, the detection of any of the GPVs is observed most frequently in ovarian cancer at 17.8–29%, followed by breast and pancreatic cancers at 5.8–9.5% and 6.7–9.3%, respectively, among all cancer patients involved in each type of cancer [47,52,82,110,111,112,113,114,115,116,117,118] (Figure 2). The frequency of GPVs observed in prostate cancer was significantly more in the USA (20.3%) than in Japan (3.0%), based on the results of MGPT analyses performed independently in the two countries. Deleterious variants of HR-related genes other than BRCA1/2 were dominant in pancreatic cancer patients as well as prostate cancer patients in the USA, whereas BRCA1/2 variants were most frequently detected as inherited predisposition factors in breast and ovarian cancers in the USA, China, and Japan.
There is also emerging discussion over the possible ethnic differences in germline variants of HR genes represented by BRCA1 and BRCA2 or ATM. The Global Alliance for Genomics and Health (GA4GH) launched a framework known as the BRCA Exchange (https://brcaexchange.org/, accessed on 20 December 2021) for international data sharing of pathogenic variants in BRCA1 and BRCA2. Another international workshop for the ATM and cancer risk has begun to collect population-based worldwide data of germline variants in ATM associated with HBOC [119]. Through those international initiatives for data sharing, it is reasonably understood that ethnic background can be taken into account for the interpretation of variants of HR-related genes and risk intervention strategies for HBOC.
MGPT can be a pervasive modality for hereditary tumor diagnosis owing to its advantages of increased throughput and reduced overall turnaround time, duplication, and cost. Although the method potentially increases the chances of encountering variants of unknown significance (VUS), optimizing MGPT at an appropriate time point for each proband and relative will be essential in the clinical practice of hereditary tumors including HR.
5. Conclusions
We have reviewed and discussed functions of representative HR molecules that are highly conserved across species and are involved in repair mechanisms, which are complex and precise against DNA double-strand breaks. HR consists of four main steps: sensing, end resection, loading of RAD51, and strand invasion. The penetrance of each HR-related gene variant varies, reflecting the distinct risks and proper intensities of cancer susceptibility and incidence of hereditary tumors. Pathogenic variants in BRCA1/2 convey the highest risk of HBOC-associated cancers. We also discussed the advantages of utilizing inherited impairment of HR, such as in the drug application for PARPi and platinum chemotherapy, the application of which can be discussed based on the molecular characteristics of HR in malignancies. MGPT is becoming a standard modality to screen possible hereditary tumor-related genes, including HR genes, with a higher potential of comprehensive screening for inherited cancer susceptibilities and an accurate diagnosis of hereditary tumors.
Author Contributions
H.Y. and A.H. have contributed to researching the literature and in drafting and editing of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partly supported by the Health Labour Sciences Research Grant (20EA1027), Foundation for Promotion of Cancer Research, Daiwa Securities Health Foundation, and Kobayashi Foundation for Cancer Research.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
Schema in figurers were designed and illustrated through kind cooperation with Reno Medical K.K. (Tokyo, Japan) (www.reno.co.jp, accessed on 26 November 2021).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ATM | ataxia telangiectasia mutated |
| ATR | ATM- and Rad3-related |
| BACH1 | BRCA1-associated C-terminal helicase 1 |
| BARD1 | BRCA1 Associated RING domain 1 |
| BRIP1 | BRCA1 interacting helicase 1 |
| CAP | College of American Pathologist |
| CHEK2 | checkpoint kinase 2 |
| CHK2 | CHEK2 gene coding checkpoint kinase 2 protein |
| CLIA | Clinical Laboratory Improvement Amendments |
| CtIP | CtBP-interacting protein |
| DSB | double strand break |
| GPV | germline pathogenic variant |
| HBOC | hereditary breast and ovarian cancer syndrome |
| HER2 | human epidermal growth factor |
| HGSC | high-grade serous carcinoma |
| HR | homologous recombination |
| HRD | homologous recombination deficiency |
| MGPT | multi-gene panel testing |
| NGS | next-generation sequencing |
| MMEJ | microhomology-mediated end-joining |
| NHEJ | non-homologous end-joining |
| MRN | MRE11-RAD50-NBS1 (or NBN) |
| NES | nuclear exporting signal |
| PALB2 | partner and localizer of BRCA2 |
| PARP | poly (ADP-ribose) polymerase |
| PARPi | poly (ADP-ribose) polymerase inhibitors |
| RPA | replication protein A |
| RRSO | risk-reducing salpingo-oophorectomy |
| TNBC | triple-negative breast cancer |
| VUS | variant of unknown significance |
References
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Xu, X. Microhomology-mediated end joining: New players join the team. Cell Biosci. 2017, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Ueki, A.; Hirasawa, A. Molecular Features and Clinical Management of Hereditary Gynecological Cancers. Int. J. Mol. Sci. 2020, 21, 9504. [Google Scholar] [CrossRef] [PubMed]
- Piombino, C.; Cortesi, L.; Lambertini, M.; Punie, K.; Grandi, G.; Toss, A. Secondary Prevention in Hereditary Breast and/or Ovarian Cancer Syndromes Other Than BRCA. J. Oncol. 2020, 2020, 6384190. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- Takaya, H.; Nakai, H.; Takamatsu, S.; Mandai, M.; Matsumura, N. Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma. Sci. Rep. 2020, 10, 2757. [Google Scholar] [CrossRef] [Green Version]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585–598. [Google Scholar] [CrossRef]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Eckert-Boulet, N.; Rothstein, R.; Lisby, M. Cell biology of homologous recombination in yeast. Methods Mol. Biol. 2011, 745, 523–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Hurov, K.; Hofmann, K.; Elledge, S.J. NBA1, a new player in the Brca1 A complex, is required for DNA damage resistance and checkpoint control. Genes Dev. 2009, 23, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Coleman, K.A.; Greenberg, R.A. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J. Biol. Chem. 2011, 286, 13669–13680. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, R. Hereditary breast and ovarian cancer (HBOC): Review of its molecular characteristics, screening, treatment, and prognosis. Breast Cancer 2020, 28, 1167–1180. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Stewart, G.S.; Maser, R.S.; Stankovic, T.; Bressan, D.A.; Kaplan, M.I.; Jaspers, N.G.; Raams, A.; Byrd, P.J.; Petrini, J.H.; Taylor, A.M. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 1999, 99, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Varon, R.; Vissinga, C.; Platzer, M.; Cerosaletti, K.M.; Chrzanowska, K.H.; Saar, K.; Beckmann, G.; Seemanova, E.; Cooper, P.R.; Nowak, N.J.; et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 1998, 93, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, R.A.; Lee, J.H.; Paull, T.T. Rad50 ATPase activity is regulated by DNA ends and requires coordination of both active sites. Nucleic Acids Res. 2017, 45, 5255–5268. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T.; Gellert, M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell 1998, 1, 969–979. [Google Scholar] [CrossRef]
- Zhao, H.; Watkins, J.L.; Piwnica-Worms, H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc. Natl. Acad. Sci. USA 2002, 99, 14795–14800. [Google Scholar] [CrossRef] [Green Version]
- Stolarova, L.; Kleiblova, P.; Janatova, M.; Soukupova, J.; Zemankova, P.; Macurek, L.; Kleibl, Z. CHEK2 Germline Variants in Cancer Predisposition: Stalemate Rather than Checkmate. Cells 2020, 9, 2675. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem. 2001, 276, 14537–14540. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Matsuoka, S.; Ballif, B.A.; Zhang, D.; Smogorzewska, A.; Gygi, S.P.; Elledge, S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007, 316, 1194–1198. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.X. BRCA1: Cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006, 34, 1416–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, H.; Hirasawa, A. Hereditary Gynecological Malignancy and Molecular Features. In Molecular Diagnosis and Targeting for Gynecologic Malignancy; Springer Nature Singapore Pte Ltd.: Singapore, 2021; pp. 145–165. [Google Scholar]
- Toss, A.; Tenedini, E.; Piombino, C.; Venturelli, M.; Marchi, I.; Gasparini, E.; Barbieri, E.; Razzaboni, E.; Domati, F.; Caggia, F.; et al. Clinicopathologic Profile of Breast Cancer in Germline ATM and CHEK2 Mutation Carriers. Genes 2021, 12, 616. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Canman, C.E.; Taya, Y.; Sakaguchi, K.; Appella, E.; Kastan, M.B. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997, 11, 3471–3481. [Google Scholar] [CrossRef] [Green Version]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Nevanlinna, H.; Bartek, J. The CHEK2 gene and inherited breast cancer susceptibility. Oncogene 2006, 25, 5912–5919. [Google Scholar] [CrossRef] [Green Version]
- Levitus, M.; Waisfisz, Q.; Godthelp, B.C.; de Vries, Y.; Hussain, S.; Wiegant, W.W.; Elghalbzouri-Maghrani, E.; Steltenpool, J.; Rooimans, M.A.; Pals, G.; et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J. Nat. Genet. 2005, 37, 934–935. [Google Scholar] [CrossRef] [PubMed]
- Ouhtit, A.; Gupta, I.; Shaikh, Z. BRIP1, a potential candidate gene in development of non-BRCA1/2 breast cancer. Front. Biosci. 2016, 8, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.B.; Wu, H.T.; Zhang, M.L.; Liu, J.; Zhang, G.J. Fanconi Anemia Pathway: Mechanisms of Breast Cancer Predisposition Development and Potential Therapeutic Targets. Front. Cell Dev. Biol. 2020, 8, 160. [Google Scholar] [CrossRef]
- Guenard, F.; Labrie, Y.; Ouellette, G.; Beauparlant, C.J.; Simard, J.; BRCAs, I.; Durocher, F. Mutational analysis of the breast cancer susceptibility gene BRIP1 /BACH1/FANCJ in high-risk non-BRCA1/BRCA2 breast cancer families. J. Hum. Genet. 2008, 53, 579. [Google Scholar] [CrossRef] [Green Version]
- Suszynska, M.; Ratajska, M.; Kozlowski, P. BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: Mutation prevalence and precise risk estimates based on a pooled analysis of ~30,000 cases. J. Ovarian Res. 2020, 13, 50. [Google Scholar] [CrossRef]
- Ali, M.; Delozier, C.D.; Chaudhary, U. BRIP-1 germline mutation and its role in colon cancer: Presentation of two case reports and review of literature. BMC Med. Genet. 2019, 20, 75. [Google Scholar] [CrossRef] [Green Version]
- Weber-Lassalle, N.; Borde, J.; Weber-Lassalle, K.; Horvath, J.; Niederacher, D.; Arnold, N.; Kaulfuss, S.; Ernst, C.; Paul, V.G.; Honisch, E.; et al. Germline loss-of-function variants in the BARD1 gene are associated with early-onset familial breast cancer but not ovarian cancer. Breast Cancer Res. 2019, 21, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef]
- Rahman, N.; Seal, S.; Thompson, D.; Kelly, P.; Renwick, A.; Elliott, A.; Reid, S.; Spanova, K.; Barfoot, R.; Chagtai, T.; et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 2007, 39, 165–167. [Google Scholar] [CrossRef] [Green Version]
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncologist 2021, 26, e1526–e1537. [Google Scholar] [CrossRef]
- Slavin, T.P.; Maxwell, K.N.; Lilyquist, J.; Vijai, J.; Neuhausen, S.L.; Hart, S.N.; Ravichandran, V.; Thomas, T.; Maria, A.; Villano, D.; et al. The contribution of pathogenic variants in breast cancer susceptibility genes to familial breast cancer risk. NPJ Breast Cancer 2017, 3, 22. [Google Scholar] [CrossRef]
- Abe, K.; Ueki, A.; Urakawa, Y.; Kitago, M.; Yoshihama, T.; Nanki, Y.; Kitagawa, Y.; Aoki, D.; Kosaki, K.; Hirasawa, A. Familial pancreatic cancer with PALB2 and NBN pathogenic variants: A case report. Hered Cancer Clin. Pr. 2021, 19, 5. [Google Scholar] [CrossRef]
- Canto, M.I.; Harinck, F.; Hruban, R.H.; Offerhaus, G.J.; Poley, J.W.; Kamel, I.; Nio, Y.; Schulick, R.S.; Bassi, C.; Kluijt, I.; et al. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013, 62, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Kitago, M.; Kitagawa, Y.; Hirasawa, A. Hereditary pancreatic cancer. Int. J. Clin. Oncol. 2021, 26, 1784–1792. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. Linking the cellular functions of BRCA genes to cancer pathogenesis and treatment. Annu. Rev. Pathol. 2009, 4, 461–487. [Google Scholar] [CrossRef] [PubMed]
- Esashi, F.; Christ, N.; Gannon, J.; Liu, Y.; Hunt, T.; Jasin, M.; West, S.C. CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 2005, 434, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef]
- Hirasawa, A.; Imoto, I.; Naruto, T.; Akahane, T.; Yamagami, W.; Nomura, H.; Masuda, K.; Susumu, N.; Tsuda, H.; Aoki, D. Prevalence of pathogenic germline variants detected by multigene sequencing in unselected Japanese patients with ovarian cancer. Oncotarget 2017, 8, 112258–112267. [Google Scholar] [CrossRef] [Green Version]
- Futagawa, M.; Yamamoto, H.; Kochi, M.; Urakawa, Y.; Sogawa, R.; Kato, F.; Okazawa-Sakai, M.; Ennishi, D.; Shinozaki, K.; Inoue, H.; et al. Retroperitoneal leiomyosarcoma in a female patient with a germline splicing variant RAD51D c.904-2A > T: A case report. Hered Cancer Clin. Pr. 2021, 19, 48. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. Cancer suppression by the chromosome custodians, BRCA1 and BRCA2. Science 2014, 343, 1470–1475. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.C.; Pharoah, P.D.; McMullan, G.; Day, N.E.; Ponder, B.A.; Easton, D. Evidence for further breast cancer susceptibility genes in addition to BRCA1 and BRCA2 in a population-based study. Genet. Epidemiol. 2001, 21, 1–18. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Ho, B.N.; Singh, A.T.K. The Evolution, Functions and Applications of the Breast Cancer Genes BRCA1 and BRCA2. Cancer Genom. Proteom. 2017, 14, 293–298. [Google Scholar] [CrossRef] [Green Version]
- Momozawa, Y.; Iwasaki, Y.; Hirata, M.; Liu, X.; Kamatani, Y.; Takahashi, A.; Sugano, K.; Yoshida, T.; Murakami, Y.; Matsuda, K.; et al. Germline Pathogenic Variants in 7636 Japanese Patients With Prostate Cancer and 12,366 Controls. J. Natl. Cancer Inst. 2020, 112, 369–376. [Google Scholar] [CrossRef] [Green Version]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Hart, S.N.; Polley, E.C.; Gnanaolivu, R.; Shimelis, H.; Lee, K.Y.; Lilyquist, J.; Na, J.; Moore, R.; Antwi, S.O.; et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA 2018, 319, 2401–2409. [Google Scholar] [CrossRef]
- Hu, C.; LaDuca, H.; Shimelis, H.; Polley, E.C.; Lilyquist, J.; Hart, S.N.; Na, J.; Thomas, A.; Lee, K.Y.; Davis, B.T.; et al. Multigene Hereditary Cancer Panels Reveal High-Risk Pancreatic Cancer Susceptibility Genes. JCO Precis. Oncol. 2018, 2, 1–28. [Google Scholar] [CrossRef]
- AlDubayan, S.H.; Giannakis, M.; Moore, N.D.; Han, G.C.; Reardon, B.; Hamada, T.; Mu, X.J.; Nishihara, R.; Qian, Z.; Liu, L.; et al. Inherited DNA-Repair Defects in Colorectal Cancer. Am. J. Hum. Genet. 2018, 102, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Kurian, A.W.; Hughes, E.; Handorf, E.A.; Gutin, A.; Allen, B.; Hartman, A.R.; Hall, M.J. Breast and Ovarian Cancer Penetrance Estimates Derived From Germline Multiple-Gene Sequencing Results in Women. JCO Precis. Oncol. 2017, 1, 1–12. [Google Scholar] [CrossRef]
- Lilyquist, J.; LaDuca, H.; Polley, E.; Davis, B.T.; Shimelis, H.; Hu, C.; Hart, S.N.; Dolinsky, J.S.; Couch, F.J.; Goldgar, D.E. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol. Oncol. 2017, 147, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [Green Version]
- Antoniou, A.; Pharoah, P.D.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, A.; et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 2003, 72, 1117–1130. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef] [Green Version]
- Brose, M.S.; Rebbeck, T.R.; Calzone, K.A.; Stopfer, J.E.; Nathanson, K.L.; Weber, B.L. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J. Natl. Cancer Inst. 2002, 94, 1365–1372. [Google Scholar] [CrossRef]
- Finch, A.; Beiner, M.; Lubinski, J.; Lynch, H.T.; Moller, P.; Rosen, B.; Murphy, J.; Ghadirian, P.; Friedman, E.; Foulkes, W.D.; et al. Salpingo-oophorectomy and the risk of ovarian, fallopian tube, and peritoneal cancers in women with a BRCA1 or BRCA2 Mutation. JAMA 2006, 296, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Levine, D.A.; Argenta, P.A.; Yee, C.J.; Marshall, D.S.; Olvera, N.; Bogomolniy, F.; Rahaman, J.A.; Robson, M.E.; Offit, K.; Barakat, R.R.; et al. Fallopian tube and primary peritoneal carcinomas associated with BRCA mutations. J. Clin. Oncol. 2003, 21, 4222–4227. [Google Scholar] [CrossRef]
- Levy-Lahad, E.; Friedman, E. Cancer risks among BRCA1 and BRCA2 mutation carriers. Brit. J. Cancer 2007, 96, 11–15. [Google Scholar] [CrossRef]
- Oh, M.; Alkhushaym, N.; Fallatah, S.; Althagafi, A.; Aljadeed, R.; Alsowaida, Y.; Jeter, J.; Martin, J.R.; Babiker, H.M.; McBride, A.; et al. The association of BRCA1 and BRCA2 mutations with prostate cancer risk, frequency, and mortality: A meta-analysis. Prostate 2019, 79, 880–895. [Google Scholar] [CrossRef]
- Nyberg, T.; Frost, D.; Barrowdale, D.; Evans, D.G.; Bancroft, E.; Adlard, J.; Ahmed, M.; Barwell, J.; Brady, A.F.; Brewer, C.; et al. Prostate Cancer Risks for Male BRCA1 and BRCA2 Mutation Carriers: A Prospective Cohort Study. Eur. Urol. 2020, 77, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Ferrone, C.R.; Levine, D.A.; Tang, L.H.; Allen, P.J.; Jarnagin, W.; Brennan, M.F.; Offit, K.; Robson, M.E. BRCA germline mutations in Jewish patients with pancreatic adenocarcinoma. J. Clin. Oncol. 2009, 27, 433–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavanagh, H.; Rogers, K.M. The role of BRCA1 and BRCA2 mutations in prostate, pancreatic and stomach cancers. Hered Cancer Clin. Pr. 2015, 13, 16. [Google Scholar] [CrossRef] [Green Version]
- Rafnar, T.; Gudbjartsson, D.F.; Sulem, P.; Jonasdottir, A.; Sigurdsson, A.; Jonasdottir, A.; Besenbacher, S.; Lundin, P.; Stacey, S.N.; Gudmundsson, J.; et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat. Genet. 2011, 43, 1104–1107. [Google Scholar] [CrossRef]
- Cybulski, C.; Wokolorczyk, D.; Jakubowska, A.; Huzarski, T.; Byrski, T.; Gronwald, J.; Masojc, B.; Deebniak, T.; Gorski, B.; Blecharz, P.; et al. Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J. Clin. Oncol. 2011, 29, 3747–3752. [Google Scholar] [CrossRef] [PubMed]
- Brandao, A.; Paulo, P.; Maia, S.; Pinheiro, M.; Peixoto, A.; Cardoso, M.; Silva, M.P.; Santos, C.; Eeles, R.A.; Kote-Jarai, Z.; et al. The CHEK2 Variant C.349A > G Is Associated with Prostate Cancer Risk and Carriers Share a Common Ancestor. Cancers 2020, 12, 3254. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Gorski, B.; Huzarski, T.; Masojc, B.; Mierzejewski, M.; Debniak, T.; Teodorczyk, U.; Byrski, T.; Gronwald, J.; Matyjasik, J.; et al. CHEK2 is a multiorgan cancer susceptibility gene. Am. J. Hum. Genet. 2004, 75, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Thompson, E.R.; Rowley, S.M.; Li, N.; McInerny, S.; Devereux, L.; Wong-Brown, M.W.; Trainer, A.H.; Mitchell, G.; Scott, R.J.; James, P.A.; et al. Panel Testing for Familial Breast Cancer: Calibrating the Tension Between Research and Clinical Care. J. Clin. Oncol. 2016, 34, 1455–1459. [Google Scholar] [CrossRef] [Green Version]
- Steffen, J.; Nowakowska, D.; Niwinska, A.; Czapczak, D.; Kluska, A.; Piatkowska, M.; Wisniewska, A.; Paszko, Z. Germline mutations 657del5 of the NBS1 gene contribute significantly to the incidence of breast cancer in Central Poland. Int. J. Cancer 2006, 119, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Gorski, B.; Debniak, T.; Gliniewicz, B.; Mierzejewski, M.; Masojc, B.; Jakubowska, A.; Matyjasik, J.; Zlowocka, E.; Sikorski, A.; et al. NBS1 is a prostate cancer susceptibility gene. Cancer Res. 2004, 64, 1215–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, S.; Schindler, D.; Hanenberg, H.; Barker, K.; Hanks, S.; Kalb, R.; Neveling, K.; Kelly, P.; Seal, S.; Freund, M.; et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat. Genet. 2007, 39, 162–164. [Google Scholar] [CrossRef]
- Antoniou, A.C.; Casadei, S.; Heikkinen, T.; Barrowdale, D.; Pylkas, K.; Roberts, J.; Lee, A.; Subramanian, D.; De Leeneer, K.; Fostira, F.; et al. Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 2014, 371, 497–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Dicks, E.; Ramus, S.J.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef] [Green Version]
- Petrucelli, N.; Daly, M.B.; Pal, T. BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef] [Green Version]
- Cortesi, L.; Toss, A.; Cucinotto, I. PARP Inhibitors for the Treatment of Ovarian Cancer. Curr. Cancer Drug Targets 2018, 18, 877–893. [Google Scholar] [CrossRef]
- Loizzi, V.; Ranieri, G.; Laforgia, M.; Gadaleta, C.D.; Gargano, G.; Kardhashi, A.; De Liso, M.; Naglieri, E.; Del Vecchio, V.; Cicinelli, E.; et al. PARP inhibitors and epithelial ovarian cancer: Molecular mechanisms, clinical development and future prospective. Oncol. Lett. 2020, 20, 90. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Burstein, H.J.; Schwartz, R.S. Molecular origins of cancer. N. Engl. J. Med. 2008, 358, 527. [Google Scholar] [CrossRef]
- Cortesi, L.; Piombino, C.; Toss, A. Germline Mutations in Other Homologous Recombination Repair-Related Genes Than BRCA1/2: Predictive or Prognostic Factors? J. Pers. Med. 2021, 11, 245. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Kasi, P.M.; Bamlet, W.R.; Ho, T.P.; Polley, E.C.; Hu, C.; Hart, S.N.; Rabe, K.G.; Boddicker, N.J.; Gnanaolivu, R.D.; et al. Effect of Germline Mutations in Homologous Recombination Repair Genes on Overall Survival of Patients with Pancreatic Adenocarcinoma. Clin. Cancer Res. 2020, 26, 6505–6512. [Google Scholar] [CrossRef]
- Nguyen, L.; Martens, J.W.M.; Van Hoeck, A.; Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef] [PubMed]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [Green Version]
- Casolino, R.; Paiella, S.; Azzolina, D.; Beer, P.A.; Corbo, V.; Lorenzoni, G.; Gregori, D.; Golan, T.; Braconi, C.; Froeling, F.E.M.; et al. Homologous Recombination Deficiency in Pancreatic Cancer: A Systematic Review and Prevalence Meta-Analysis. J. Clin. Oncol. 2021, 39, 2617–2631. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Norquist, B.; Lacchetti, C.; Armstrong, D.; Grisham, R.N.; Goodfellow, P.J.; Kohn, E.C.; Levine, D.A.; Liu, J.F.; Lu, K.H.; et al. Germline and Somatic Tumor Testing in Epithelial Ovarian Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 1222–1245. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Perol, D.; Gonzalez-Martin, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Maenpaa, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Jonsson, P.; Bandlamudi, C.; Cheng, M.L.; Srinivasan, P.; Chavan, S.S.; Friedman, N.D.; Rosen, E.Y.; Richards, A.L.; Bouvier, N.; Selcuklu, S.D.; et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature 2019, 571, 576–579. [Google Scholar] [CrossRef]
- Hosey, A.M.; Gorski, J.J.; Murray, M.M.; Quinn, J.E.; Chung, W.Y.; Stewart, G.E.; James, C.R.; Farragher, S.M.; Mulligan, J.M.; Scott, A.N.; et al. Molecular basis for estrogen receptor alpha deficiency in BRCA1-linked breast cancer. J. Natl. Cancer Inst. 2007, 99, 1683–1694. [Google Scholar] [CrossRef] [Green Version]
- Miao, K.; Lei, J.H.; Valecha, M.V.; Zhang, A.; Xu, J.; Wang, L.; Lyu, X.; Chen, S.; Miao, Z.; Zhang, X.; et al. NOTCH1 activation compensates BRCA1 deficiency and promotes triple-negative breast cancer formation. Nat. Commun. 2020, 11, 3256. [Google Scholar] [CrossRef] [PubMed]
- Sonderstrup, I.M.H.; Jensen, M.R.; Ejlertsen, B.; Eriksen, J.O.; Gerdes, A.M.; Kruse, T.A.; Larsen, M.J.; Thomassen, M.; Laenkholm, A.V. Subtypes in BRCA-mutated breast cancer. Hum. Pathol. 2019, 84, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spurdle, A.B.; Couch, F.J.; Parsons, M.T.; McGuffog, L.; Barrowdale, D.; Bolla, M.K.; Wang, Q.; Healey, S.; Schmutzler, R.; Wappenschmidt, B.; et al. Refined histopathological predictors of BRCA1 and BRCA2 mutation status: A large-scale analysis of breast cancer characteristics from the BCAC, CIMBA, and ENIGMA consortia. Breast Cancer Res. 2014, 16, 3419. [Google Scholar] [CrossRef] [Green Version]
- Limaiem, F.; Mlika, M. Medullary Breast Carcinoma. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://pubmed.ncbi.nlm.nih.gov/31194432/ (accessed on 23 November 2021).
- Taylor, R.A.; Fraser, M.; Rebello, R.J.; Boutros, P.C.; Murphy, D.G.; Bristow, R.G.; Risbridger, G.P. The influence of BRCA2 mutation on localized prostate cancer. Nat. Rev. Urol. 2019, 16, 281–290. [Google Scholar] [CrossRef]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef]
- Swisher, E.M.; Sakai, W.; Karlan, B.Y.; Wurz, K.; Urban, N.; Taniguchi, T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008, 68, 2581–2586. [Google Scholar] [CrossRef] [Green Version]
- Waks, A.G.; Cohen, O.; Kochupurakkal, B.; Kim, D.; Dunn, C.E.; Buendia Buendia, J.; Wander, S.; Helvie, K.; Lloyd, M.R.; Marini, L.; et al. Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Ann. Oncol. 2020, 31, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Cook-Deegan, R.; Niehaus, A. After Myriad: Genetic Testing in the Wake of Recent Supreme Court Decisions about Gene Patents. Curr. Genet. Med. Rep. 2014, 2, 223–241. [Google Scholar] [CrossRef] [Green Version]
- Tung, N.; Battelli, C.; Allen, B.; Kaldate, R.; Bhatnagar, S.; Bowles, K.; Timms, K.; Garber, J.E.; Herold, C.; Ellisen, L.; et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 2015, 121, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Buys, S.S.; Sandbach, J.F.; Gammon, A.; Patel, G.; Kidd, J.; Brown, K.L.; Sharma, L.; Saam, J.; Lancaster, J.; Daly, M.B. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 2017, 123, 1721–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momozawa, Y.; Iwasaki, Y.; Parsons, M.T.; Kamatani, Y.; Takahashi, A.; Tamura, C.; Katagiri, T.; Yoshida, T.; Nakamura, S.; Sugano, K.; et al. Germline pathogenic variants of 11 breast cancer genes in 7,051 Japanese patients and 11,241 controls. Nat. Commun. 2018, 9, 4083. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Meng, H.; Yao, L.; Lv, M.; Bai, J.; Zhang, J.; Wang, L.; Ouyang, T.; Li, J.; Wang, T.; et al. Germline Mutations in Cancer Susceptibility Genes in a Large Series of Unselected Breast Cancer Patients. Clin. Cancer Res. 2017, 23, 6113–6119. [Google Scholar] [CrossRef] [Green Version]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Shao, D.; Li, L.; Wu, M.; Ma, S.; Tan, X.; Zhong, S.; Guo, F.; Wang, Z.; Ye, M. Germline and somatic mutations of multi-gene panel in Chinese patients with epithelial ovarian cancer: A prospective cohort study. J. Ovarian Res. 2019, 12, 80. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Chittenden, A.B.; Morales-Oyarvide, V.; Rubinson, D.A.; Dunne, R.F.; Kozak, M.M.; Qian, Z.R.; Welch, M.W.; Brais, L.K.; Da Silva, A.; et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet. Med. 2019, 21, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Mizukami, K.; Iwasaki, Y.; Kawakami, E.; Hirata, M.; Kamatani, Y.; Matsuda, K.; Endo, M.; Sugano, K.; Yoshida, T.; Murakami, Y.; et al. Genetic characterization of pancreatic cancer patients and prediction of carrier status of germline pathogenic variants in cancer-predisposing genes. EBioMedicine 2020, 60, 103033. [Google Scholar] [CrossRef]
- Nicolosi, P.; Ledet, E.; Yang, S.; Michalski, S.; Freschi, B.; O’Leary, E.; Esplin, E.D.; Nussbaum, R.L.; Sartor, O. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol. 2019, 5, 523–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesueur, F.; Easton, D.F.; Renault, A.L.; Tavtigian, S.V.; Bernstein, J.L.; Kote-Jarai, Z.; Eeles, R.A.; Plaseska-Karanfia, D.; Feliubadalo, L.; Spanish ATM Working Group; et al. First international workshop of the ATM and cancer risk group (4–5 December 2019). Fam. Cancer 2021, 1–17. [Google Scholar] [CrossRef]
Figure 1.
DNA double-strand breaks (DSBs) repair by homologous recombination (HR) and key molecules (MRN complex, ATM, CHK2, BRCA1, PALB2, BARD1, BRIP1, BRCA2, RAD51, RAD51C, and RAD51D). Part 1 (overall HR pathway against DSBs). In response to DSBs or stalled replication fork (not shown), the initial step for HR is the sensing of damaged sites by the MRN complex, which is comprised of MRE11, RAD50, and Nijmegen breakage syndrome protein 1 (NBS1 or, alternatively, NBN), followed by the resection procedure. Either side of DNA is lysed from 5′ to 3′ by MRE11 (shown like “PAC-MAN” in this figure) with its exonuclease and endonuclease activities and BRCA1 enzymatic activities, forming an overhanging 3′ single-stranded DNA (ssDNA) end which can invade homologous template. RAD51 is recruited by the BRCA1–PALB2–BRCA2 effector complex and is loaded on ssDNA to form RAD51–ssDNA nucleofilament. Recombinase RAD51 synthesizes DNA using sister chromatids as a template, extending to the second DNA end, which is terminated by D-loop capture (not shown), and forms a double Holliday junction (not shown), resulting in non-crossover or crossover outcomes via resolution. Parts 2–4 (magnified view of BRCA1–PALB2–BRCA2 complex and molecular associates). In response to DSBs, a serine/threonine kinase ATM, another sensor molecule of DSBs, is recruited with checkpoint activation and phosphorylation of checkpoint kinase 2 (CHK2) to recruit the BRCA complex (Part 2). BRCA1 (1863 amino acids (a.a.)) is phosphorylated by ATM or ATR (ATR works when stalled replication fork occurs) at several motifs containing serine residues. Another serine residue (S988), located in the center of BRCA1, is phosphorylated by CHK2. Phosphorylated BRCA1 is recruited and accumulated at the damaged sites. BRCA1 functions as an adaptor for BRCA2 recruitment through the linking mediator of PALB2. BRCA1 functions by forming a heterodimeric complex with BARD1 via the RING finger domain at the N-terminus of BRCA1 and with BRIP1 through two BRCT domains at the carboxyl-terminus of BRCA1. PALB2 binds to BRCA1 at the coiled-coil domain via the N-terminal residue of PALB2 (Part 3). The N-terminus of BRCA2 interacts with PALB2 through its carboxyl terminus. BRCA2 (3418 amino acids (a.a.)) contains eight conserved motifs called BRC repeats in the middle part encoded by exon 11 to which RAD51s are bound. The carboxyl-terminus of BRCA2 contains a TR2 domain, another binding site for RAD51. BRCA2 serine 3291 (S3,291) phosphorylation, which is required for binding of RAD51 to BRCA2, is regulated by cyclin-dependent kinase 2 (CDK2). RAD51 paralog complexes (RAD51B–RAD51C–RAD51D-XRCC2 (BCDX2) and RAD51C–XRCC3 (CX3)) interact with RAD51 under the regulation of the BRCA1–PALB2–BRCA2 complex and may promote RAD51 nucleofilament formation (Part 4).
Figure 1.
DNA double-strand breaks (DSBs) repair by homologous recombination (HR) and key molecules (MRN complex, ATM, CHK2, BRCA1, PALB2, BARD1, BRIP1, BRCA2, RAD51, RAD51C, and RAD51D). Part 1 (overall HR pathway against DSBs). In response to DSBs or stalled replication fork (not shown), the initial step for HR is the sensing of damaged sites by the MRN complex, which is comprised of MRE11, RAD50, and Nijmegen breakage syndrome protein 1 (NBS1 or, alternatively, NBN), followed by the resection procedure. Either side of DNA is lysed from 5′ to 3′ by MRE11 (shown like “PAC-MAN” in this figure) with its exonuclease and endonuclease activities and BRCA1 enzymatic activities, forming an overhanging 3′ single-stranded DNA (ssDNA) end which can invade homologous template. RAD51 is recruited by the BRCA1–PALB2–BRCA2 effector complex and is loaded on ssDNA to form RAD51–ssDNA nucleofilament. Recombinase RAD51 synthesizes DNA using sister chromatids as a template, extending to the second DNA end, which is terminated by D-loop capture (not shown), and forms a double Holliday junction (not shown), resulting in non-crossover or crossover outcomes via resolution. Parts 2–4 (magnified view of BRCA1–PALB2–BRCA2 complex and molecular associates). In response to DSBs, a serine/threonine kinase ATM, another sensor molecule of DSBs, is recruited with checkpoint activation and phosphorylation of checkpoint kinase 2 (CHK2) to recruit the BRCA complex (Part 2). BRCA1 (1863 amino acids (a.a.)) is phosphorylated by ATM or ATR (ATR works when stalled replication fork occurs) at several motifs containing serine residues. Another serine residue (S988), located in the center of BRCA1, is phosphorylated by CHK2. Phosphorylated BRCA1 is recruited and accumulated at the damaged sites. BRCA1 functions as an adaptor for BRCA2 recruitment through the linking mediator of PALB2. BRCA1 functions by forming a heterodimeric complex with BARD1 via the RING finger domain at the N-terminus of BRCA1 and with BRIP1 through two BRCT domains at the carboxyl-terminus of BRCA1. PALB2 binds to BRCA1 at the coiled-coil domain via the N-terminal residue of PALB2 (Part 3). The N-terminus of BRCA2 interacts with PALB2 through its carboxyl terminus. BRCA2 (3418 amino acids (a.a.)) contains eight conserved motifs called BRC repeats in the middle part encoded by exon 11 to which RAD51s are bound. The carboxyl-terminus of BRCA2 contains a TR2 domain, another binding site for RAD51. BRCA2 serine 3291 (S3,291) phosphorylation, which is required for binding of RAD51 to BRCA2, is regulated by cyclin-dependent kinase 2 (CDK2). RAD51 paralog complexes (RAD51B–RAD51C–RAD51D-XRCC2 (BCDX2) and RAD51C–XRCC3 (CX3)) interact with RAD51 under the regulation of the BRCA1–PALB2–BRCA2 complex and may promote RAD51 nucleofilament formation (Part 4).

Figure 2.
Prevalence of BRCA and non-BRCA in HRD-related hereditary tumors. The proportion of inheritance backgrounds due to cancer susceptibility gene alterations in each type of malignancy was summed as proportion rates (%), and is shown in circles on the human body figure. The summed rates (%) are based on the data of multi-gene panel testings (MGPTs), performed and analyzed in each country independently [47,52,111,112,113,114,115,116,117,118]. The proportions for the types of each gene alteration are categorized under any of these four groups: BRCA1, BRCA2, HR-related genes (non-BRCA1/2), or others (MMR, HOXB13, TP53, etc.), whose germline variants were determined as pathogenic/deleterious, and are shown in pie charts. Ovarian cancer includes fallopian tube and primary peritoneal carcinomas.
Figure 2.
Prevalence of BRCA and non-BRCA in HRD-related hereditary tumors. The proportion of inheritance backgrounds due to cancer susceptibility gene alterations in each type of malignancy was summed as proportion rates (%), and is shown in circles on the human body figure. The summed rates (%) are based on the data of multi-gene panel testings (MGPTs), performed and analyzed in each country independently [47,52,111,112,113,114,115,116,117,118]. The proportions for the types of each gene alteration are categorized under any of these four groups: BRCA1, BRCA2, HR-related genes (non-BRCA1/2), or others (MMR, HOXB13, TP53, etc.), whose germline variants were determined as pathogenic/deleterious, and are shown in pie charts. Ovarian cancer includes fallopian tube and primary peritoneal carcinomas.


{kind=link}
{kind=link}
Table 1.
Representative HR-related genes, functions, and susceptibility to cancer types due to germline variants.
Table 1.
Representative HR-related genes, functions, and susceptibility to cancer types due to germline variants.
| HR-Related Genes | Molecular Functions | Functions in HR | Susceptible Organs | Risk Estimates | References | |
|---|---|---|---|---|---|---|
| ATM | serine-threonine kinase/checkpoint activation (G2-M and G1-S checkpoints)/p53 activation | BRCA1 phosphorylation | Breast (luminal B-like/HER2-negative), Ovary, Pancreas Prostate, Colon | Breast cancer (2.8–3.3), Ovarian cancer (0.9–2.4), Pancreatic cancer (5.7–9.0), Prostate cancer (2.9), Colorectal cancer (2.8) | [40,52,53,54,55,56,57,58,59] | |
| BRCA1 (FANCS) | mediator/adaptor/enzymatic/cell cycle regulator (G2-M checkpoint control) | BRCT domain-mediated phospho-protein interactions | Breast (TNBC), Ovary (HGSC), Pancreas, Prostate | Breast cancer # (46–87% vs. 12%), Ovarian cancer # (39–63% vs. 1–2%), Pancreatic cancer # (1–3% vs. 0.50%), Prostate cancer # (8.6% by age 65 vs. 6% through age 69) | [54,60,61,62,63,64,65,66,67,68,69,70] | |
| BARD1 | constitutive RING-mediated heterodimerization with BRCA1 | forming BRCA1-BARD1 E3 ubiquitin ligase/interacting with BRCA1 through RING domain | Breast(early-age onset), Ovary | Breast cancer (1.9–3.2), Ovarian cancer (4.2) | [36,40,53,57,59] | |
| BRCA2 (FANCD1) | mediator of RAD51 | recombination mediator/RAD51 binding | Breast, Ovary (HGSC), Pancreas, Prostate, Skin (potential risk for Malignant Melanoma) | Breast cancer # (38–84% vs. 12%), Ovarian cancer # (16.5–27% vs. 1–2%), Pancreatic cancer # ( 2–7% vs. 0.50%), Prostate cancer # (15% by age 65; 20% lifetime vs. 6% through age 69) | [54,60,61,62,64,65,66,67,68,69,70] | |
| BRIP1 (FANCJ) | 5′ to 3′ DNA helicase/binds to BRCA1/phosphorylated following DNA damage | interacting with BRCA1 through BRCT domain/genome integrity/tumor suppressive | Breast, Ovary, Pancreas, Prostate | Breast cancer (1.6), Ovarian cancer (2.4–8.1), Pancreatic cancer (2.7) | [53,57,58,71] | |
| CHEK2 | serine/threonine kinase (ATM-mediated) | BRCA1 phosphorylation | Breast (luminal A-like), Prostate, Colon | Breast cancer (2.68), Prostate cancer (ethnic differences in CHEK2 variant frequencies) | [21,72,73,74] | |
| MRN complex | MRE11 (MRE11A) | enzymatic/nuclease | MRE11, endonuclease; 3′–5′ exonuclease; cooperate with CtIP to initiate DSB resection | Breast | Breast cancer (0.9–9.0) | [40,53,75] |
| RAD50 | structural/ATPase | RAD50, structural maintenance of chromosomes SMC-like protein | – | – | [53,58,59,75] | |
| NBS1 (NBN) | adaptor/checkpoint | NBS1 (NBN), phospho-protein and ATM kinase interactions | Breast, Ovary, Prostate | Breast cancer (3.2), Ovarian cancer (1.9), Prostate cancer (3.9) | [57,76,77] | |
| PALB2 (FANCN) | scaffold/mediator/partner for BRCA2 stability/nuclear localization | complex formation/linking BRCA1 and BRCA2 | Breast, Ovary, Pancreas, Prostate, Colon, Kidney (Wilm’s tumor), CNS (Medulloblastoma) | Breast cancer (2.3–9.5), Ovarian cancer (2.9–4.4), Pancreatic cancer (2.3–14.8), Prostate cancer (0.4), Colorectal cancer (4.9) | [6,53,54,55,56,57,58,59,75,78,79] | |
| RAD51C | RAD51 paralog | interacting with RAD51/forming subcomplex (BCDX2 complex) | Breast, Ovary, Pancreas | Breast cancer (0.4–1.4), Ovarian cancer (3.4–5.1) | [40,53,57,58,59,80] | |
| RAD51D | RAD51 paralog | interacting with RAD51/forming subcomplex (BCDX2 complex) | Breast, Ovary, Pancreas | Breast cancer (3.1–8.3), Ovarian cancer(4.8–10.9) | [40,53,57,58,59] | |
| RAD51 | recombinase | final effector of HR/nucleofilament formation/strand invasion | Not specified | – | – | |
| #: Estimated lifetime cumulative risks for the indicated malignancy due to pathogenic germline variants versus general population risks are shown as risk estimates by referring to GeneReviews® [Internet] https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK1247/ (accessed on 19 November 2021) and [81]. | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yamamoto, H.; Hirasawa, A. Homologous Recombination Deficiencies and Hereditary Tumors. Int. J. Mol. Sci. 2022, 23, 348. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010348
AMA Style
Yamamoto H, Hirasawa A. Homologous Recombination Deficiencies and Hereditary Tumors. International Journal of Molecular Sciences. 2022; 23(1):348. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010348
Chicago/Turabian StyleYamamoto, Hideki, and Akira Hirasawa. 2022. "Homologous Recombination Deficiencies and Hereditary Tumors" International Journal of Molecular Sciences 23, no. 1: 348. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010348
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.