
Attenuation of Adverse Postinfarction Left Ventricular Remodeling with Empagliflozin Enhances Mitochondria-Linked Cellular Energetics and Mitochondrial Biogenesis

 , ,
, ,
Abstract
:1. Introduction
2. Results
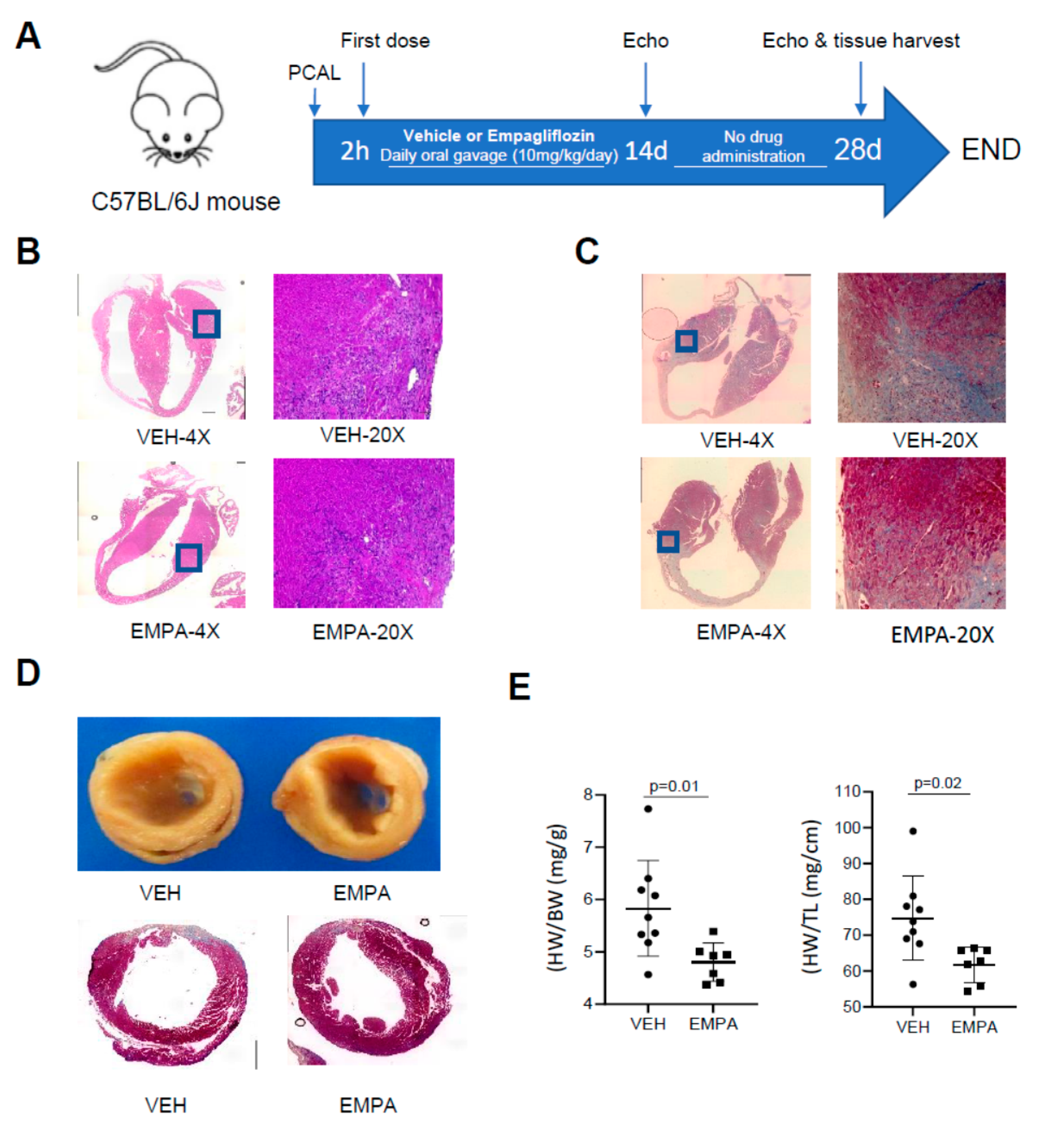
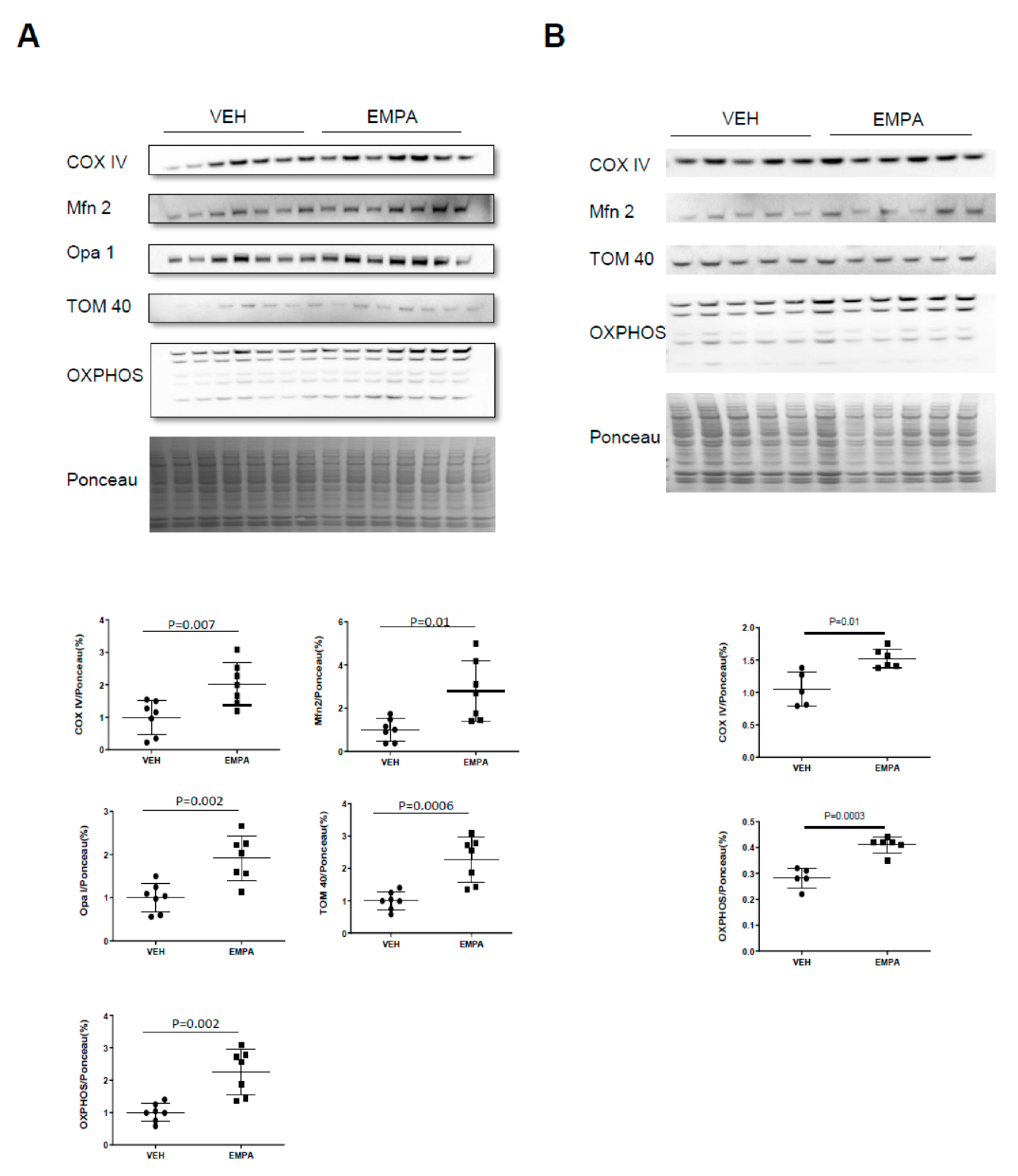
2.1. The Effect of EMPA on Postinfarction Adverse LV Remodeling in WT Mice and Immune Cell Infiltration
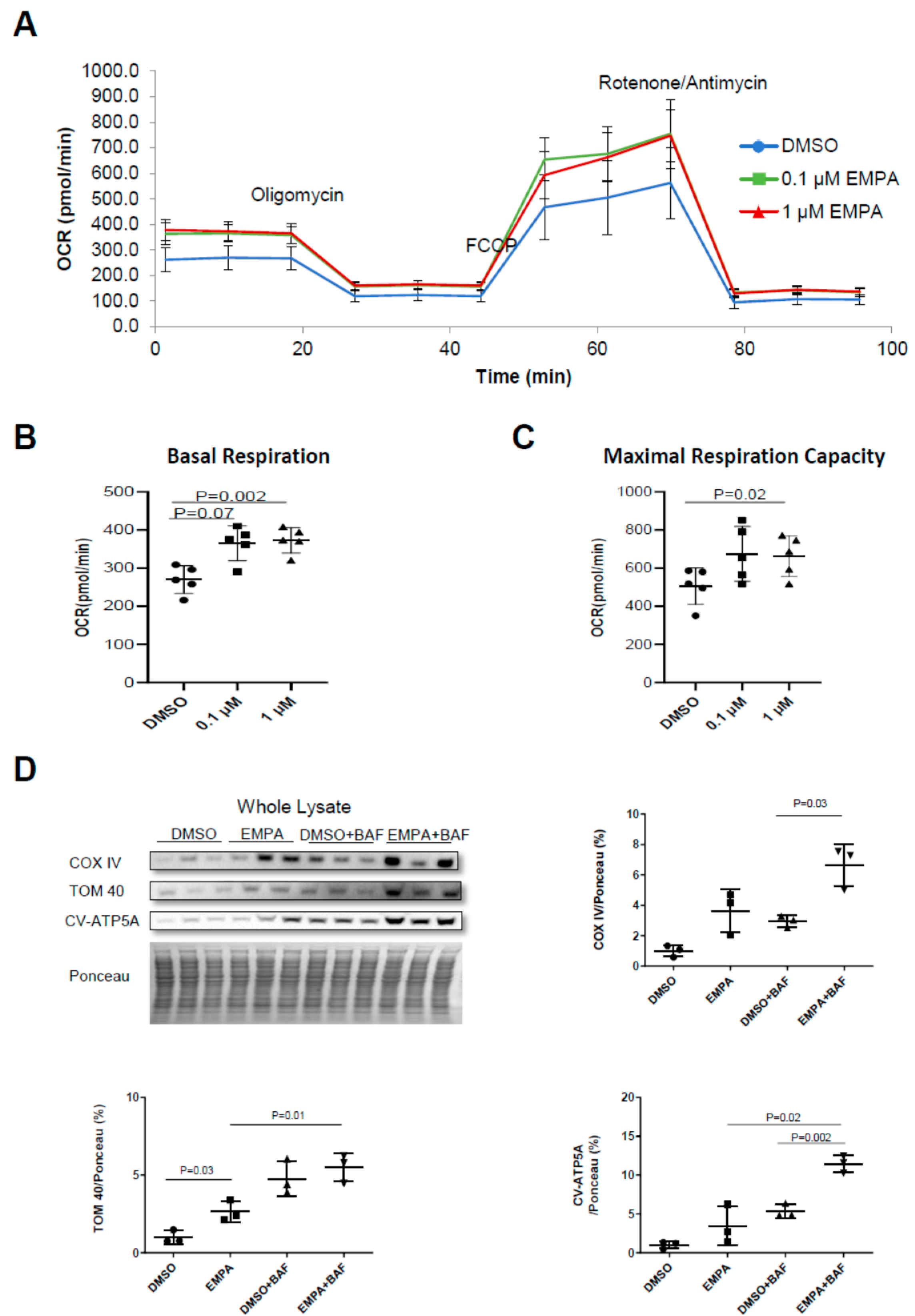
2.2. The Effect of EMPA on Mitochondrial Energetics and Content in H9C2 Cardiac Cells
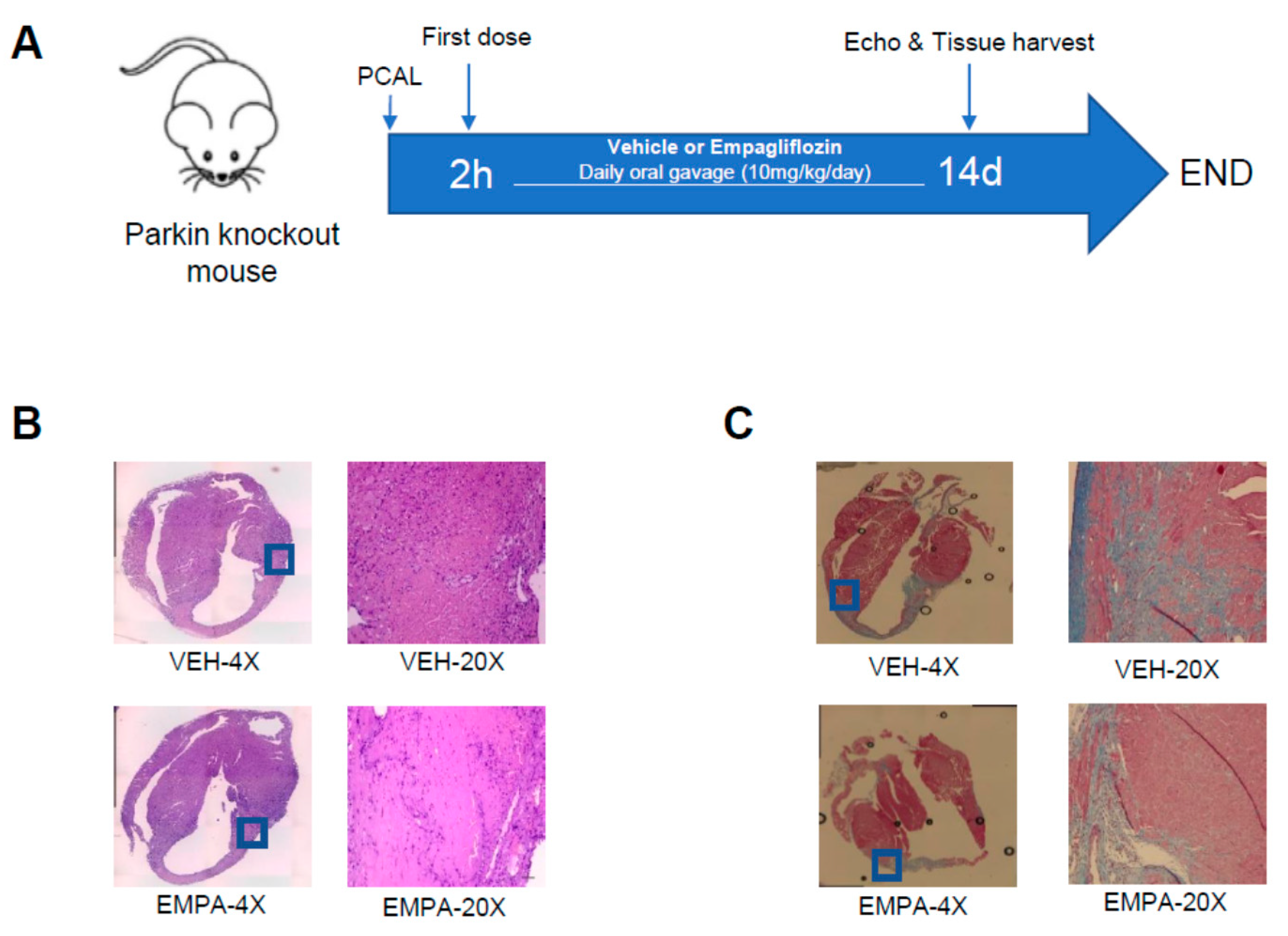
2.3. Effect of EMPA on Postinfarction Adverse LV Remodeling and Immune Cell Infiltration in Mitophagy-Impaired PKO Mice
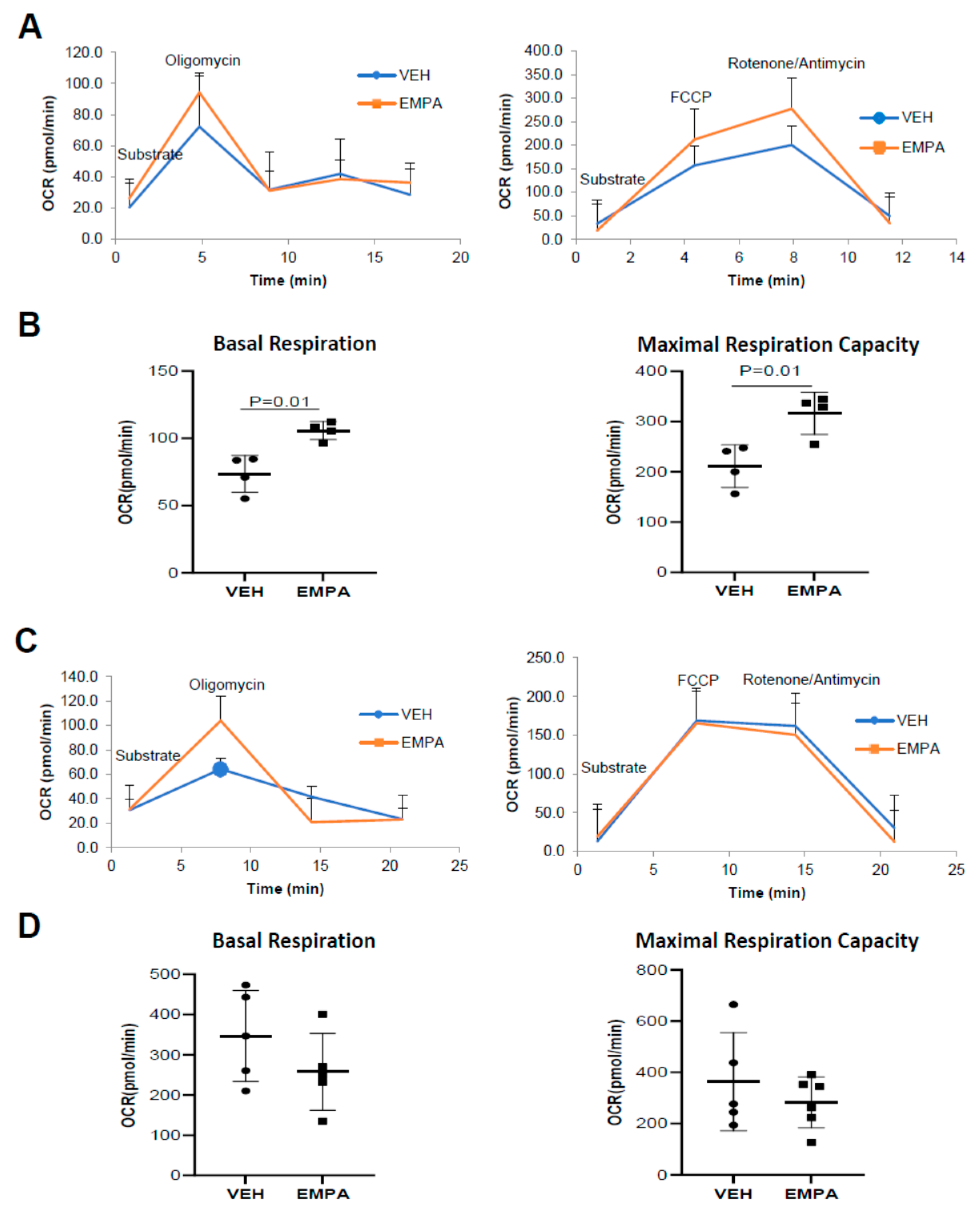
2.4. The Effect of EMPA on Isolated Mitochondria Obtained from WT Mice and PKO Mice with Impaired Mitophagy
3. Materials and Methods
3.1. Animals
3.2. Animal Protocol for In Vivo Wild-Type and PKO Mouse Studies
3.3. Echocardiography
3.4. Histology for Immune Cell Infiltration and Fibrosis Post-MI
3.5. Effect of EMPA on Mitophagy and Mitochondrial Biogenesis on Differentiated H9C2 Cells
3.6. Subcellular Fractionation of Heart Tissues and Differentiated H9C2 Cells
3.7. Western Blot Analysis
3.8. Seahorse Mitochondria Stress Test (Respirometry)
3.9. Statistical Analysis
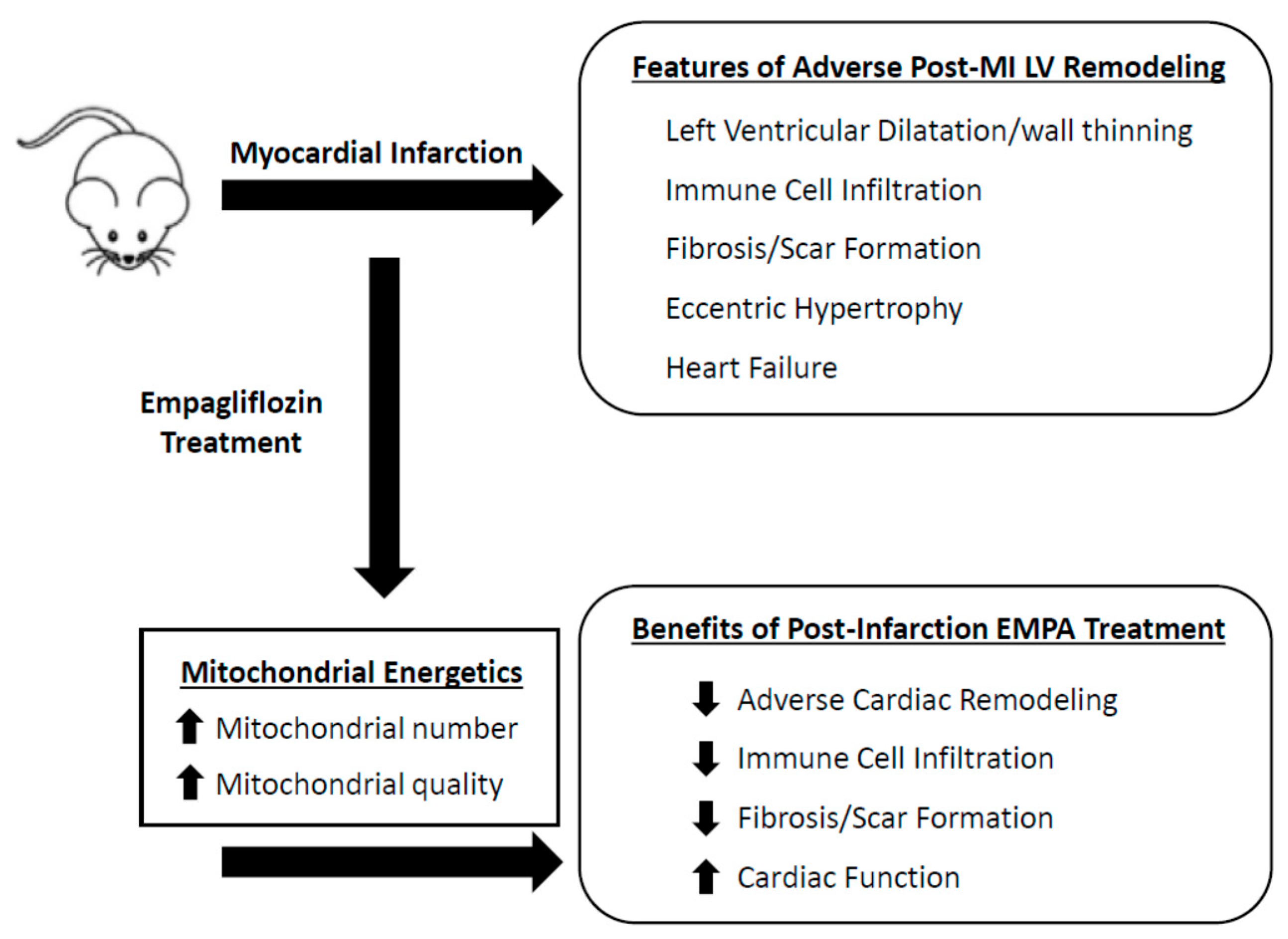
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bhatt, A.S.; Ambrosy, A.P.; Velazquez, E.J. Adverse Remodeling and Reverse Remodeling After Myocardial Infarction. Curr. Cardiol. Rep. 2017, 19, 71. [Google Scholar] [CrossRef] [PubMed]
- Galli, A.; Lombardi, F. Postinfarct Left Ventricular Remodelling: A Prevailing Cause of Heart Failure. Cardiol. Res. Pract. 2016, 2016, 2579832. [Google Scholar] [CrossRef] [Green Version]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Blaha, M.J. Executive summary: Heart disease and stroke statistics—2014 update: A report from the American Heart Association. Circulation 2014, 129, 399–410. [Google Scholar] [CrossRef]
- Dai, W.; Hale, S.; Kloner, R.A. Delayed therapeutic hypothermia protects against the myocardial no-reflow phenomenon independently of myocardial infarct size in a rat ischemia/reperfusion model. Int. J. Cardiol. 2017, 236, 400–404. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Verma, S.; Mazer, C.D.; Al-Omran, M.; Inzucchi, S.E.; Fitchett, D.; Hehnke, U.; George, J.T.; Zinman, B. Cardiovascular Outcomes and Safety of Empagliflozin in Patients With Type 2 Diabetes Mellitus and Peripheral Artery Disease: A Subanalysis of EMPA-REG OUTCOME. Circulation 2018, 137, 405–407. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Mazer, C.D.; Fitchett, D.; Inzucchi, S.E.; Pfarr, E.; George, J.T.; Zinman, B. Empagliflozin reduces cardiovascular events, mortality and renal events in participants with type 2 diabetes after coronary artery bypass graft surgery: Subanalysis of the EMPA-REG OUTCOME(R) randomised trial. Diabetologia 2018, 61, 1712–1723. [Google Scholar] [CrossRef] [Green Version]
- Butler, J.; Packer, M.; Greene, S.J.; Fiuzat, M.; Anker, S.D.; Anstrom, K.J.; Carson, P.E.; Cooper, L.B.; Fonarow, G.C.; Hernandez, A.F.; et al. Heart Failure End Points in Cardiovascular Outcome Trials of Sodium Glucose Cotransporter 2 Inhibitors in Patients With Type 2 Diabetes Mellitus: A Critical Evaluation of Clinical and Regulatory Issues. Circulation 2019, 140, 2108–2118. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; Fulcher, G.; Erondu, N.; Desai, M.; Shaw, W.; Law, G.; Walton, M.K.; Rosenthal, N.; et al. Optimizing the analysis strategy for the CANVAS Program: A prespecified plan for the integrated analyses of the CANVAS and CANVAS-R trials. Diabetes Obes. Metab. 2017, 19, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaduganathan, M.; Januzzi, J.L., Jr. Preventing and Treating Heart Failure with Sodium-Glucose Co-Transporter 2 Inhibitors. Am. J. Cardiol. 2019, 124 (Suppl. 1), S20–S27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytvyn, Y.; Bjornstad, P.; Udell, J.A.; Lovshin, J.A.; Cherney, D.Z. Response by Lytvyn et al to Letter Regarding Article, “Sodium Glucose Cotransporter-2 Inhibition in Heart Failure: Potential Mechanisms, Clinical Applications, and Summary of Clinical Trials”. Circulation 2018, 137, 1984–1985. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Zinman, B.; Wanner, C.; Ferrari, R.; Fitchett, D.; Hantel, S.; Espadero, R.-M.; Woerle, H.-J.; Broedl, U.C.; Johansen, O.E. SGLT-2 inhibitors and cardiovascular risk: Proposed pathways and review of ongoing outcome trials. Diabetes Vasc. Dis. Res. 2015, 12, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Hsia, D.S.; Grove, O.; Cefalu, W.T. An update on sodium-glucose co-transporter-2 inhibitors for the treatment of diabetes mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2016, 24, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Radlinger, B.; Hornsteiner, F.; Folie, S.; Salvenmoser, W.; Haubner, B.J.; Schuetz, T.; Haas, S.; Ress, C.; Adolph, T.E.; Salzmann, K.; et al. Cardioprotective effects of short-term empagliflozin treatment in db/db mice. Sci. Rep. 2020, 10, 19686. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; Antonio, R.S.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Flores, E.; Garcia-Ropero, A.; Sanz, J.; Hajjar, R.J.; et al. Empagliflozin Ameliorates Adverse Left Ventricular Remodeling in Nondiabetic Heart Failure by Enhancing Myocardial Energetics. J. Am. Coll. Cardiol. 2019, 73, 1931–1944. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Minoshima, S.; Mizuno, Y.; Shimizu, N. Molecular cloning, gene expression, and identification of a splicing variant of the mouse parkin gene. Mamm. Genome 2000, 11, 417–421. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Vives-Bauza, C.; de Vries, R.L.; Tocilescu, M.; Przedborski, S. PINK1/Parkin direct mitochondria to autophagy. Autophagy 2010, 6, 315–316. [Google Scholar] [CrossRef] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Andres, A.M.; Hernandez, G.; Lee, P.; Huang, C.; Ratliff, E.P.; Sin, J.; Thornton, C.A.; Damasco, M.V.; Gottlieb, R.A. Mitophagy Is Required for Acute Cardioprotection by Simvastatin. Antioxid. Redox Signal. 2014, 21, 1960–1973. [Google Scholar] [CrossRef]
- Feng, Y.; Zhao, J.; Hou, H.; Zhang, H.; Jiao, Y.; Wang, J.; Wang, Y.; Sun, Y. WDR26 promotes mitophagy of cardiomyocytes induced by hypoxia through Parkin translocation. Acta Biochim. Biophys. Sin. 2016, 48, 1075–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Andres, A.M.; Ratliff, E.P.; Hernandez, G.; Lee, P.; Gottlieb, R.A. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS ONE 2011, 6, e20975. [Google Scholar] [CrossRef]
- Kubli, D.A.; Zhang, X.; Lee, Y.; Hanna, R.A.; Quinsay, M.N.; Nguyen, C.K.; Jimenez, R.; Petrosyan, S.; Murphy, A.N.; Gustafsson, A.B. Parkin Protein Deficiency Exacerbates Cardiac Injury and Reduces Survival following Myocardial Infarction. J. Biol. Chem. 2013, 288, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Pan, S.-S. Parkin Mediates Mitophagy to Participate in Cardioprotection Induced by Late Exercise Preconditioning but Bnip3 Does Not. J. Cardiovasc. Pharmacol. 2018, 71, 303–316. [Google Scholar] [CrossRef]
- Yang, F.; Liu, Y.-H.; Yang, X.-P.; Xu, J.; Kapke, A.; Carretero, O.A. Myocardial Infarction and Cardiac Remodelling in Mice. Exp. Physiol. 2002, 87, 547–555. [Google Scholar] [CrossRef]
- Germano, J.D.F.; Huang, C.; Sin, J.; Song, Y.; Tucker, K.C.; Taylor, D.J.R.; Saadaeijahromi, H.; Stotland, A.; Piplani, H.; Gottlieb, R.A.; et al. Intermittent Use of a Short-Course Glucagon-like Peptide-1 Receptor Agonist Therapy Limits Adverse Cardiac Remodeling via Parkin-dependent Mitochondrial Turnover. Sci. Rep. 2020, 10, 8284. [Google Scholar] [CrossRef]
- Cannon, C.P.; Pratley, R.; Dagogo-Jack, S.; Mancuso, J.; Huyck, S.; Masiukiewicz, U.; Charbonnel, B.; Frederich, R.; Gallo, S.; Cosentino, F.; et al. Cardiovascular Outcomes with Ertugliflozin in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef]
- Sawicki, K.T.; Ben-Sahra, I.; McNally, E.M. SGLT2 Inhibition on Cardiac Mitochondrial Function: Searching for a Sweet Spot. J. Am. Heart. Assoc. 2021, 10, e021949. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- Ravassa, S.; Zudaire, A.; Díez, J. GLP-1 and cardioprotection: From bench to bedside. Cardiovasc. Res. 2012, 94, 316–323. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, M.; Kuno, A.; Yano, T.; Miki, T.; Oshima, H.; Sato, T.; Nakata, K.; Kimura, Y.; Tanno, M.; Miura, T. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol. Rep. 2018, 6, e13741. [Google Scholar] [CrossRef]
- Lee, W.-C.; Chau, Y.-Y.; Ng, H.-Y.; Chen, C.-H.; Wang, P.-W.; Liou, C.-W.; Lin, T.-K.; Chen, J.-B. Empagliflozin Protects HK-2 Cells from High Glucose-Mediated Injuries via a Mitochondrial Mechanism. Cells 2019, 8, 1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Kim, S.H.; Kang, J.M.; Heo, J.H.; Kim, D.-J.; Park, S.H.; Sung, M.; Kim, J.; Oh, J.; Yang, D.H.; et al. Empagliflozin attenuates diabetic tubulopathy by improving mitochondrial fragmentation and autophagy. Am. J. Physiol. Physiol. 2019, 317, F767–F780. [Google Scholar] [CrossRef]
- Liu, X.; Xu, C.; Xu, L.; Li, X.; Sun, H.; Xue, M.; Li, T.; Yu, X.; Sun, B.; Chen, L. Empagliflozin improves diabetic renal tubular injury by alleviating mitochondrial fission via AMPK/SP1/PGAM5 pathway. Metabolism 2020, 111, 154334. [Google Scholar] [CrossRef] [PubMed]
- Seefeldt, J.M.; Lassen, T.R.; Hjortbak, M.V.; Jespersen, N.R.; Kvist, F.; Hansen, J.; Bøtker, H.E. Cardioprotective effects of empagliflozin after ischemia and reperfusion in rats. Sci. Rep. 2021, 11, 9544. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Meng, L.; Lee, S.; Tse, G.; Gong, M.; Zhang, Z.; Zhao, J.; Zhao, Y.; Li, G.; Liu, T. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2019, 18, 165. [Google Scholar] [CrossRef] [Green Version]
- Thirunavukarasu, S.; Jex, N.; Chowdhary, A.; Hassan, I.U.; Straw, S.; Craven, T.C.; Gorecka, M.; Broadbent, D.; Swoboda, P.; Witte, K.K.; et al. Empagliflozin Treatment is Associated With Improvements in Cardiac Energetics and Function and Reductions in Myocardial Cellular Volume in Patients With Type 2 Diabetes. Diabetes 2021, 70, db210270. [Google Scholar] [CrossRef]
- Yurista, S.; Silljé, H.H.; Oberdorf-Maass, S.U.; Schouten, E.; Giani, M.G.P.; Hillebrands, J.; Van Goor, H.; Van Veldhuisen, D.J.; De Boer, R.A.; Westenbrink, B.D. Sodium–glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur. J. Heart Fail. 2019, 21, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Croteau, D.; Luptak, I.; Chambers, J.M.; Hobai, I.; Panagia, M.; Pimentel, D.R.; Siwik, D.A.; Qin, F.; Colucci, W.S. Effects of Sodium-Glucose Linked Transporter 2 Inhibition With Ertugliflozin on Mitochondrial Function, Energetics, and Metabolic Gene Expression in the Presence and Absence of Diabetes Mellitus in Mice. J. Am. Heart Assoc. 2021, 10, e019995. [Google Scholar] [CrossRef]
- Makrecka-Kuka, M.; Korzh, S.; Videja, M.; Vilks, K.; Cirule, H.; Kuka, J.; Dambrova, M.; Liepinsh, E. Empagliflozin Protects Cardiac Mitochondrial Fatty Acid Metabolism in a Mouse Model of Diet-Induced Lipid Overload. Cardiovasc. Drugs Ther. 2020, 34, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Yu, K.; Liang, L.; Liu, Y.; Song, F.; Ge, Q.; Fang, X.; Yu, T.; Huang, Z.; Jiang, L.; et al. Sodium–Glucose Co-Transporter 2 Inhibition With Empagliflozin Improves Cardiac Function After Cardiac Arrest in Rats by Enhancing Mitochondrial Energy Metabolism. Front. Pharmacol. 2021, 12, 758080. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Bilsen, M.; van Nieuwenhoven, F.A.; van der Vusse, G.J. Metabolic remodelling of the failing heart: Beneficial or detrimental? Cardiovasc. Res. 2009, 81, 420–428. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 14 Day | 28 Day | |||||
|---|---|---|---|---|---|---|
| WT-VEH(7) | WT-EMPA(8) | p Value | WT-VEH(6) | WT-EMPA(7) | p Value | |
| HR (bpm) | 451 ± 70.48 | 429 ± 49.65 | 0.49 | 440 ± 71.57 | 397 ± 35.10 | 0.19 |
| EF (%) | 38.71 ± 5.76 | 50.42 ± 8.34 | 0.01 | 34.40 ± 8.32 | 47.85 ± 11.16 | 0.03 |
| FS (%) | 18.70 ± 3.02 | 25.74 ± 5.21 | 0.01 | 16.51 ± 4.37 | 24.26 ± 6.53 | 0.03 |
| LV Mass (mg) | 152.52 ± 21.22 | 156.42 ± 57.44 | 0.87 | 136.15 ± 34.31 | 127.01 ± 32.33 | 0.63 |
| LV Mass (Corrected) (mg) | 122.02 ± 16.98 | 125.13 ± 45.95 | 0.87 | 136.15 ± 34.31 | 127.01 ± 32.33 | 0.63 |
| LV Vol; d (μL) | 93.87 ± 14.75 | 97.00 ± 25.34 | 0.78 | 119.96 ± 61.41 | 100.99 ± 36.56 | 0.51 |
| LV Vol; s (μL) | 58.18 ± 14.61 | 48.65 ± 17.09 | 0.27 | 81.43 ± 51.59 | 54.54 ± 26.76 | 0.25 |
| LVID; d (mm) | 4.52 ± 0.30 | 4.57 ± 0.48 | 0.83 | 4.93 ± 0.97 | 4.61 ± 0.72 | 0.51 |
| LVID; s (mm) | 3.68 ± 0.38 | 3.40 ± 0.48 | 0.23 | 4.14 ± 0.99 | 3.52 ± 0.75 | 0.22 |
| LVPW; d (mm) | 0.92 ± 0.10 | 0.88 ± 0.13 | 0.54 | 0.86 ± 0.20 | 0.95 ± 0.18 | 0.41 |
| LVPW; s (mm) | 1.01 ± 0.12 | 1.15 ± 0.14 | 0.05 | 1.01 ± 0.21 | 1.20 ± 0.17 | 0.10 |
| PKO-VEH(7) | PKO-EMPA(9) | p Value | |
|---|---|---|---|
| HR (bpm) | 410 ± 35.29 | 385 ± 12.65 | 0.24 |
| EF (%) | 36.28 ± 7.75 | 46.58 ± 10.78 | 0.04 |
| FS (%) | 17.70 ± 7.75 | 23.44 ± 6.34 | 0.05 |
| LV Mass (mg) | 144.52 ± 39.27 | 145.2 ± 42.29 | 0.60 |
| LV Mass (Corrected) (mg) | 115.62 ± 31.42 | 116.16 ± 33.83 | 0.60 |
| LV Vol; d (μL) | 98.53 ± 35.39 | 99.04 ± 39.54 | 0.60 |
| LV Vol; s (μL) | 64.84 ± 27.38 | 56.23 ± 33.19 | 0.29 |
| LVID; d (mm) | 4.56 ± 0.74 | 4.57 ± 0.75 | 0.56 |
| LVID; s (mm) | 3.78 ± 0.80 | 3.54 ± 0.86 | 0.26 |
| LVPW; d (mm) | 0.83 ± 0.27 | 0.79 ± 0.17 | 0.63 |
| LVPW; s (mm) | 1.04 ± 0.27 | 1.08 ± 0.26 | 0.77 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.; Huang, C.; Sin, J.; Germano, J.d.F.; Taylor, D.J.R.; Thakur, R.; Gottlieb, R.A.; Mentzer, R.M., Jr.; Andres, A.M. Attenuation of Adverse Postinfarction Left Ventricular Remodeling with Empagliflozin Enhances Mitochondria-Linked Cellular Energetics and Mitochondrial Biogenesis. Int. J. Mol. Sci. 2022, 23, 437. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010437
Song Y, Huang C, Sin J, Germano JdF, Taylor DJR, Thakur R, Gottlieb RA, Mentzer RM Jr., Andres AM. Attenuation of Adverse Postinfarction Left Ventricular Remodeling with Empagliflozin Enhances Mitochondria-Linked Cellular Energetics and Mitochondrial Biogenesis. International Journal of Molecular Sciences. 2022; 23(1):437. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010437
Chicago/Turabian StyleSong, Yang, Chengqun Huang, Jon Sin, Juliana de F. Germano, David J. R. Taylor, Reetu Thakur, Roberta A. Gottlieb, Robert M. Mentzer, Jr., and Allen M. Andres. 2022. "Attenuation of Adverse Postinfarction Left Ventricular Remodeling with Empagliflozin Enhances Mitochondria-Linked Cellular Energetics and Mitochondrial Biogenesis" International Journal of Molecular Sciences 23, no. 1: 437. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010437