
Role of Leptin and Adiponectin in Endometrial Cancer

 , , and
, , and 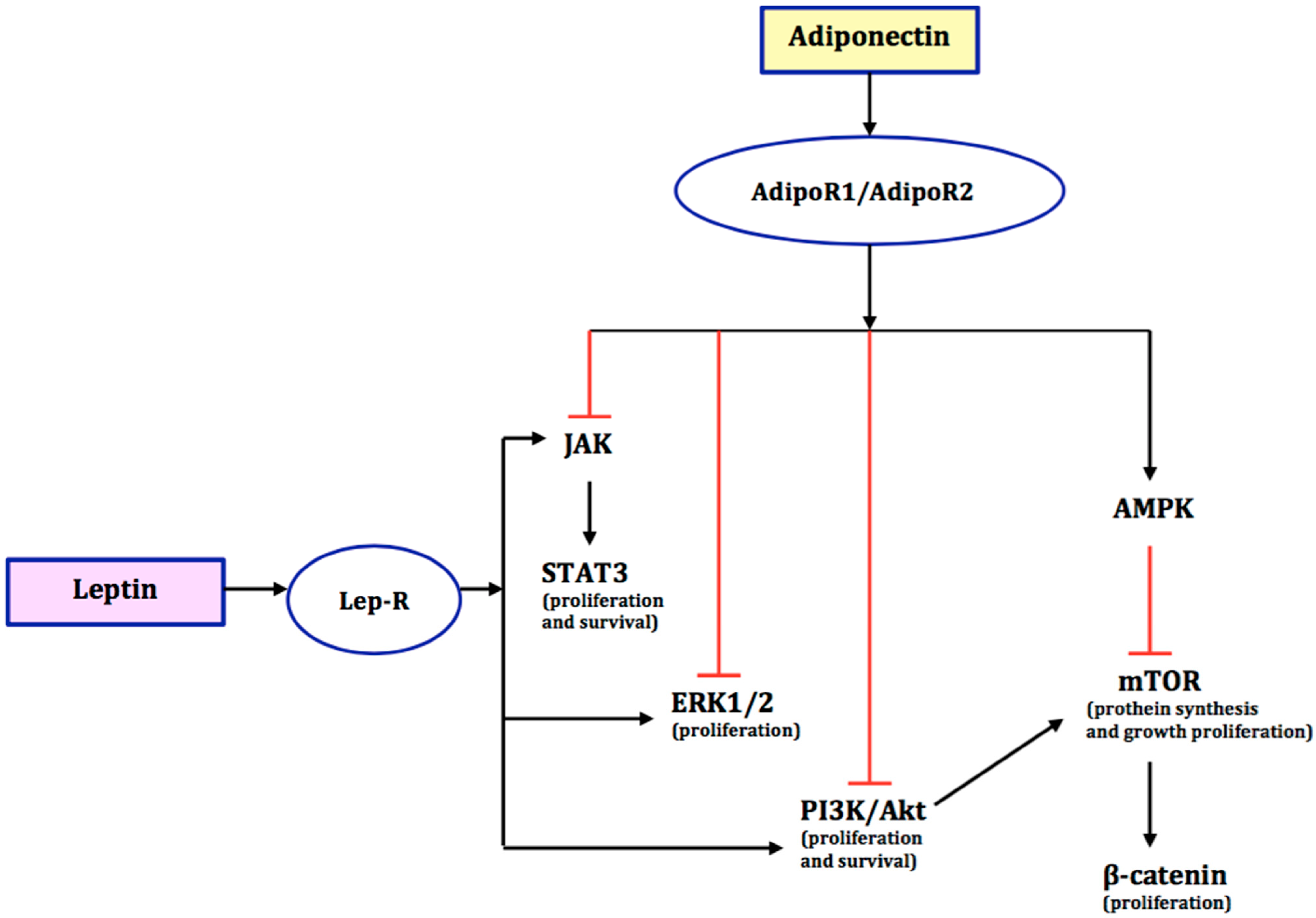{kind=link}
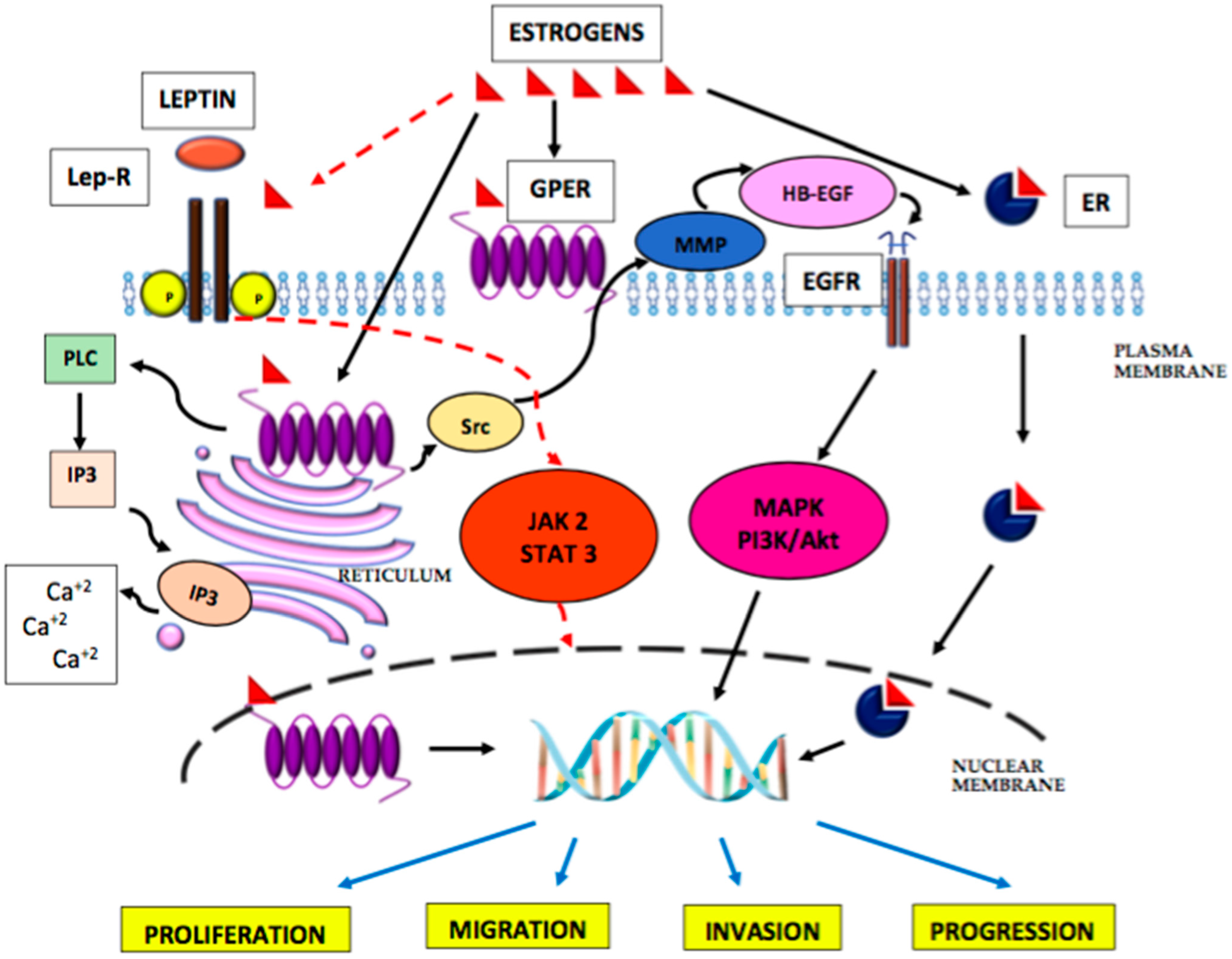{kind=link}
Abstract
:1. Introduction
2. Leptin and Adiponectin
2.1. Leptin
2.2. Adiponectin
3. Leptin Effect on the Development of Endometrial Cancer
4. The Role of Adiponectin in the Development of Endometrial Cancer
5. The Role of Oestrogens in the Development of Endometrial Cancer
6. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization; International Agency for Research on Cancer (IARC). Global Cancer Observatory. Available online: https://gco.iarc.fr (accessed on 20 February 2022).
- Ali, A.T. Reproductive factors and risk of endometrial cancer. Int. J. Gynecol. Cancer 2014, 21, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, V.W.; Yang, H.P.; Pike, M.C.; McCann, S.E.; Yu, H.; Xiang, Y.B.; Wolk, A.; Wentzensen, N.; Weiss, N.S.; Webb, P.M.; et al. Type I and II endometrial cancers: Have they different risk factors? J. Clin. Oncol. 2013, 31, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Perez-Hernandez, A.I.; Catalan, V.; Gomez-Ambrosi, J.; Rodríguez, A.; Frühbeck, G. Mechanisms linking excess adiposity and carcinogenesis promotion. Front. Endocrinol. 2014, 5, 65. [Google Scholar]
- Lee, C.H.; Woo, Y.C.; Wang, Y.; Yeung, C.Y.; Xu, A.; Lam, K.S. Obesity, adipokines and cancer: An update. Clin. Endocrinol. (Oxf) 2015, 83, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Guadagni, F.; Roselli, M.; Martini, F.; Spila, A.; Riondino, S.; D’Alessandro, R.; Del Monte, G.; Formica, V.; Laudisi, A.; Portarena, I.; et al. Prognostic Significance of Serum Adipokine Levels in Colorectal Cancer Patients. Anticancer Res. 2009, 29, 3321–3327. [Google Scholar]
- Chen, D.C.; Chung, Y.F.; Yeh, Y.T.; Chaung, H.C.; Kuo, F.C.; Fu, O.Y.; Chen, H.Y.; Hou, M.F.; Yuan, S.S.F. Serum adiponectin and leptin levels in Taiwanese breast cancer patients. Cancer Lett. 2006, 237, 109–114. [Google Scholar] [CrossRef]
- Ellis, P.E.; Barron, G.A.; Bermano, G. Adipocytokines and their relationship to endometrial cancer risk: A systematic review and meta-analysis. Gyn. Oncol. 2020, 158, 507–516. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Margetic, S.; Gazzola, C.; Pegg, G.G.; Hill, R.A. Leptin: A review of its peripheral actions and interactions. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 1407–1433. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Vilariño-García, T.; Sánchez-Margalet, V. Role of Leptin in Inflammation and Vice Versa. Int. J. Mol. Sci. 2020, 21, 5887. [Google Scholar] [CrossRef] [PubMed]
- Gruzdeva, O.; Borodkina, D.; Uchasova, E.; Dyleva, Y.; Barbarash, O. Leptin resistance: Underlying mechanisms and diagnosis. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Morley, T.S.; Kim, M.; Clegg, D.J.; Scherer, P.E. Obesity and cancer—Mechanisms underlying tumour progression and recurrence. Nat. Rev. Endocrinol. 2014, 10, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boroń, D.; Nowakowski, R.; Grabarek, B.O.; Zmarzły, N.; Opławski, M. Expression Pattern of Leptin and Its Receptors in Endometrioid Endometrial Cancer. J. Clin. Med. 2021, 10, 2787. [Google Scholar] [CrossRef]
- Ahima, R.S. Adipose tissue as an endocrine organ. Obesity (Silver Spring) 2006, 14, 242–249. [Google Scholar] [CrossRef]
- Bjursell, M.; Ahnmark, A.; Bohlooly, Y.M.; William-Olsson, L.; Rhedin, M.; Peng, X.R.; Ploj, K.; Gerdin, A.K.; Arnerup, G.; Elmgren, A.; et al. Opposing Effects of Adiponectin Receptors 1 and 2 on Energy Metabolism. Diabetes 2007, 56, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Kikani, C.K.; Riojas, R.A.; Langlais, P.; Wang, L.; Ramos, F.J.; Fang, Q.; Christ-Roberts, C.Y.; Hong, J.Y.; Kim, R.Y.; et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat. Cell. Biol. 2006, 8, 516–523. [Google Scholar] [CrossRef]
- Chen, J.; Tan, B.; Karteris, E.; Zervou, S.; Digby, J.; Hillhouse, E.; Vatish, M.; Randeva, H.S. Secretion of adiponectin by human placenta: Differential modulation of adiponectin and its receptors by cytokines. Diabetologia 2006, 49, 1292–1302. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, N.; Takazawa, Y.; Maeda, D.; Hibiya, T.; Tanaka, M.; Iwabu, M.; Okada-Iwabu, M.; Yamauchi, T.; Kadowaki, T.; Fukayama, M. Expression levels of adiponectin receptors are decreased in human endometrial adenocarcinoma tissues. Int. J. Gynecol. Pathol. 2012, 31, 352–357. [Google Scholar] [CrossRef]
- Yamauchi, T.; Hara, K.; Kubota, N.; Terauchi, Y.; Tobe, K.; Froguel, P.; Nagai, R.; Kadowaki, T. Dual roles of adiponectin/Acrp30 in vivo as an anti-diabetic and anti-atherogenic adipokine. Curr. Drug Targets-Immune Endocr. Metab. Disord. 2003, 3, 243–254. [Google Scholar] [CrossRef]
- Wang, Y.; Lam, K.S.; Xu, J.Y.; Lu, G.; Xu, L.Y.; Cooper, G.J.; Xu, A. Adiponectin inhibits cell proliferation by interacting with several growth factors in an oligomerization-dependent manner. J. Biol. Chem. 2005, 280, 18341–18347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Iwabu, M.; Yamauchi, T.; Okada-Iwabu, M.; Sato, K.; Nakagawa, T.; Funata, M.; Yamaguchi, M.; Namiki, S.; Nakayama, R.; Tabata, M. Adiponectin and AdipoR1 regulate PGC-1α and mitochondria by Ca2+ and AMPK/SIRT1. Nature 2010, 464, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Bub, J.D.; Uzuki, M.; Iwamoto, Y. Adiponectin activates c-Jun NH2-terminal kinase and inhibits signal transducer and activator of transcription 3. Biochem. Biophys. Res. Commun. 2005, 333, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.; Robinson, F.; Beers Gibson, T.; Xu, B.E.; Karandikar, M.; Berman, K.; Cobb, M.H. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr. Rev. 2001, 22, 153–183. [Google Scholar] [PubMed] [Green Version]
- Kadowaki, T.; Yamauchi, T. Adiponectin and adiponectin receptors. Endocr. Rev. 2005, 26, 439–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Saha, A.K.; Xiang, X.; Ruderman, N.B. AMPK, the metabolic syndrome and cancer. Trends Pharmacol. Sci. 2005, 26, 69–76. [Google Scholar] [CrossRef]
- Rattan, R.; Giri, S.; Singh, A.K.; Singh, I. 5-Aminoimi-dazole-4-carboxamide-1-beta-Dribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J. Biol. Chem. 2005, 280, 582–593. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Alessi, D.R. LKB1 and AMPK and the cancer-metabolism link-ten years after. BMC Biol. 2013, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Gormand, A.; Henriksson, E.; Ström, K.; Jensen, T.E.; Sakamoto, K.; Göransson, O. Regulation of AMP-activated protein kinase by LKB1 and CaMKK in adipocytes. J. Cell Biochem. 2011, 112, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. The LKB1-AMPK pathway-friend or foe in cancer? Cancer Cell 2013, 23, 131–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, J.M.; Svensson, R.U.; Lou, H.J.; Winslow, M.M.; Turk, B.E.; Shaw, R.J. An AMPK-independent signaling pathway downstream of the LKB1 tumor suppressor controls Snail1 and metastatic potential. Mol. Cell 2014, 55, 436–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieudonne, M.N.; Bussiere, M.; Dos Santos, E.; Leneveu, M.C.; Giudicelli, Y.; Pecquery, R. Adiponectin mediates antiproliferative and apoptotic responses in human MCF7 breast cancer cells. Biochem. Biophys. Res. Commun. 2006, 345, 271–279. [Google Scholar] [CrossRef]
- Ouchi, N.; Kihara, S.; Arita, Y.; Okamoto, Y.; Maeda, K.; Kuriyama, H.; Hotta, K.; Nishida, M.; Takahashi, M.; Muraguchi, M. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-κB signaling through a cAMP-dependent pathway. Circulation 2000, 102, 1296–1301. [Google Scholar] [CrossRef]
- Matsuzawa, Y. Establishment of a Concept of Visceral Fat Syndrome and Discovery of Adiponectin. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Sivridis, E.; Giatromanolaki, A.; Gatter, K.C.; Harris, A.L.; Koukourakis, M.I. Association of hypoxia-inducible factors 1alpha and 2alpha with activated angiogenic pathways and prognosis in patients with endometrial carcinoma. Cancer 2002, 95, 1055–1063. [Google Scholar] [CrossRef]
- Grosfeld, A.; Andre, J.; Hauguel-de Mouzon, S.; Berra, E.; Pouysségur, J.; Guerre-Millo, M. Hypoxia-inducible factor 1 transactivates the human leptin gene promoter. J. Biol. Chem. 2002, 277, 42953–42957. [Google Scholar] [CrossRef] [Green Version]
- Ambrosini, G.; Nath, A.K.; Sierra-Honigmann, M.R.; Flores-Riveros, J. Transcriptional activation of the human leptin gene in response to hypoxia: Involvement of hypoxiainducible factor 1. J. Biol. Chem. 2002, 277, 34601–34609. [Google Scholar] [CrossRef] [Green Version]
- Koda, M.; Sulkowska, M.; Wincewicz, A.; Kanczuga-Koda, L.; Musiatowicz, B.; Szymanska, M.; Sulkowski, S. Expression of leptin, leptin receptor, and hypoxia-inducible factor 1 alpha in human endometrial cancer. Ann. N. Y. Acad. Sci. 2007, 1095, 90–98. [Google Scholar] [CrossRef]
- Wincewicz, A.; Koda, M.; Sulkowska, M.; Kanczuga-Koda, L.; Sulkowski, S. Comparison of STAT3 with HIF-1alpha, Ob and ObR expressions in human endometrioid adenocarcinomas. Tissue Cell 2008, 40, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.Y.; Sun, D.; Liu, X.Y.; Pan, Y.; Liang, H. STAT-3 correlates with lymph node metastasis and cell survival in gastric cancer. World J. Gastroenterol. 2010, 16, 5380–5387. [Google Scholar] [CrossRef] [PubMed]
- Badr, G.; Mohany, M.; Abu-Tarboush, F. Thymoquinone decreases F-actin polymerization and the proliferation of human multiple myeloma cells by suppressing STAT3 phosphorylation and Bcl2/Bcl-XL expression. Lipids Health Dis. 2011, 10, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Grande, F.; Neamati, N. Small molecule inhibitors of STAT3 signaling pathway. Curr. Cancer Drug Targets 2007, 7, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Tian, J.; Lv, Y.; Shi, F.; Kong, F.; Shi, H.; Zhao, L. Leptin induces functional activation of cyclooxygenase-2 through JAK2/STAT3, MAPK/ERK, and PI3K/AKT pathways in human endometrial cancer cells. Cancer Sci. 2009, 100, 389–395. [Google Scholar] [CrossRef]
- Chen, C.L.; Hsieh, F.C.; Lieblein, J.C.; Brown, J.; Chan, C.; Wallace, J.A.; Cheng, G.; Hall, B.M.; Lin, J. Stat3 activation in human endometrial and cervical cancers. Br. J. Cancer 2007, 96, 591–599. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, L.; Li, C.; Ai, H. Correlation analysis between the expressions of leptin and its receptor (ObR) and clinicopathology in endometrial cancer. Cancer Biomark. 2014, 14, 353–359. [Google Scholar] [CrossRef]
- Cymbaluk-Płoska, A.; Chudecka-Głaz, A.; Jagodzińska, A.; Pius-Sadowska, E.; Sompolska-Rzechuła, A.; Machaliński, B.; Menkiszak, J. Evaluation of biologically active substances promoting the development of or protecting against endometrial cancer. Onco. Targets Ther. 2018, 11, 1363–1372. [Google Scholar] [CrossRef] [Green Version]
- Lipsey, C.C.; Harbuzariu, A.; Daley-Brown, D.; Gonzalez-Perez, R.R. Oncogenic role of leptin and Notch interleukin-1 leptin crosstalk outcome in cancer. World J. Methodol. 2016, 6, 43–55. [Google Scholar] [CrossRef]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef] [Green Version]
- Jonusiene, V.; Sasnauskiene, A. Notch and Endometrial Cancer. Adv. Exp. Med. Biol. 2021, 1287, 47–57. [Google Scholar] [PubMed]
- Mitsuhashi, Y.; Horiuchi, A.; Miyamoto, T.; Kashima, H.; Suzuki, A.; Shiozawa, T. Prognostic significance of Notch signalling molecules and their involvement in the invasiveness of endometrial carcinoma cells. Histopathology 2012, 60, 826–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhailik, A.; Mazella, J.; Liang, S.; Tseng, L. Notch ligand-dependent gene expression in human endometrial stromal cells. Biochem. Biophys. Res. Commun. 2009, 388, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Cobellis, L.; Caprio, F.; Trabucco, E.; Mastrogiacomo, A.; Coppola, G.; Manente, L.; Colacurci, N.; De Falco, M.; De Luca, A. The pattern of expression of Notch protein members in normal and pathological endometrium. J. Anat. 2008, 213, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Aoki, D.; Susumu, N.; Udagawa, Y.; Nozawa, S. Imbalanced expression of TAN-1 and human Notch4 in endometrial cancers. Int. J. Oncol. 2000, 17, 1131–1139. [Google Scholar] [CrossRef]
- Mori, M.; Miyamoto, T.; Ohno, S.; Miyake, Y.; Sakaguchi, T.; Ohno, E. Diagnostic utility of notch-1 immu- nocytochemistry in endometrial cytology. Acta Cytol. 2012, 56, 166–170. [Google Scholar] [CrossRef]
- Sasnauskiene, A.; Jonusiene, V.; Krikstaponiene, A.; Butkyte, S.; Dabkeviciene, D.; Kanopiene, D.; Kazbariene, B.; Didziapetriene, J. NOTCH1, NOTCH3, NOTCH4, and JAG2 protein levels in human endometrial cancer. Medicina 2014, 50, 14–18. [Google Scholar] [CrossRef]
- Townsend, M.H.; Ence, Z.E.; Felsted, A.M.; Parker, A.C.; Piccolo, S.R.; Robison, R.A.; O’Neill, K.L. Potential new biomarkers for endometrial cancer. Cancer Cell Int. 2019, 19, 19. [Google Scholar] [CrossRef]
- Jonusiene, V.; Sasnauskiene, A.; Lachej, N.; Kanopiene, D.; Dabkeviciene, D.; Sasnauskiene, S.; Kazbariene, B.; Didziapetriene, J. Down-regulated expression of Notch signaling molecules in human endometrial cancer. Med. Oncol. 2013, 30, 438. [Google Scholar] [CrossRef]
- Williams, E.; Villar-Prados, A.; Bowser, J.; Broaddus, R.; Gladden, A.B. Loss of polarity alters proliferation and differentiation in low-grade endometrial cancers by disrupting Notch signaling. PLoS ONE 2017, 12, e0189081. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, K.; Liu, Z.; Wang, T.; Shi, F.; Zhang, Y.; Su, J.; Jia, Y. Upregulated delta-like protein 3 expression is a diagnostic and prognostic marker in endometrial cancer: A retrospective study. Medicine 2018, 97, e13442. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Gonzalez-Perez, R.R. Notch, IL-1 and Leptin Crosstalk Outcome (NILCO) Is Critical for Leptin- Induced Proliferation, Migration and VEGF/VEGFR-2 Expression in Breast Cancer. PLoS ONE 2011, 6, e21467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbuzariu, A.; Rampoldi, A.; Daley-Brown, D.S.; Candelaria, P.; Harmon, T.L.; Lipsey, C.C.; Beech, D.J.; Quarshie, A.; Ilies, G.O.; Gonzalez-Perez, R.R. Leptin-Notch signaling axis is involved in pancreatic cancer progression. Oncotarget 2017, 8, 7740–7752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbuzariu, A.; Gonzalez-Perez, R.R. Leptin-Notch axis impairs 5-fluorouracil effects on pancreatic cancer. Oncotarget 2018, 9, 18239–18253. [Google Scholar] [CrossRef] [Green Version]
- Carino, C.; Olawaiye, A.B.; Cherfils, S.; Serikawa, T.; Lynch, M.P.; Rueda, B.R.; Gonzalez, R.R. Leptin regulation of proangiogenic molecules in benign and cancerous endometrial cells. Int. J. Cancer 2008, 123, 2782–2790. [Google Scholar] [CrossRef] [Green Version]
- Battle, M.; Gillespie, C.; Quarshie, A.; Lanier, V.; Harmon, T.; Wilson, K.; Torroella-Kouri, M.; Gonzalez-Perez, R.R. Obesity Induced a Leptin-Notch Signaling Axis in Breast Cancer. Int. J. Cancer 2014, 134, 1605–1616. [Google Scholar] [CrossRef] [Green Version]
- Daley-Brown, D.; Harbuzariu, A.; Kurian, A.A.; Oprea-Ilies, G.; Gonzalez-Perez, R.R. Leptin-induced Notch and IL-1 signaling crosstalk in endometrial adenocarcinoma is associated with invasiveness and chemoresistance. World J. Clin. Oncol. 2019, 10, 222–233. [Google Scholar] [CrossRef]
- Yunusova, N.V.; Kondakova, I.V.; Kolomiets, L.A.; Afanas’ ev, S.G.; Chernyshova, A.L.; Kudryavtsev, I.V.; Tsydenova, A.A. Molecular targets for the therapy ofcancer associated with metabolic syndrome (transcriptionand growth factors). Asia-Pac. J. Clin. Oncol. 2018, 14, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Kelesidis, I.; Kelesidis, T.; Mantzoros, C.S. Adiponectin and cancer: A systematic review. Br. J. Cancer 2006, 94, 1221–1225. [Google Scholar] [CrossRef]
- Cust, A.E.; Kaaks, R.; Friedenreich, C.; Bonnet, F.; Laville, M.; Lukanova, A.; Rinaldi, S.; Dossus, L.; Slimani, N.; Lundin, E.; et al. Plasma adiponectin levels and endometrial cancer risk in pre- and postmenopausal women. J. Clin. Endocrinol. Metab. 2007, 92, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wen, K.; Han, X.; Liu, R.; Qu, Q. Adiponectin mediates antiproliferative and apoptotic responses in endometrial carcinoma by the AdipoRs/AMPK pathway. Gynecol. Oncol. 2015, 137, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Ashizawa, N.; Yahata, T.; Quan, J.; Adachi, S.; Yoshihara, K.; Tanaka, K. Serum leptin-adiponectin ratio and endometrial cancer risk in postmenopausal female subject. Gynecol. Oncol. 2010, 119, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Morielli, A.R.; Kokts-Porietis, R.L.; Benham, J.L. Associations of insulin resistance and inflammatory biomarkers with endometrial cancer survival: The Alberta endometrial cancer cohort study. Cancer Med. 2022, 11, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Shi, H.; Zhao, Z.; Wang, S.; Zhou, S.; Mu, Y.; Ding, N.; Lai, Y.; Zhao, A.Z.; Cheng, L.; et al. Adiponectin deficiency promotes endometrial carcinoma pathogenesis and development via activation of mitogen-activated protein kinase. J. Pathol. 2022, 257, 146–157. [Google Scholar] [CrossRef]
- Taliaferro-Smith, L.; Nagalingam, A.; Zhong, D.; Zhou, W.; Saxena, N.K.; Sharma, D. LKB1 is required for adiponectin-mediated modulation of AMPK-S6K axis and inhibition of migration and invasion of breast cancer cells. Oncogene 2009, 28, 2621–2633. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.S.; Chamberland, J.P.; Aronis, K.; Tseleni-Balafouta, S.; Mantzoros, C.S. Direct role of adiponectin and adiponectin receptors in endometrial cancer: In vitro and ex vivo studies in humans. Mol. Cancer Ther. 2011, 10, 2234–2243. [Google Scholar] [CrossRef] [Green Version]
- Barbe, A.; Bongrani, A.; Mellouk, N.; Estienne, A.; Kurowska, P.; Grandhaye, J.; Elfassy, Y.; Levy, R.; Rak, A.; Froment, P.; et al. Mechanisms of adiponectin action in fertility: An overview from gametogenesis to gestation in humans and animal models in normal and pathological conditions. Int. J. Mol. Sci. 2019, 20, 1526. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Yan, Q.; Zhang, Z.; Du, G.; Wan, X. Acrp30 inhibits leptin-induced metastasis by downregulating the JAK/STAT3 pathway via AMPK activation in aggressive SPEC-2 endometrial cancer cells. Oncol. Rep. 2012, 27, 1488–1496. [Google Scholar] [CrossRef]
- Maybin, J.A.; Critchley, H.O.D. Menstrual physiology: Implications for endometrial pathology and beyond. Hum. Reprod. Update 2015, 21, 748–761. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.J.; Erickson, G.F. Morphology and Physiology of the Ovary; Endotext; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Anckaert, E.; Jank, A.; Petzold, J.; Rohsmann, F.; Paris, R.; Renggli, M.; Schönfeld, K.; Schiettecatte, J.; Kriner, M. Extensive monitoring of the natural menstrual cycle using the serum biomarkers estradiol, luteinizing hormone and progesterone. Pract. Lab. Med. 2021, 25, e00211. [Google Scholar] [CrossRef]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Meseguer, A.; Puche, C.; Cabero, A. Sex steroid biosynthesis in white adipose tissue. Horm. Metab. Res. 2002, 34, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, M.; Decorby, K.; Choi, B.C. The association between obesity and cancer risk: A meta-analysis of observational studies from 1985 to 2011. Prev. Med. 2013, 2013, 680536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, A.C.; Blanchard, Z.; Maurer, K.A.; Gertz, J. Estrogen signaling in endometrial cancer: A key oncogenic pathway with several open questions. Horm. Cancer 2019, 10, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyster, K.M. Estrogen receptors methods and protocols methods in molecular biology 1366. Methods Mol. Biol. 2016, 18–19. [Google Scholar]
- Pakdel, F. Molecular pathways of estrogen receptor action. Int. J. Mol. Sci. 2018, 19, 2591. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Huang, Z.Y.; Xu, X.L.; Li, J.; Fu, X.W.; Deng, S.L. Estrogen receptor function: Impact on the human endometrium. Front. Endocrinol. 2022, 1, 113. [Google Scholar] [CrossRef]
- Samarnthai, N.; Hall, K.; Yeh, I.T. Molecular profiling of endometrial malignancies. Obstet. Gynecol. Int. 2010, 2010, 162363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, P.A.; Critchley, H.O.D.; Williams, A.R.W.; Arends, M.J.; Saunders, P.T.K. New concepts for an old problem: The diagnosis of endometrial hyperplasia. Hum. Reprod. Update 2017, 23, 232–254. [Google Scholar] [CrossRef]
- Shen, F.; Gao, Y.; Ding, J.; Chen, Q. Is the positivity of estrogen receptor or progesterone receptor different between type 1 and type 2 endometrial cancer? Oncotarget 2017, 8, 506–511. [Google Scholar] [CrossRef]
- Wan, J.; Gao, Y.; Zeng, K.; Yin, Y.; Zhao, M.; Wei, J.; Chen, Q. The levels of the sex hormones are not different between type 1 and type 2 endometrial cancer. Sci. Rep. 2016, 6, 39744. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Saji, S.; Mäkinen, S.; Cheng, G.; Jensen, E.V.; Warner, M.; Gustafsson, J.Å. Estrogen receptor (ER) beta, a modulator of ER alpha in the uterus. Proc. Natl. Acad. Sci. USA 2000, 97, 5936–5941. [Google Scholar] [CrossRef] [Green Version]
- Paul, M.; Cholewa, K.; Mazurek, U.; Witek, A.; Wilczok, T. Estrogen receptor beta delta 6 (ER Beta Delta 6) isoform in human endometrial hyperplasia and adenocarcinoma. Cancer Investig. 2004, 22, 211–218. [Google Scholar] [CrossRef]
- Smuc, T.; Rizner, T.L. Aberrant pre-receptor regulation of estrogen and progesterone action in endometrial cancer. Mol. Cell Endocrinol. 2009, 301, 74–82. [Google Scholar] [CrossRef]
- Skrzypczak, M.; Bieche, I.; Szymczak, S.; Tozlu, S.; Lewandowski, S.; Girault, I.; Radwanska, K.; Szczylik, C.; Jakowicki, J.A.; Lidereau, R.; et al. Evaluation of mRNA expression of estrogen receptor beta and its isoforms in human normal and neoplastic endometrium. Int. J. Cancer 2004, 110, 783–787. [Google Scholar] [CrossRef]
- Häring, J.; Skrzypczak, M.; Stegerer, A.; Lattrich, C.; Weber, F.; Goerse, R.; Ortmann, O.; Treeck, O. Estrogen receptor beta transcript variants associate with oncogene expression in endometrial cancer. Int. J. Mol. Med. 2012, 29, 1127–1136. [Google Scholar] [CrossRef]
- Hewitt, S.C.; Korach, K.S. Estrogen receptors: Structure, mechanisms and function. Rev. Endocr. Metab. Disord. 2002, 3, 193–200. [Google Scholar] [CrossRef]
- Tang, Z.R.; Zhang, R.; Lian, Z.X.; Deng, S.L.; Yu, K. Estrogen-Receptor Expression and Function in Female Reproductive Disease. Cells 2019, 8, 1123. [Google Scholar] [CrossRef] [Green Version]
- Prossnitz, E.R.; Arterburn, J.B. International union of basic and clinical pharmacology. XCVII. G protein–coupled estrogen receptor and its pharmacologic modulators. Pharmacol. Rev. 2015, 67, 505–540. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.; Yin, Y.; Zhao, M.; Shen, F.; Chen, M.; Chen, Q. The positivity of G- protein-coupled receptor-30 (GPR 30), an alternative estrogen receptor is not different between type 1 and type 2 endometrial cancer. Oncotarget 2017, 8, 90897–90904. [Google Scholar] [CrossRef] [Green Version]
- Carlson, M.J.; Thiel, K.W.; Leslie, K.K. Past, present, and future of hormonal therapy in recurrent endometrial cancer. Int. J. Women’s Health 2014, 6, 429–435. [Google Scholar]
- Onstad, M.A.; Schmandt, R.E.; Lu, K.H. Addressing the role of obesity in endometrial cancer risk, prevention, and treatment. J. Clin. Oncol. 2016, 34, 4225–4230. [Google Scholar] [CrossRef]
- Shetty, A.; Venkatesh, T.; Tsutsumi, R.; Suresh, P.S. Gene expression changes and promoter methylation with the combined effects of estradiol and leptin in uterine tissue of the ovariectomized mice model of menopause. Mol. Biol. Rep. 2019, 47, 151–168. [Google Scholar] [CrossRef]
- Tanos, P.; Dimitriou, S.; Gullo, G.; Tanos, V. Biomolecular and Genetic Prognostic Factors That Can Facilitate Fertility-Sparing Treatment (FST) Decision Making in Early Stage Endometrial Cancer (ES-EC): A Systematic Review. Int. J. Mol. Sci. 2022, 23, 2653. [Google Scholar] [CrossRef]
- Rosa, V.L.; Valenti, G.; Sapia, F.; Gullo, G.; María, A.V.; Rapisarda, C. Psychological impact of gynecological diseases: The importance of a multidisciplinary approach. It. J. Gynaecol. Obstet 2018, 30, 2. [Google Scholar]
- Cavaliere, A.F.; Perelli, F.; Zaami, S.; D’Indinosante, M.; Turrini, I.; Giusti, M.; Gullo, G.; Vizzielli, G.; Mattei, A.; Scambia, G.; et al. Fertility Sparing Treatments in Endometrial Cancer Patients: The Potential Role of the New Molecular Classification. Int. J. Mol. Sci. 2021, 22, 12248. [Google Scholar] [CrossRef]
- Gullo, G.; Etrusco, A.; Cucinella, G.; Perino, A.; Chiantera, V.; Laganà, A.S.; Tomaiuolo, R.; Vitagliano, A.; Giampaolino, P.; Noventa, M.; et al. Fertility-Sparing Approach in Women Affected by Stage I and Low-Grade Endometrial Carcinoma: An Updated Overview. Int. J. Mol. Sci. 2021, 22, 11825. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Słabuszewska-Jóźwiak, A.; Lukaszuk, A.; Janicka-Kośnik, M.; Wdowiak, A.; Jakiel, G. Role of Leptin and Adiponectin in Endometrial Cancer. Int. J. Mol. Sci. 2022, 23, 5307. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105307
Słabuszewska-Jóźwiak A, Lukaszuk A, Janicka-Kośnik M, Wdowiak A, Jakiel G. Role of Leptin and Adiponectin in Endometrial Cancer. International Journal of Molecular Sciences. 2022; 23(10):5307. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105307
Chicago/Turabian StyleSłabuszewska-Jóźwiak, Aneta, Aron Lukaszuk, Marta Janicka-Kośnik, Artur Wdowiak, and Grzegorz Jakiel. 2022. "Role of Leptin and Adiponectin in Endometrial Cancer" International Journal of Molecular Sciences 23, no. 10: 5307. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105307