
Structure-Based Design of an RNase Chimera for Antimicrobial Therapy

 , ,
, , 
Abstract
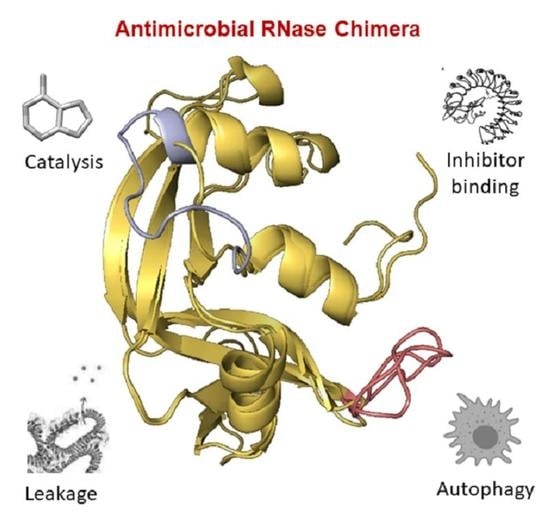
:
1. Introduction
2. Results
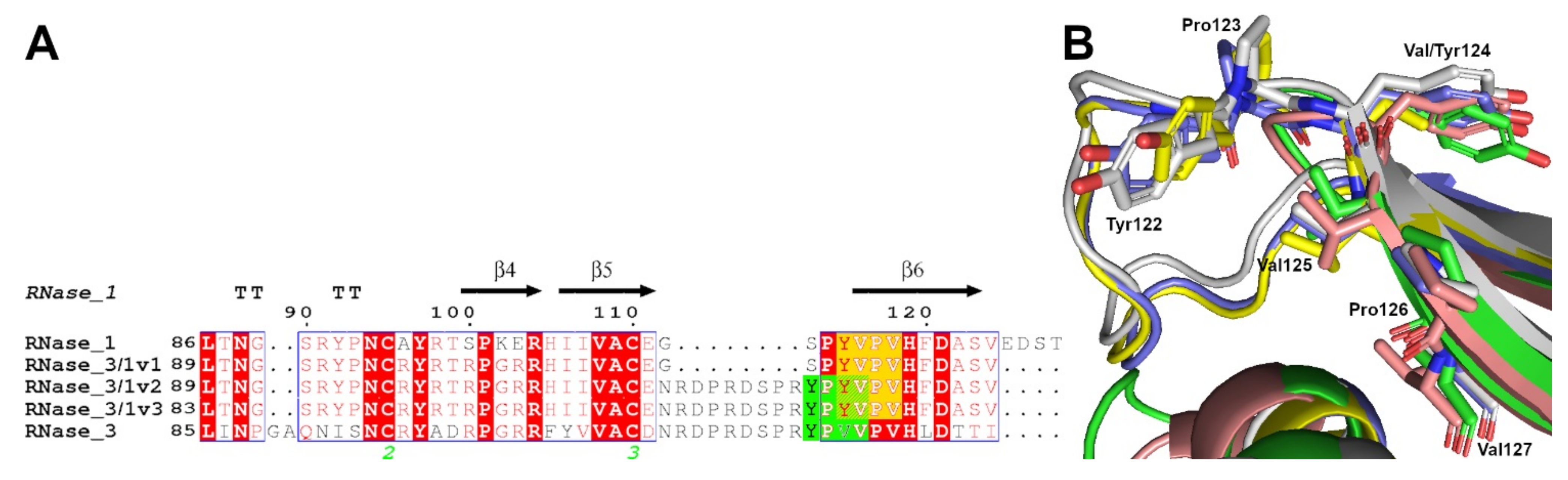
2.1. RNases 3/1-v2 and v3 Rational Design
2.2. Catalytic Activity
2.3. Bactericidal Activity and Cytotoxicity
2.4. Lipopolysaccharide Binding and Liposome Leakage Activities
2.5. Interaction with the RNase Inhibitor (RI)
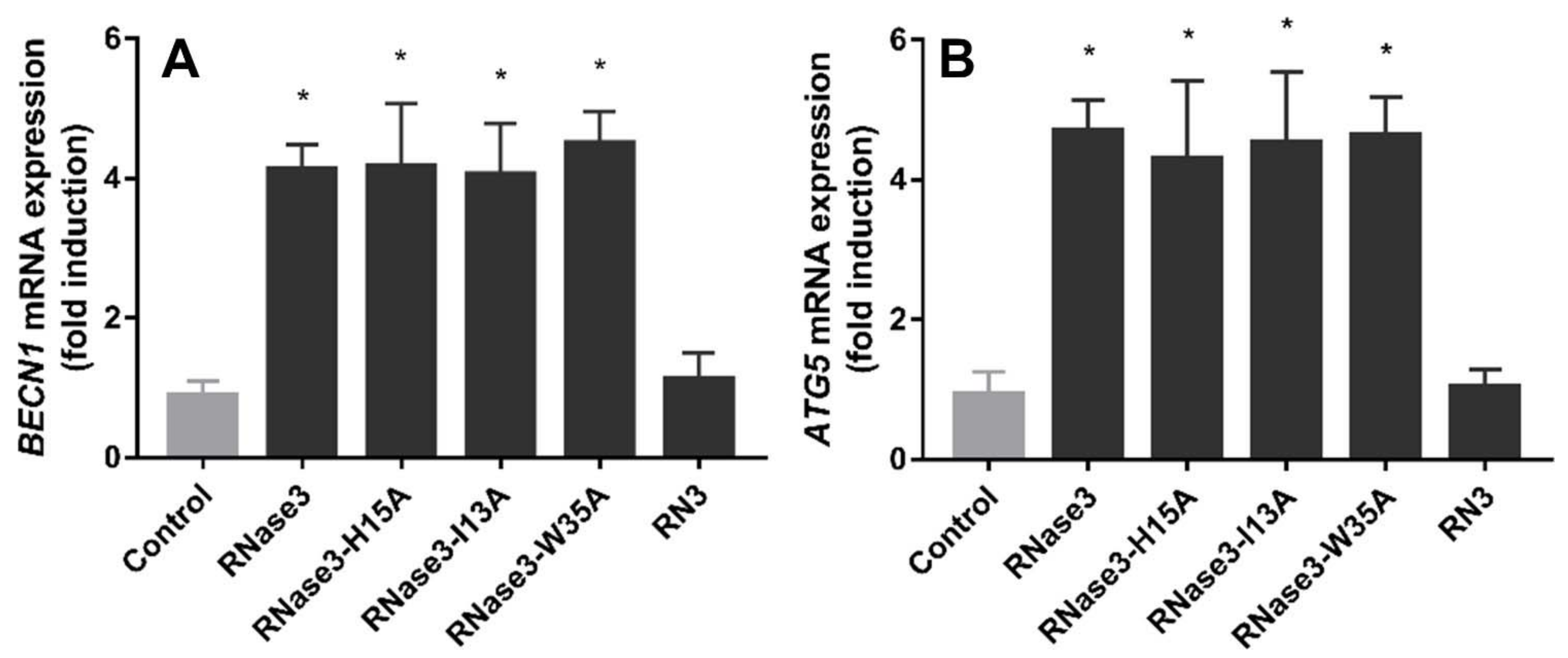
2.6. RNase 3/1 Autophagy Induction
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Protein Expression and Purification
4.3. Mutagenesis and Construction of RNase 3/1 Chimera
4.4. Molecular Dynamic Simulations
4.5. Circular Dichroism (CD)
4.6. Activity Staining Gel
4.7. Minimum Bactericidal Concentration (MBC) Determination
4.8. Cytotoxicity Assay
4.9. LPS Binding Assay
4.10. Liposome Preparation
4.11. Liposome Leakage
4.12. Spectrophotometric Kinetic Analysis
4.13. Ribonuclease Inhibitor Activity Assay
4.14. Macrophage Cell Culture, Infection and Intracellular MIC Determination
4.15. Real-Time qPCR Assays
4.16. Acridine Orange Staining
4.17. Intracellular Aggregates Formation Measurement by Thioflavin-S Staining
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cuchillo, C.M.; Nogués, M.V.; Raines, R.T. Bovine Pancreatic Ribonuclease: Fifty Years of the First Enzymatic Reaction Mechanism. Biochemistry 2011, 50, 7835–7841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomax, J.E.; Eller, C.H.; Raines, R.T. Comparative functional analysis of ribonuclease 1 homologs: Molecular insights into evolving vertebrate physiology. Biochem. J. 2017, 474, 2219–2233. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, E.; D’Alessio, G. The success of the RNase scaffold in the advance of biosciences and in evolution. Gene 2007, 406, 8–12. [Google Scholar] [CrossRef]
- Rosenberg, H.F. RNase A ribonucleases and host defense: An evolving story. J. Leukoc. Biol. 2008, 83, 1079–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goncalves, K.A.; Silberstein, L.; Li, S.; Severe, N.; Hu, M.G.; Yang, H.; Scadden, D.T.; Hu, G.-F. Angiogenin promotes hematopoietic regeneration by dichotomously regulating quiescence of stem and progenitor cells. Cell 2016, 166, 894–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, L.V.; Stappenbeck, T.S.; Hong, C.V.; Gordon, J.I. Angiogenins: A new class of microbicidal proteins involved in innate immunity. Nat. Immunol. 2003, 4, 269–273. [Google Scholar] [CrossRef]
- Koczera, P.; Martin, L.; Marx, G.; Schuerholz, T. The ribonuclease a superfamily in humans: Canonical RNases as the buttress of innate immunity. Int. J. Mol. Sci. 2016, 17, 1278. [Google Scholar] [CrossRef]
- Lu, L.; Li, J.; Moussaoui, M.; Boix, E. Immune modulation by human secreted RNases at the extracellular space. Front. Immunol. 2018, 9, 1012. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, S. The eight human “canonical” ribonucleases: Molecular diversity, catalytic properties, and special biological actions of the enzyme proteins. FEBS Lett. 2010, 584, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Liao, W.L.; Lu, Q.J.; Li, C.G.; Yuan, Y.; Xu, Z.Y.; Huang, S.D.; Chen, H.Z. ANG promotes proliferation and invasion of the cell of lung squamous carcinoma by directly up-regulating HMGA2. J. Cancer 2016, 7, 862–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreras, E.; Boix, E.; Rosenberg, H.F.; Cuchillo, C.M.; Nogués, M.V. Both aromatic and cationic residues contribute to the membrane-lytic and bactericidal activity of eosinophil cationic protein. Biochemistry 2003, 42, 6636–6644. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, H.F. Recombinant human eosinophil cationic protein. Ribonuclease activity is not essential for cytotoxicity. J. Biol. Chem. 1995, 270, 7876–7881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrent, M.; Andreu, D.; Nogués, V.M.; Boix, E. Connecting peptide physicochemical and antimicrobial properties by a rational prediction model. PLoS ONE 2011, 6, e16968. [Google Scholar] [CrossRef]
- Torrent, M.; Cuyás, E.; Carreras, E.; Navarro, S.; López, O.; De La Maza, A.; Nogués, M.V.; Reshetnyak, Y.K.; Boix, E. Topography studies on the membrane interaction mechanism of the eosinophil cationic protein. Biochemistry 2007, 46, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Kopfnagel, V.; Wagenknecht, S.; Harder, J.; Hofmann, K.; Kleine, M.; Buch, A.; Sodeik, B.; Werfel, T. RNase 7 strongly promotes TLR9-mediated DNA sensing by human plasmacytoid dendritic cells. J. Investig. Dermatol. 2018, 138, 872–881. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Wei, R.; Prats-Ejarque, G.; Goetz, M.; Wang, G.; Torrent, M.; Boix, E. Human RNase3 immune modulation by catalytic-dependent and independent modes in a macrophage-cell line infection model. Cell. Mol. Life Sci. 2021, 78, 2963–2985. [Google Scholar] [CrossRef]
- Rademacher, F.; Dreyer, S.; Kopfnagel, V.; Gläser, R.; Werfel, T.; Harder, J. The antimicrobial and immunomodulatory function of RNase 7 in skin. Front. Immunol. 2019, 10, 2553. [Google Scholar] [CrossRef] [Green Version]
- Phipps, S.; Lam, C.E.; Mahalingam, S.; Newhouse, M.; Ramirez, R.; Rosenberg, H.F.; Foster, P.S.; Matthaei, K.I. Eosinophils contribute to innate antiviral immunity and promote clearance of respiratory syncytial virus. Blood 2007, 110, 1578–1586. [Google Scholar] [CrossRef] [Green Version]
- Salazar, V.A.; Arranz-Trullén, J.; Navarro, S.; Blanco, J.A.; Sánchez, D.; Moussaoui, M.; Boix, E. Exploring the mechanisms of action of human secretory RN ase 3 and RN ase 7 against Candida albicans. MicrobiologyOpen 2016, 5, 830–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Arranz-Trullén, J.; Prats-Ejarque, G.; Pulido-Gomez, D.; Bhakta, S.; Boix, E. Human antimicrobial RNases inhibit intracellular bacterial growth and induce autophagy in mycobacteria-infected macrophages. Front. Immunol. 2019, 10, 1–17. [Google Scholar] [CrossRef]
- Prats-Ejarque, G.; Li, J.; Ait-Ichou, F.; Lorente, H.; Boix, E. Testing a human antimicrobial RNase chimera against bacterial resistance. Front. Microbiol. 2019, 10, 1357. [Google Scholar] [CrossRef]
- Carreras, E.; Boix, E.; Navarro, S.; Rosenberg, H.F.; Cuchillo, C.M.; Nogués, M.V. Surface-exposed amino acids of eosinophil cationic protein play a critical role in the inhibition of mammalian cell proliferation. Mol. Cell. Biochem. 2005, 272, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, D.; Moussaoui, M.; Carreras, E.; Torrent, M.; Nogués, V.; Boix, E. Mapping the eosinophil cationic protein antimicrobial activity by chemical and enzymatic cleavage. Biochimie 2011, 93, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; de la Torre, B.G.; Nogués, V.M.; Andreu, D.; Boix, E. Bactericidal and membrane disruption activities of the eosinophil cationic protein are largely retained in an N-terminal fragment. Biochem. J. 2009, 421, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrent, M.; Odorizzi, F.; Nogués, M.V.; Boix, E. Eosinophil cationic protein aggregation: Identification of an N-terminus amyloid prone region. Biomacromolecules 2010, 11, 1983–1990. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Pulido, D.; Valle, J.; Nogués, M.V.; Andreu, D.; Boix, E.; Pulido-Gomez, D. Ribonucleases as a host-defence family: Evidence of evolutionarily conserved antimicrobial activity at the N-terminus. Biochem. J. 2013, 456, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doucet, N.; Watt, E.D.; Loria, J.P. The flexibility of a distant loop modulates active site motion and product release in ribonuclease A. Biochemistry 2009, 48, 7160–7168. [Google Scholar] [CrossRef] [Green Version]
- Gagné, D.; French, R.L.; Narayanan, C.; Simonović, M.; Agarwal, P.K.; Doucet, N. Perturbation of the conformational dynamics of an active-site loop alters enzyme activity. Structure 2015, 23, 2256–2266. [Google Scholar] [CrossRef] [Green Version]
- Boix, E.; Salazar, V.A.; Torrent, M.; Pulido-Gomez, D.; Nogués, M.V.; Moussaoui, M. Structural determinants of the eosinophil cationic protein antimicrobial activity. Biol. Chem. 2012, 393, 801–815. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. 2016, 86, 2.9.1–2.9.37. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, 320–324. [Google Scholar] [CrossRef] [Green Version]
- Fowler, C.B.; Evers, D.L.; O’Leary, T.J.; Mason, J.T. Antigen retrieval causes protein unfolding: Evidence for a linear epitope model of recovered immunoreactivity. J. Histochem. Cytochem. 2011, 59, 366–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrent, M.; Navarro, S.; Moussaoui, M.; Nogués, M.V.; Boix, E. Eosinophil cationic protein high-affinity binding to bacteria-wall lipopolysaccharides and peptidoglycans. Biochemistry 2008, 47, 3544–3555. [Google Scholar] [CrossRef] [PubMed]
- Rutkoski, T.J.; Raines, R.T. Evasion of ribonuclease inhibitor as a determinant of ribonuclease cytotoxicity. Curr. Pharm. Biotechnol. 2008, 9, 185–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomax, J.E.; Bianchetti, C.M.; Chang, A.; Phillips, G.N.; Fox, B.G.; Raines, R.T. Functional evolution of ribonuclease inhibitor: Insights from birds and reptiles. J. Mol. Biol. 2014, 426, 3041–3056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venge, P.; Byström, J.; Carlson, M.; Hâkansson, L.; Karawacjzyk, M.; Peterson, C.; Sevéus, L.; Trulson, A. Eosinophil cationic protein (ECP): Molecular and biological properties and the use of ECP as a marker of eosinophil activation in disease. Clin. Exp. Allergy 1999, 29, 1172–1186. [Google Scholar] [CrossRef]
- Torrent, M.; Pulido-Gomez, D.; Nogués, M.V.; Boix, E. Exploring new biological functions of amyloids: Bacteria cell agglutination mediated by host protein aggregation. PLoS Pathog. 2012, 8, e1003005. [Google Scholar] [CrossRef] [Green Version]
- Birgisdottir, Å.B.; Lamark, T.; Johansen, T. The LIR motif—crucial for selective autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar] [CrossRef] [Green Version]
- Popelka, H.; Klionsky, D.J. Analysis of the native conformation of the LIR/AIM motif in the Atg8/LC3/GABARAP-binding proteins. Autophagy 2015, 11, 2153–2159. [Google Scholar] [CrossRef]
- Torrent, M.; Pulido, D.; de la Torre, B.G.; García-Mayoral, M.F.; Nogueés, M.V.; Bruix, M.; Andreu, D.; Boix, E.; Pulido-Gomez, D. Refining the eosinophil cationic protein antibacterial pharmacophore by rational structure minimization. J. Med. Chem. 2011, 54, 5237–5244. [Google Scholar] [CrossRef] [PubMed]
- Pulido, D.; Prats-Ejarque, G.; Villalba, C.; Albacar, M.; Moussaoui, M.; Andreu, D.; Volkmer, R.; Torrent, M.; Boix, E. Positional scanning library applied to the human eosinophil cationic protein/RNase3 N-terminus reveals novel and potent anti-biofilm peptides. Eur. J. Med. Chem. 2018, 152, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.P.; Hoang, T.T.; Ressler, V.T.; Raines, R.T. Human angiogenin is a potent cytotoxin in the absence of ribonuclease inhibitor. RNA 2018, 24, 1018–1027. [Google Scholar] [CrossRef] [Green Version]
- Boix, E.; Nikolovski, Z.; Moiseyev, G.P.; Rosenberg, H.F.; Cuchillo, C.M.; Nogués, M.V. Kinetic and product distribution analysis of human eosinophil cationic protein indicates a subsite arrangement that favors exonuclease-type activity. J. Biol. Chem. 1999, 274, 15605–15614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomax, J.E.; Eller, C.H.; Raines, R.T. Rational design and evaluation of mammalian ribonuclease cytotoxins. Methods Enzymol. 2012, 502, 273–290. [Google Scholar] [CrossRef] [Green Version]
- Boix, E. Eosinophil Cationic Protein. In Methods in Enzymology; Nicholson, A.W., Ed.; Academic Press: San Diego, CA, USA, 2001; Volume 341, pp. 287–305. [Google Scholar]
- Boix, E.; Blanco, J.A.; Nogués, M.V.; Moussaoui, M. Nucleotide binding architecture for secreted cytotoxic endoribonucleases. Biochimie 2013, 95, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Kurten, R.C. Sorting motifs in receptor trafficking. Adv. Drug Deliv. Rev. 2003, 55, 1405–1419. [Google Scholar] [CrossRef]
- Ohno, H.; Aguilar, R.C.; Yeh, D.; Taura, D.; Saito, T.; Bonifacino, J.S. The medium subunits of adaptor complexes recognize distinct but overlapping sets of tyrosine-based sorting signals. J. Biol. Chem. 1998, 273, 25915–25921. [Google Scholar] [CrossRef] [Green Version]
- Ohno, H.; Stewart, J.; Fournier, M.C.; Bosshart, H.; Rhee, I.; Miyatake, S.; Saito, T.; Gallusser, A.; Kirchhausen, T.; Bonifacino, J.S. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science 1995, 269, 1872–1875. [Google Scholar] [CrossRef] [Green Version]
- Noda, N.N.; Ohsumi, Y.; Inagaki, F. Atg8-family interacting motif crucial for selective autophagy. FEBS Lett. 2010, 584, 1379–1385. [Google Scholar] [CrossRef] [Green Version]
- Johansen, T.; Lamark, T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef] [PubMed]
- Rozenknop, A.; Rogov, V.V.; Rogova, N.Y.; Löhr, F.; Güntert, P.; Dikic, I.; Dötsch, V. Characterization of the interaction of GABARAPL-1 with the LIR motif of NBR1. J. Mol. Biol. 2011, 410, 477–487. [Google Scholar] [CrossRef]
- Ostendorf, T.; Zillinger, T.; Andryka, K.; Schlee-Guimaraes, T.M.; Schmitz, S.; Marx, S.; Bayrak, K.; Linke, R.; Salgert, S.; Wegner, J.; et al. Immune sensing of synthetic, bacterial, and protozoan RNA by toll-like receptor 8 requires coordinated processing by RNase T2 and RNase 2. Immunity 2020, 52, 591–605.e6. [Google Scholar] [CrossRef] [PubMed]
- Greulich, W.; Wagner, M.; Gaidt, M.M.; Stafford, C.; Cheng, Y.; Linder, A.; Carell, T.; Hornung, V. TLR8 is a sensor of RNase T2 degradation products. Cell 2019, 179, 1264–1275.e13. [Google Scholar] [CrossRef] [PubMed]
- Palmer, I.; Wingfield, P.T. Preparation and Extraction of Insoluble (Inclusion-Body) Proteins from Escherichia coli. In Current Protocols in Protein Science; John Wiley & Sons: Hoboken, NJ, USA, 2004; pp. 6.3.1–6.3.18. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Páll, S.; Hess, B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 2013, 184, 2641–2650. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Debye, P. Näherungsformeln für die Zylinderfunktionen für große Werte des Arguments und unbeschränkt veränderliche Werte des Index. Math. Ann. 1909, 67, 535–558. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Yang, J.T.; Wu, C.S.C.; Martinez, H.M. Calculation of protein conformation from circular dichroism. Methods Enzymol. 1986, 130, 208–269. [Google Scholar] [CrossRef]
- Bravo, J.; Fernandez, E.; Ribó, M.; Dellorens, R.; Cuchillo, C.M. A versatile negative-staining ribonuclease zymogram. Anal. Biochem. 1994, 219, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Pulido, D.; Arranz-Trullén, J.; Prats-Ejarque, G.; Veláquez, D.; Torrent, M.; Moussaoui, M.; Boix, E. Insights into the antimicrobial mechanism of action of human RNase6: Structural determinants for bacterial cell agglutination and membrane permeation. Int. J. Mol. Sci. 2016, 17, 552. [Google Scholar] [CrossRef] [Green Version]
- Prats-Ejarque, G.; Arranz-Trullén, J.; Blanco, J.A.; Pulido, D.; Nogués, M.V.; Moussaoui, M.; Boix, E. The first crystal structure of human RNase 6 reveals a novel substrate-binding and cleavage site arrangement. Biochem. J. 2016, 473, 1523–1536. [Google Scholar] [CrossRef] [Green Version]
- Boix, E.; Wu, Y.; Vasandani, V.M.; Saxena, S.K.; Ardelt, W.; Ladner, J.; Youle, R.J. Role of the N Terminus in RNase A Homologues: Differences in Catalytic Activity, Ribonuclease Inhibitor Interaction and Cytotoxicity. J. Mol. Biol. 1996, 257, 992–1007. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, C.; Cordani, M.; Gotte, G.; Picone, D.; Donadelli, M. Onconase induces autophagy sensitizing pancreatic cancer cells to gemcitabine and activates Akt/mTOR pathway in a ROS-dependent manner. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espargaró, A.; Sabate, R.; Ventura, S. Thioflavin-S staining coupled to flow cytometry. A screening tool to detect in vivo protein aggregation. Mol. BioSyst. 2012, 8, 2839–2844. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Relative Catalytic Activity against Dinucleotides | |||||
|---|---|---|---|---|---|
| RNase 1 | RNase 3 | RNase 3/1-v1 | RNase 3/1-v2 | RNase 3/1-v3 | |
| CpA | 641 | 22 | 100 | 37 | 35 |
| UpA | 940 | 1 | 100 | 73 | 130 |
| UpG | 124 | N.D. | 100 | 23 | 85 |
| MBC100 (µM) | |||||
|---|---|---|---|---|---|
| RNase 1 | RNase 3/1-v1 | RNase 3/1-v2 | RNase 3/1-v3 | RNase 3 | |
| E. coli | >20 | 6.25 ± 2.17 | 6.25 ± 2.17 | 0.78 ± 0.27 | 1.88 ± 0.88 |
| A. baumannii | >20 | 6.25 ± 2.17 | 16.67 ± 2.89 | 1.56 ± 1.89 | 0.6 ± 0.07 |
| P. aeruginosa | 18.33 ± 2.89 | 3.13 ± 1.08 | 18.33 ± 2.89 | 1.25 ± 0.54 | 0.6 ± 0.07 |
| Cell Toxicity (IC50) (µM) | |||||
| RNase 1 | RNase 3/1-v11 | RNase 3/1-v2 | RNase 3/1-v3 | RNase 3 | |
| HepG2 cells | N.D. | N.D. | N.D. | N.D. | 134.66 ± 0.95 |
| LPS Binding (LBC50) (µM) | ||||
|---|---|---|---|---|
| RNase 1 | RNase 3/1-v1 | RNase 3/1-v2 | RNase 3/1-v3 | RNase 3 |
| 1.50 ± 0.28 | 0.59 ± 0.02 | 0.66 ± 0.08 | 0.42 ± 0.07 | 0.38 ± 0.03 |
| Liposome Leakage | ||||
|---|---|---|---|---|
| 1 h Exposure | 24 h Exposure | |||
| LC20 (µM) | LC50 (µM) | LC20 (µM) | LC50 (µM) | |
| RNase 1 | 42.73 | N. D. | 16.8 | N. D. |
| (27.02–57.29) | (4.28–37.31) | |||
| RNase 3/1-v1 | N. D. | N. D. | 5.766 | N. D. |
| (2.19–15.33) | ||||
| RNase 3/1-v2 | 24.33 | 34.19 | 2.916 | 12.35 |
| (21.39–27.42) | (31.39–37.29) | (2.13–3.9) | (10.03–15.36) | |
| RNase 3/1-v3 | 26.74 | 60.19 | 1.028 | 4.541 |
| (22.89–30.87) | (52.39–73.5) | (0.43–2.07) | (2.7–8.07) | |
| RNase 3 | 5.53 | 12.85 | 1.027 | 1.682 |
| (4.17–7.17) | (10.92–15.09) | (0.49–1.66) | (1.14–2.35) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prats-Ejarque, G.; Lorente, H.; Villalba, C.; Anguita, R.; Lu, L.; Vázquez-Monteagudo, S.; Fernández-Millán, P.; Boix, E. Structure-Based Design of an RNase Chimera for Antimicrobial Therapy. Int. J. Mol. Sci. 2022, 23, 95. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010095
Prats-Ejarque G, Lorente H, Villalba C, Anguita R, Lu L, Vázquez-Monteagudo S, Fernández-Millán P, Boix E. Structure-Based Design of an RNase Chimera for Antimicrobial Therapy. International Journal of Molecular Sciences. 2022; 23(1):95. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010095
Chicago/Turabian StylePrats-Ejarque, Guillem, Helena Lorente, Clara Villalba, Raúl Anguita, Lu Lu, Sergi Vázquez-Monteagudo, Pablo Fernández-Millán, and Ester Boix. 2022. "Structure-Based Design of an RNase Chimera for Antimicrobial Therapy" International Journal of Molecular Sciences 23, no. 1: 95. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010095