
Self-Renewal and Cancers of the Gastric Epithelium: An Update and the Role of the Lectin TFF1 as an Antral Tumor Suppressor


Abstract
: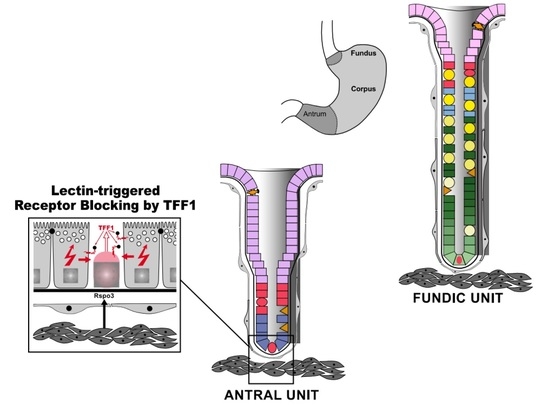
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Gastric Self-Renewal from Stem and Precursor Cells
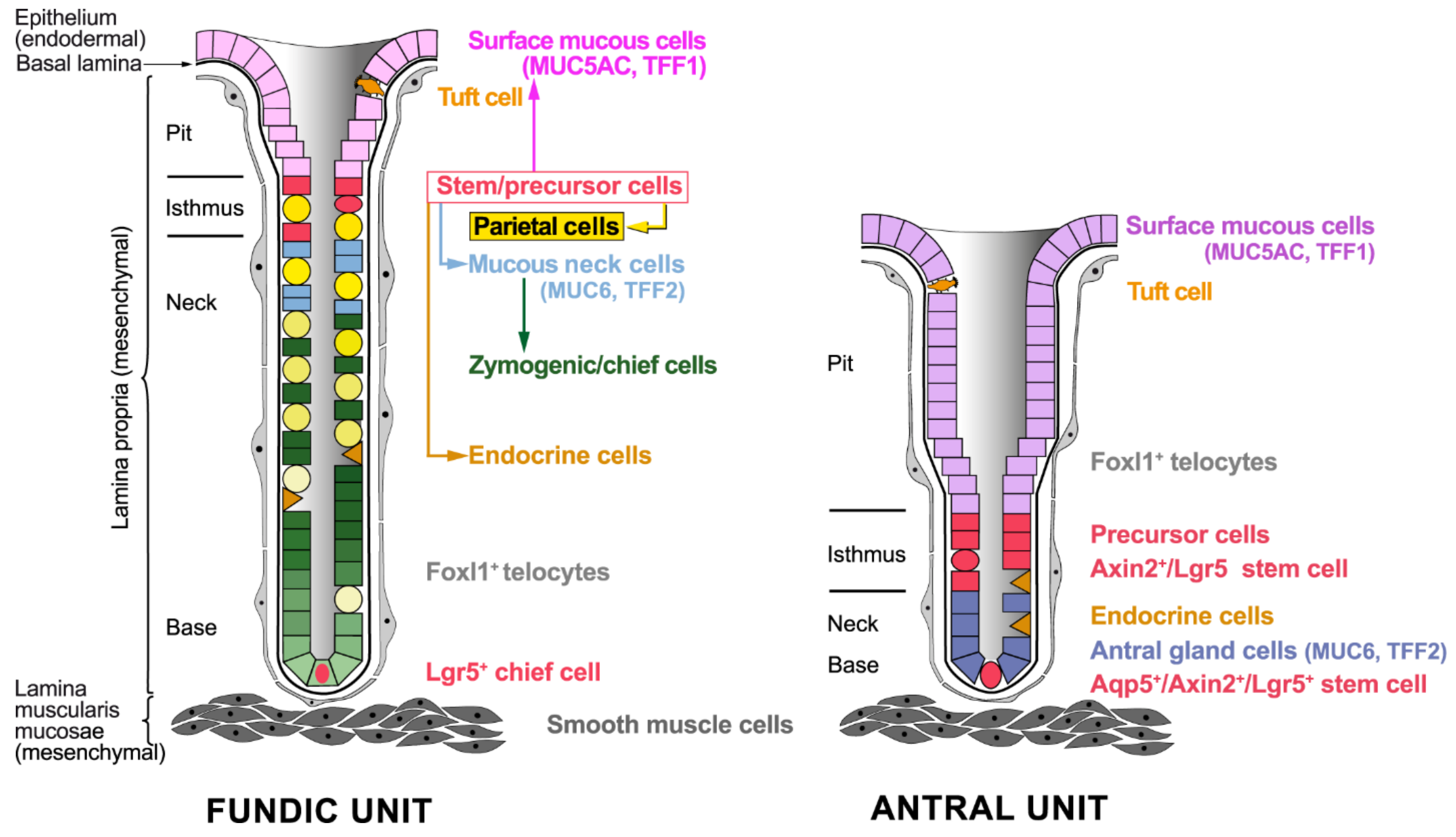
2.1. Cellular Architecture of the Gastric Mucosa
2.2. Stem Cells of the Gastric Epithelium
2.2.1. Stem Cells in the Fundic Units
2.2.2. Stem Cells in the Antral Units
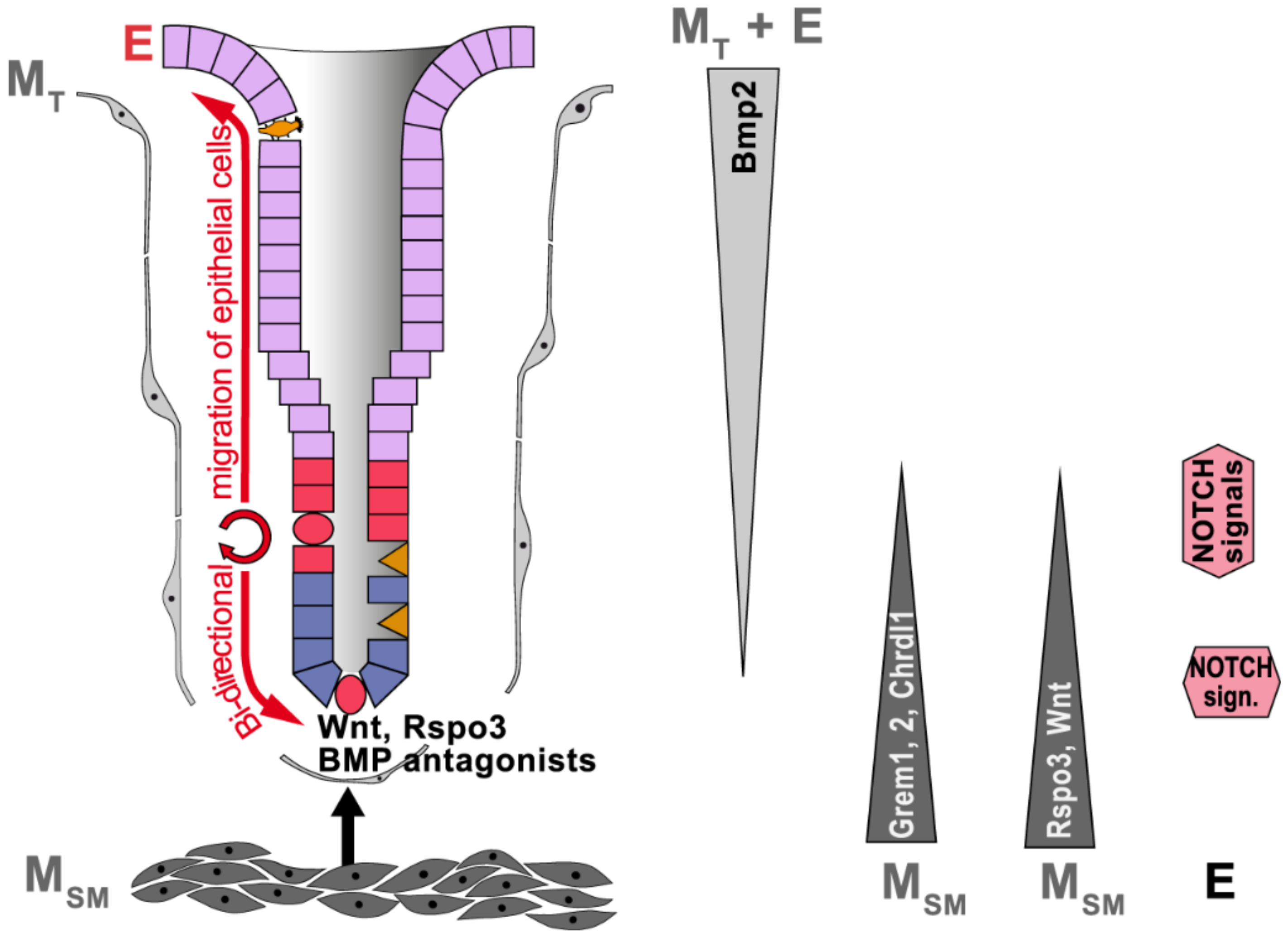
2.3. Gastric Stem Cell Niches, Reciprocal Epithelial–Mesenchymal Interactions
3. Gastric Cancers and Their Pre-Cancerous Lesions
3.1. Helicobacter pylori and Its Colonization of Gastric Glands
3.2. Fundic Intestinal-Type Adenocarcinomas: Atrophic Gastritis, Metaplasias, Inflammation
3.3. Diffuse Gastric Adenocarcinomas
3.4. Neoplasms of the Gastric Antrum
3.5. Mechanisms of Field Cancerization: Monoclonal Conversion, Gland Fission
4. TFF1 Is an Antral Tumor Suppressor
4.1. The Secretory Lectin Trefoil Factor Family 1 (TFF1)
4.2. Results from Tff1KO Mice
4.3. Lineage Tracing Studies
4.4. TFF1 and Human Gastric Cancer
4.5. Possible Molecular Function(s) of Tff1: Lectin-Triggered Receptor Activation/Blocking
5. Conclusions and Medical Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A4GNT | α1,4-N-acetylglucosaminyltransferase |
| AGC | Antral gland cell |
| AR | Amphiregulin |
| BMP | Bone morphogenetic protein |
| EGF | Epidermal growth factor |
| ER | Endoplasmic reticulum |
| FCGBP | IgG Fc binding protein |
| FOX | Forkhead box |
| GC | Gastric cancer |
| GKN | Gastrokine |
| IL | Interleukin |
| ILC2 | Group 2 innate lymphoid cell |
| IM | Intestinal metaplasia |
| MNC | Mucous neck cell |
| PAS | Periodic acid–Schiff |
| PPI | Proton pump inhibitor |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SHH | Sonic hedgehog |
| SMC | Surface mucous cell |
| SPEM | Spasmolytic polypeptide expressing metaplasia |
| TFF | Trefoil factor family |
| TGF | Transforming growth factor |
| vENCC | Vagal enteric neural crest cell |
References
- Kim, T.H.; Shivdasani, R.A. Stomach development, stem cells and disease. Development 2016, 143, 554–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, W. Gastric stem cells: Of flies and men. Cell Cycle 2011, 10, 1186–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, W. Regeneration of the gastric mucosa and its glands from stem cells. Curr. Med. Chem. 2008, 15, 3133–3144. [Google Scholar] [CrossRef] [PubMed]
- Bartfeld, S.; Koo, B.K. Adult gastric stem cells and their niches. Wiley Interdisc. Rev. Dev. Biol. 2017, 6, e261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, W. Self-renewal of the gastric epithelium from stem and progenitor cells. Front. Biosci. 2013, 5, 720–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karam, S.M. Lineage commitment and maturation of epithelial cells in the gut. Front. Biosci. 1999, 4, D286–D298. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, W. Current Status on Stem Cells and Cancers of the Gastric Epithelium. Int. J. Mol. Sci. 2015, 16, 19153–19169. [Google Scholar] [CrossRef]
- Le Guen, L.; Marchal, S.; Faure, S.; de Santa Barbara, P. Mesenchymal-epithelial interactions during digestive tract development and epithelial stem cell regeneration. Cell. Mol. Life Sci. 2015, 72, 3883–3896. [Google Scholar] [CrossRef] [Green Version]
- McCracken, K.W.; Wells, J.M. Mechanisms of embryonic stomach development. Sem. Cell Dev. Biol. 2017, 66, 36–42. [Google Scholar] [CrossRef]
- Brabletz, S.; Schmalhofer, O.; Brabletz, T. Gastrointestinal stem cells in development and cancer. J. Pathol. 2009, 217, 307–317. [Google Scholar] [CrossRef]
- Goldenring, J.R.; Mills, J.C. Cellular Plasticity, Reprogramming, and Regeneration: Metaplasia in the Stomach and Beyond. Gastroenterology 2022, 162, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, W. Trefoil Factor Family (TFF) Peptides and Their Diverse Molecular Functions in Mucus Barrier Protection and More: Changing the Paradigm. Int. J. Mol. Sci. 2020, 21, 4535. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, W. Trefoil Factor Family (TFF) Peptides and Their Links to Inflammation: A Re-evaluation and New Medical Perspectives. Int. J. Mol. Sci. 2021, 22, 4909. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, O.; Chenard, M.P.; Masson, R.; Linares, J.; Dierich, A.; LeMeur, M.; Wendling, C.; Tomasetto, C.; Chambon, P.; Rio, M.C. Gastric mucosa abnormalities and tumorigenesis in mice lacking the pS2 trefoil protein. Science 1996, 274, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Tomasetto, C.; Rio, M.C. Pleiotropic effects of Trefoil Factor 1 deficiency. Cell. Mol. Life Sci. 2005, 62, 2916–2920. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Fox, J.G.; Gonda, T.; Worthley, D.L.; Muthupalani, S.; Wang, T.C. Mouse models of gastric cancer. Cancers 2013, 5, 92–130. [Google Scholar] [CrossRef] [Green Version]
- Willet, S.G.; Mills, J.C. Stomach Organ and Cell Lineage Differentiation: From Embryogenesis to Adult Homeostasis. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 546–559. [Google Scholar] [CrossRef] [Green Version]
- Powell, D.W.; Pinchuk, I.V.; Saada, J.I.; Chen, X.; Mifflin, R.C. Mesenchymal cells of the intestinal lamina propria. Annu. Rev. Physiol. 2011, 73, 213–237. [Google Scholar] [CrossRef] [Green Version]
- Leedham, S.J.; Brittan, M.; Preston, S.L.; McDonald, S.A.; Wright, N.A. The stomach periglandular fibroblast sheath: All present and correct. Gut 2006, 55, 295–296. [Google Scholar]
- Kaestner, K.H. The Intestinal Stem Cell Niche: A Central Role for Foxl1-Expressing Subepithelial Telocytes. Cell. Mol. Gastroenterol. Hepathol. 2019, 8, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.R.; Engevik, A.C.; Madorsky, T.; Belmont, E.; Stier, M.T.; Norlander, A.E.; Pilkinton, M.A.; McDonnell, W.J.; Weis, J.A.; Jang, B.; et al. Group 2 Innate Lymphoid Cells Coordinate Damage Response in the Stomach. Gastroenterology 2020, 159, 2077–2091. [Google Scholar] [CrossRef] [PubMed]
- Busada, J.T.; Peterson, K.N.; Khadka, S.; Xu, X.; Oakley, R.H.; Cook, D.N.; Cidlowski, J.A. Glucocorticoids and Androgens Protect From Gastric Metaplasia by Suppressing Group 2 Innate Lymphoid Cell Activation. Gastroenterology 2021, 161, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Karam, S.; Leblond, C.P. Origin and migratory pathways of the eleven epithelial cell types present in the body of the mouse stomach. Microsc. Res. Technol. 1995, 31, 193–214. [Google Scholar] [CrossRef] [PubMed]
- Karam, S.M.; Straiton, T.; Hassan, W.M.; Leblond, C.P. Defining epithelial cell progenitors in the human oxyntic mucosa. Stem Cells 2003, 21, 322–336. [Google Scholar] [CrossRef]
- Patel, S.; Rew, D.A.; Taylor, I.; Potten, C.S.; Owen, C.; Roberts, S.A. Study of the proliferation in human gastric mucosa after in vivo bromodeoxyuridine labelling. Gut 1993, 34, 893–896. [Google Scholar] [CrossRef] [Green Version]
- Kouznetsova, I.; Kalinski, T.; Meyer, F.; Hoffmann, W. Self-renewal of the human gastric epithelium: New insights from expression profiling using laser microdis. Mol. Biosyst. 2011, 7, 1105–1112. [Google Scholar] [CrossRef]
- Hoffmann, W. Stem cells, self-renewal and cancer of the gastric epithelium. Curr. Med. Chem. 2012, 19, 5975–5983. [Google Scholar] [CrossRef]
- Karam, S.M. A focus on parietal cells as a renewing cell population. World J. Gastroenterol. 2010, 16, 538–546. [Google Scholar] [CrossRef] [Green Version]
- Kornberg, T.B. Barcoding Hedgehog for intracellular transport. Sci. Signal. 2011, 4, pe44. [Google Scholar] [CrossRef] [Green Version]
- Konstantinou, D.; Bertaux-Skeirik, N.; Zavros, Y. Hedgehog signaling in the stomach. Curr. Opin. Pharmacol. 2016, 31, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Merchant, J.L.; Ding, L. Hedgehog Signaling Links Chronic Inflammation to Gastric Cancer Precursor Lesions. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 201–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engevik, A.C.; Kaji, I.; Goldenring, J.R. The Physiology of the Gastric Parietal Cell. Physiol. Rev. 2020, 100, 573–602. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.S.; Sigal, M. The Role of Wnt and R-spondin in the Stomach During Health and Disease. Biomedicines 2019, 7, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Fink, J.; Jörg, D.J.; Lee, E.; Yum, M.K.; Chatzeli, L.; Merker, S.R.; Josserand, M.; Trendafilova, T.; Andersson-Rolf, A.; et al. Defining the Identity and Dynamics of Adult Gastric Isthmus Stem Cells. Cell Stem Cell 2019, 25, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, J.; Douchi, D.; Myint, K.; Mon, N.N.; Yamamura, A.; Kohu, K.; Heng, D.L.; Chen, S.; Mawan, N.A.; Nuttonmanit, N.; et al. Iqgap3-Ras axis drives stem cell proliferation in the stomach corpus during homeostatis and repair. Gut 2021, 70, 1833–1846. [Google Scholar] [CrossRef]
- Nienhüser, H.; Kim, W.; Malagola, E.; Ruan, T.; Valenti, G.; Middelhoff, M.; Bass, A.; Der, C.J.; Hayakawa, Y.; Wang, T.C. Mist1+ gastric isthmus stem cells are regulated by Wnt5a and expand in response to injury and inflammation in mice. Gut 2021, 70, 654–665. [Google Scholar] [CrossRef]
- Leushacke, M.; Tan, S.H.; Wong, A.; Swathi, Y.; Hajamohideen, A.; Tan, L.T.; Goh, J.; Wong, E.; Denil, S.; Murakami, K.; et al. Lgr5-expressing chief cells drive epithelial regeneration and cancer in the oxyntic stomach. Nat. Cell Biol. 2017, 19, 774–786. [Google Scholar] [CrossRef]
- Stange, D.E.; Koo, B.K.; Huch, M.; Sibbel, G.; Basak, O.; Lyubimova, A.; Kujala, P.; Bartfeld, S.; Koster, J.; Geahlen, J.H.; et al. Differentiated Troy+ chief cells act as reserve stem cells to generate all lineages of the stomach epithelium. Cell 2013, 155, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Barker, N.; Huch, M.; Kujala, P.; van de Wetering, M.; Snippert, H.J.; van Es, J.H.; Sato, T.; Stange, D.E.; Begthel, H.; van den Born, M.; et al. Lgr5+ve stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell 2010, 6, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Sigal, M.; Logan, C.Y.; Kapalczynska, M.; Mollenkopf, H.J.; Berger, H.; Wiedenmann, B.; Nusse, R.; Amieva, M.R.; Meyer, T.F. Stromal R-spondin orchestrates gastric epithelial stem cells and gland homeostasis. Nature 2017, 548, 451–455. [Google Scholar] [CrossRef]
- Tan, S.H.; Swathi, Y.; Tan, S.; Goh, J.; Seishima, R.; Murakami, K.; Oshima, M.; Tsuji, T.; Phuah, P.; Tan, L.T.; et al. AQP5 enriches for stem cells and cancer origins in the distal stomach. Nature 2020, 578, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Leushacke, M.; Ng, A.; Galle, J.; Loeffler, M.; Barker, N. Lgr5+ gastric stem cells divide symmetrically to effect epithelial homeostasis in the pylorus. Cell Rep. 2013, 5, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, K.; Sarkar, A.; Yram, M.A.; Polo, J.M.; Bronson, R.; Sengupta, S.; Seandel, M.; Geijsen, N.; Hochedlinger, K. Sox2+ adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 2011, 9, 317–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, Y.; Jin, G.; Wang, H.; Chen, X.; Westphalen, C.B.; Asfaha, S.; Renz, B.W.; Ariyama, H.; Dubeykovskaya, Z.A.; Takemoto, Y.; et al. CCK2R identifies and regulates gastric antral stem cell states and carcinogenesis. Gut 2015, 64, 544–553. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, T.; Fukuda, A.; Araki, O.; Ogawa, S.; Hanyu, Y.; Matsumoto, Y.; Yamaga, Y.; Nakanishi, Y.; Kawada, K.; Sakai, Y.; et al. Bmi1 marks gastric stem cells located in the isthmus in mice. J. Pathol. 2019, 248, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Sigal, M.; del Mar Reinés, M.; Müllerke, S.; Fischer, C.; Kapalczynska, M.; Berger, H.; Bakker, E.R.M.; Mollenkopf, H.-J.; Rothenberg, M.E.; Wiedenmann, B.; et al. R-spondin-3 induces secretory, antimicrobial Lgr5+ cells in the stomach. Nat. Cell Biol. 2019, 21, 812–823. [Google Scholar] [CrossRef]
- Shyer, A.; Huycke, T.R.; Lee, C.L.; Mahadevan, L.; Tabin, C.J. Bending Gradients: How the Intestinal Stem Cell Gets Its Home. Cell 2015, 161, 569–580. [Google Scholar] [CrossRef] [Green Version]
- Samuelson, L.C. Intestinal-niche conundrum solved. Nature 2018, 558, 380–381. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, N.; Manieri, E.; Storm, E.E.; Saadatpour, A.; Luoma, A.M.; Kapoor, V.N.; Madha, S.; Gaynor, L.T.; Cox, C.; Keerthivasan, S.; et al. Distinct Mesenchymal Cell Populations Generate the Essential Intestinal BMP Signaling Gradient. Cell Stem Cell 2020, 26, 391–402. [Google Scholar] [CrossRef]
- Aoki, R.; Shoshkes-Carmel, M.; Gao, N.; Shin, S.; May, C.L.; Golson, M.L.; Zahm, A.M.; Ray, M.; Wiser, C.L.; Wright, C.V.; et al. Foxl1-expressing mesenchymal cells constitute the intestinal stem cell niche. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Shoshkes-Carmel, M.; Wang, Y.J.; Wangensteen, K.J.; Tóth, B.; Kondo, A.; Massasa, E.E.; Itzkovitz, S.; Kaestner, K.H. Subepithelial telocytes are an important source of Wnts that supports intestinal crypts. Nature 2018, 557, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.A.; Coates, P.J.; Ansari, B.; Hopwood, D. Regulation of cell number in the mammalian gastrointestinal tract: The importance of apoptosis. J. Cell Sci. 1994, 107, 3569–3577. [Google Scholar] [CrossRef] [PubMed]
- Worthley, D.L.; Churchill, M.; Compton, J.T.; Tailor, Y.; Rao, M.; Si, Y.; Levin, D.; Schwartz, M.G.; Uygur, A.; Hayakawa, Y.; et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 2015, 160, 269–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katano, T.; Ootani, A.; Mizoshita, T.; Tanida, S.; Tsukamoto, H.; Ozeki, K.; Kataoka, H.; Joh, T. Gastric mesenchymal myofibroblasts maintain stem cell activity and proliferation of murine gastric epithelium in vitro. Am. J. Pathol. 2015, 185, 798–807. [Google Scholar] [CrossRef]
- Biau, S.; Jin, S.; Fan, C.M. Gastrointestinal defects of the Gas1 mutant involve dysregulated Hedgehog and Ret signaling. Biol. Open 2013, 2, 144–155. [Google Scholar] [CrossRef] [Green Version]
- Zagami, C.; Papp, D.; Daddi, A.A.; Boccellato, F. Morphogen signals shaping the gastric glands in health and disease. Int. J. Mol. Sci. 2022, 23, 3632. [Google Scholar] [CrossRef]
- Scoville, D.H.; Sato, T.; He, X.C.; Li, L. Current view: Intestinal stem cells and signaling. Gastroenterology 2008, 134, 849–864. [Google Scholar] [CrossRef]
- De Lau, W.; Peng, W.C.; Gros, P.; Clevers, H. The R-spondin/Lgr5/Rnf43 module: Regulator of Wnt signal strength. Genes Develop. 2014, 28, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Shivdasani, R.A. Notch signaling in stomach epithelial stem cell homeostasis. J. Exp. Med. 2011, 208, 677–688. [Google Scholar] [CrossRef] [Green Version]
- Demitrack, E.S.; Gifford, G.B.; Keeley, T.M.; Carulli, A.J.; VanDussen, K.L.; Thomas, D.; Giordano, T.J.; Liu, Z.; Kopan, R.; Samuelson, L.C. Notch signaling regulates gastric antral LGR5 stem cell function. EMBO J. 2015, 34, 2522–2536. [Google Scholar] [CrossRef] [Green Version]
- Gifford, G.B.; Demitrack, E.S.; Keeley, T.M.; Tam, A.; La Cunza, N.; Dedhia, P.H.; Spence, J.R.; Simeone, D.M.; Saotome, I.; Louvi, A.; et al. Notch 1 and Notch 2 receptors regulate mouse and human gastric antral epithelial cell homeostasis. Gut 2017, 66, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Demitrack, E.S.; Samuelson, L.C. Notch as a Driver of Gastric Epithelial Cell Proliferation. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 323–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, Y.; Ariyama, H.; Stancikova, J.; Sakitani, K.; Asfaha, S.; Renz, B.W.; Dubeykovskaya, Z.A.; Shibata, W.; Wang, H.; Westphalen, C.B.; et al. Mist1 Expressing Gastric Stem Cells Maintain the Normal and Neoplastic Gastric Epithelium and Are Supported by a Perivascular Stem Cell Niche. Cancer Cell 2015, 28, 800–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccellato, F.; Woelffling, S.; Imai-Matsushima, A.; Sanchez, G.; Goosmann, C.; Schmid, M.; Berger, H.; Morey, P.; Denecke, C.; Ordemann, J.; et al. Polarised epithelial monolayers of the gastric mucosa reveal insights into mucosal homeostasis and defence against infection. Gut 2019, 68, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Fei, L.; Yin, W.C.; Coquenlorge, S.; Rao-Bhatia, A.; Zhang, X.; Shi, S.S.W.; Lee, J.H.; Hahn, N.A.; Rizvi, W.; et al. Single cell and genetic analyses reveal conserved populations and signaling mechanisms of gastrointestinal stromal niches. Nat. Commun. 2020, 11, 334. [Google Scholar] [CrossRef]
- Saqui-Salces, M.; Merchant, J.L. Hedgehog signaling and gastrointestinal cancer. Biochim. Biophys. Acta 2010, 1803, 786–795. [Google Scholar] [CrossRef] [Green Version]
- Wölffling, S.; Daddi, A.A.; Imai-Matsushima, A.; Fritsche, K.; Goosmann, C.; Traulsen, J.; Lisle, R.; Schmid, M.; Reines-Benassar, M.D.M.; Pfannkuch, L.; et al. EGF and BMPs Govern Differentiation and Patterning in Human Gastric Glands. Gastroenterology 2021, 161, 623–636. [Google Scholar] [CrossRef]
- Todisco, A. Regulation of Gastric Metaplasia, Dysplasia, and Neoplasia by Bone Morphogenetic Protein Signaling. Cell. Mol. Gastroenterol. Hepathol. 2017, 3, 339–347. [Google Scholar] [CrossRef] [Green Version]
- Roy, S.A.B.; Allaire, J.M.; Ouellet, C.; Maloum-Rami, F.; Pomerleau, V.; Lemieux, É.; Babeu, J.P.; Rousseau, J.; Paquet, M.; Garde-Granger, P.; et al. Loss of mesenchymal bone morphogenetic protein signaling leads to development of reactive stroma and initiation of the gastric neoplastic cascade. Sci. Rep. 2016, 6, 32759. [Google Scholar] [CrossRef] [Green Version]
- Zavros, Y.; Merchant, J.L. The immune microenvironment in gastric adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2022. [Google Scholar] [CrossRef]
- Kapalczynska, M.; Lin, M.; Maertzdorf, J.; Heuberger, J.; Muellerke, S.; Zuo, X.; Vidal, R.; Shureiqi, I.; Fischer, A.-S.; Sauer, S.; et al. BMP feed-forward loop promotes terminal differentiation in gastric glands and is interrupted by H. pylori-driven inflammation. Nat. Commun. 2022, 13, 1577. [Google Scholar] [CrossRef] [PubMed]
- Coffey, R.J.; Romano, M.; Goldenring, J. Roles for transforming growth factor-alpha in the stomach. J. Clin. Gastroenterol. 1995, 21 (Suppl. 1), S36–S39. [Google Scholar] [PubMed]
- Ogawa, K.; Saeki, N.; Igura, Y.; Hayashi, Y. Complementary expression and repulsive signaling suggest that EphB2 and ephrin-B1 are possibly involved in epithelial boundary formation at the squamocolumnar junction in the rodent stomach. Histochem. Cell Biol. 2013, 140, 659–675. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Aihara, E.; Kenny, S.; Yang, L.; Li, J.; Varro, A.; Montrose, M.H.; Shroyer, N.F.; Wang, T.C.; Shivdasani, R.A.; et al. Indian Hedgehog mediates gastrin-induced proliferation in stomach of adult mice. Gastroenterology 2014, 147, 655–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweiger, P.J.; Jensen, K.B. Modeling human disease using organotypic cultures. Curr. Opin. Cell Biol. 2016, 43, 22–29. [Google Scholar] [CrossRef]
- Eicher, A.K.; Berns, H.M.; Wells, J.M. Translating Developmental Principles to Generate Human Gastric Organoids. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Sáenz, J.B.; Mills, J.C. Biological techniques: Stomach growth in a dish. Nature 2017, 541, 160–161. [Google Scholar] [CrossRef]
- Schlaermann, P.; Toelle, B.; Berger, H.; Schmidt, S.C.; Glanemann, M.; Ordemann, J.; Bartfeld, S.; Mollenkopf, H.J.; Meyer, T.F. A novel human gastric primary cell culture system for modelling Helicobacter pylori infection in vitro. Gut 2016, 65, 202–213. [Google Scholar] [CrossRef] [Green Version]
- Fatehullah, A.; Terakado, Y.; Sagiraju, S.; Tan, T.L.; Sheng, T.; Tan, S.H.; Murakami, K.; Swathi, Y.; Ang, N.; Rajarethinam, R.; et al. A tumour-resident Lgr5+ stem-cell-like pool drives the establishment and progression of advanced gastric cancers. Nat. Cell Biol. 2021, 23, 1299–1313. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Del Arco, C.D.; Medina, L.O.; Munoz, L.E.; de las Heras, S.G.G.; Acenero, M.J.F. Is there still a place for conventional histopathology in the age of molecular medicine? Laurén classification, inflammatory infiltration and other current topics in gastric cancer diagnosis and prognosis. Histol. Histopathol. 2021, 36, 587–613. [Google Scholar]
- Correa, P.; Piazuelo, M.B. The gastric precancerous cascade. J. Dig. Dis. 2012, 13, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.G.; Wang, T.C. Inflammation, atrophy, and gastric cancer. J. Clin. Investig. 2007, 117, 60–69. [Google Scholar] [CrossRef]
- Carneiro, F. Hereditary gastric cancer. Pathologe 2012, 33 (Suppl. 2), 231–234. [Google Scholar] [CrossRef] [PubMed]
- Milne, A.N.; Carneiro, F.; O’Morain, C.; Offerhaus, G.J. Nature meets nurture: Molecular genetics of gastric cancer. Hum. Genet. 2009, 126, 615–628. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Kusters, J.; van Vliet, A.H.M.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar] [CrossRef] [Green Version]
- Amieva, M.R.; El-Omar, E.M. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology 2008, 134, 306–323. [Google Scholar] [CrossRef] [Green Version]
- Giroux, V.; Rustgi, A.R. Metaplasia: Tissue injury adaptation and a precursor to the dysplasia-cancer sequence. Nat. Rev. Cancer 2017, 17, 594–604. [Google Scholar] [CrossRef]
- Sigal, M.; Rothenberg, M.E.; Logan, C.Y.; Lee, J.Y.; Honaker, R.W.; Cooper, R.L.; Passarelli, B.; Camorlinga, M.; Bouley, D.M.; Alvarez, G.; et al. Helicobacter pylori Activates and Expands Lgr5+ Stem Cells Through Direct Colonization of the Gastric Glands. Gastroenterology 2015, 148, 1392–1404. [Google Scholar] [CrossRef]
- Amieva, M.; Peek, R.M., Jr. Pathobiology of Helicobacter pylori-induced Gastric Cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, W. Trefoil factor family (TFF) peptides and their different roles in the mucosal innate immune defense and more: An update. Curr. Med. Chem. 2021, 28, 7387–7399. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, J. Dual Roles of Gastric Gland Mucin-specific O-glycans in Prevention of Gastric Cancer. Acta Histochem. Cytochem. 2014, 47, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Wang, P.; Hoshino, H.; Ito, Y.; Kobayashi, M.; Nakayama, J.; Seeberger, P.H.; Fukuda, M. Alpha1,4GlcNAc-capped mucin-type O-glycan inhibits cholesterol alpha-glucosyltransferase from Helicobacter pylori and suppresses H. pylori growth. Glycobiology 2008, 18, 549–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karasawa, F.; Shiota, A.; Goso, Y.; Kobayashi, M.; Sato, Y.; Masumoto, J.; Fujiwara, M.; Yokosawa, S.; Muraki, T.; Miyagawa, S.; et al. Essential role of gastric gland mucin in preventing gastric cancer in mice. J. Clin. Investig. 2012, 122, 923–934. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, W. TFF2, a MUC6-binding lectin stabilizing the gastric mucus barrier and more. Int. J. Oncol. 2015, 47, 806–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heuer, F.; Stürmer, R.; Heuer, J.; Kalinski, T.; Lemke, A.; Meyer, F.; Hoffmann, W. Different Forms of TFF2, A Lectin of the Human Gastric Mucus Barrier: In Vitro Binding Studies. Int. J. Mol. Sci. 2019, 20, 5871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.G.; Rogers, A.B.; Whary, M.T.; Ge, Z.; Ohtani, M.; Jones, E.K.; Wang, T.C. Accelerated Progression of Gastritis to Dysplasia in the Pyloric Antrum of TFF2-/- C57BL6 x Sv129 Helicobacter pylori-Infected Mice. Am. J. Pathol. 2007, 171, 1520–1528. [Google Scholar] [CrossRef] [Green Version]
- Petersen, C.P.; Mills, J.C.; Goldenring, J.R. Murine models of gastric corpus preneoplasia. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, P.H.; Lee, J.R.; Joshi, V.; Playford, R.J.; Poulsom, R.; Wright, N.A.; Goldenring, J.R. Identification of a metaplastic cell lineage associated with human gastric adenocarcinoma. Lab. Investig. 1999, 79, 639–646. [Google Scholar]
- Jeong, H.; Lee, B.; Kim, K.H.; Cho, S.Y.; Cho, Y.; Park, J.; Lee, Y.; Oh, Y.; Hwang, B.R.; Jang, A.R.; et al. WFDC2 Promotes Spasmolytic Polypeptide-Expressing Metaplasia Through the Up-Regulation of IL33 in Response to Injury. Gastroenterology 2021, 161, 953–967. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Fox, J.G.; Wang, T.C. Isthmus Stem Cells Are the Origins of Metaplasia in the Gastric Corpus. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 89–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noto, C.N.; Hoft, S.G.; Bockerstett, K.A.; Jackson, N.M.; Ford, E.L.; Vest, L.S.; DiPaolo, R.J. IL13 Acts Directly on Gastric Epithelial Cells to Promote Metaplasia Development During Chronic Gastritis. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 623–642. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.T.; O’Neal, R.L.; Coffey, R.J.; Finke, P.E.; Barker, N.; Goldenring, J.R. Spasmolytic polypeptide-expressing metaplasia (SPEM) in the gastric oxyntic mucosa does not arise from Lgr5-expressing cells. Gut 2012, 61, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Willet, S.G.; Lewis, M.A.; Miao, Z.F.; Liu, D.; Radyk, M.D.; Cunningham, R.L.; Burclaff, J.; Sibbel, G.; Lo, H.G.; Blanc, V.; et al. Regenerative proliferation of differentiated cells by mTORC1-dependent paligenosis. EMBO J. 2018, 37, e98311. [Google Scholar] [CrossRef]
- Sáenz, J.B.; Vargas, N.; Cho, C.J.; Mills, J.C. Regulation of the double-stranded RNA response through ADAR1 licenses metaplastic reprogramming in gastric epithelium. JCI Insight 2022, 7, 153511. [Google Scholar] [CrossRef]
- Petersen, C.P.; Meyer, A.R.; de Salvo, C.; Choi, E.; Schlegel, C.; Petersen, A.; Engevik, A.C.; Prasad, N.; Levy, S.E.; Peebles, R.S.; et al. A signalling cascade of IL-33 to IL-13 regulates metaplasia in the mouse stomach. Gut 2018, 67, 805–817. [Google Scholar] [CrossRef]
- Nam, K.T.; Lee, H.J.; Mok, H.; Romero-Gallo, J.; Crowe, J.E., Jr.; Peek, R.M., Jr.; Goldenring, J.R. Amphiregulin-deficient mice develop spasmolytic polypeptide expressing metaplasia and intestinal metaplasia. Gastroenterology 2009, 136, 1288–1296. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.T.; Lee, H.J.; Sousa, J.F.; Weis, V.G.; O’Neal, R.L.; Finke, P.E.; Romero-Gallo, J.; Shi, G.; Mills, J.C.; Peek, R.M., Jr.; et al. Mature chief cells are cryptic progenitors for metaplasia in the stomach. Gastroenterology 2010, 139, 2028–2037. [Google Scholar] [CrossRef] [Green Version]
- Waghray, M.; Zavros, Y.; Saqui-Salces, M.; El-Zaatari, M.; Alamelumangapuram, C.B.; Todisco, A.; Eaton, K.A.; Merchant, J.L. Interleukin-1β promotes gastric atrophy through suppression of Sonic Hedgehog. Gastroenterology 2010, 138, 562–572. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Ogle, S.A.; Schumacher, M.A.; Orr-Asman, M.A.; Miller, M.L.; Lertkowit, N.; Varro, A.; Hollande, F.; Zavros, Y. Loss of parietal cell expression of Sonic hedgehog induces hypergastrinemia and hyperproliferation of surface mucous cells. Gastroenterology 2010, 138, 550–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karam, S.M.; Forte, J.G. Inhibiting gastric H+-K+-ATPase activity by omeprazole promotes degeneration and production of parietal cells. Am. J. Physiol. 1994, 266, G745–G758. [Google Scholar] [CrossRef] [PubMed]
- Brusselaers, N.; Wahlin, K.; Engstrand, L.; Lagergren, J. Maintenance therapy with proton pump inhibitors and risk of gastric cancer: A nationwide population-based cohort study in Sweden. BMJ Open 2017, 7, e017739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, W.; Takabayashi, H.; Yang, Y.; Mao, M.; Hibdon, E.S.; Samuelson, L.C.; Eaton, K.A.; Todisco, A. Regulation of gastric Lgr5+ve cell homeostasis by bone morphogenetic protein (BMP) signaling and inflammatory stimuli. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 523–538. [Google Scholar] [CrossRef] [Green Version]
- Waldum, H.; Mjones, P. Time to classify tumors of the stomach and the kidneys according to cell of origin. Int. J. Mol. Sci. 2021, 22, 13386. [Google Scholar] [CrossRef]
- Choi, W.-T.; Brown, I.; Ushiku, T.; Yozu, M.; Setia, N.; Srivastava, A.; Johcilla, M.; Pai, R.K.; Gill, R.M.; Fukayama, M.; et al. Gastric pyloric gland adenoma: A multicenter clinicopathological study of 67 cases. Histopathology 2018, 72, 1007–1014. [Google Scholar] [CrossRef]
- Takita, M.; Ohata, K.; Inamoto, R.; Kurebayashi, M.; Takayanagi, S.; Kimoto, Y.; Suzuki, Y.; Ishii, R.; Ono, K.; Negishi, R.; et al. Endoscopic and histological features of Helicobacter pylori-negative differentiated gastric adenocarcinoma arising in the antrum. JGH Open 2021, 5, 470–477. [Google Scholar] [CrossRef]
- McDonald, S.A.; Greaves, L.C.; Gutierrez-Gonzalez, L.; Rodriguez-Justo, M.; Deheragoda, M.; Leedham, S.J.; Taylor, R.W.; Lee, C.Y.; Preston, S.L.; Lovell, M.; et al. Mechanisms of field cancerization in the human stomach: The expansion and spread of mutated gastric stem cells. Gastroenterology 2008, 134, 500–510. [Google Scholar] [CrossRef]
- Gutierrez-Gonzalez, L.; Graham, T.A.; Rodriguez-Justo, M.; Leedham, S.J.; Novelli, M.R.; Gay, L.J.; Ventayol-Garcia, T.; Green, A.; Mitchell, I.; Stoker, D.L.; et al. The clonal origins of dysplasia from intestinal metaplasia in the human stomach. Gastroenterology 2011, 140, 1251–1260. [Google Scholar] [CrossRef] [Green Version]
- Rio, M.C.; Bellocq, J.P.; Daniel, J.Y.; Tomasetto, C.; Lathe, R.; Chenard, M.P.; Batzenschlager, A.; Chambon, P. Breast cancer-associated pS2 protein: Synthesis and secretion by normal stomach mucosa. Science 1988, 241, 705–708. [Google Scholar] [CrossRef]
- Ribieras, S.; Tomasetto, C.; Rio, M.C. The pS2/TFF1 trefoil factor, from basic research to clinical applications. Biochim. Biophys. Acta 1998, 1378, F61–F77. [Google Scholar] [CrossRef]
- Heuer, J.; Heuer, F.; Stürmer, R.; Harder, S.; Schlüter, H.; Braga Emidio, N.; Muttenthaler, M.; Jechorek, D.; Meyer, F.; Hoffmann, W. The Tumor Suppressor TFF1 Occurs in Different Forms and Interacts with Multiple Partners in the Human Gastric Mucus Barrier: Indications for Diverse Protective Functions. Int. J. Mol. Sci. 2020, 21, 2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Znalesniak, E.B.; Salm, F.; Hoffmann, W. Molecular Alterations in the Stomach of Tff1-Deficient Mice: Early Steps in Antral Carcinogenesis. Int. J. Mol. Sci. 2020, 21, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stürmer, R.; Reising, J.; Hoffmann, W. The TFF Peptides xP1 and xP4 Appear in Distinctive Forms in the Xenopus laevis Gastric Mucosa: Indications for Different Protective Functions. Int. J. Mol. Sci. 2019, 20, 6052. [Google Scholar] [CrossRef] [Green Version]
- Thim, L. Trefoil peptides: From structure to function. Cell. Mol. Life Sci. 1997, 53, 888–903. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, W. Trefoil Factor Family (TFF) Peptides. Encyclopedia 2021, 1, 974–987. [Google Scholar] [CrossRef]
- Fra, A.M.; Fagioli, C.; Finazzi, D.; Sitia, R.; Alberini, C.M. Quality control of ER synthesized proteins: An exposed thiol group as a three-way switch mediating assembly, retention and degradation. EMBO J. 1993, 12, 4755–4761. [Google Scholar] [CrossRef]
- Braga Emidio, N.; Baik, H.; Lee, D.; Stürmer, R.; Heuer, J.; Elliott, A.G.; Blaskovich, M.A.T.; Haupenthal, K.; Tegtmeyer, N.; Hoffmann, W.; et al. Chemical synthesis of human trefoil factor 1 (TFF1) and its homodimer provides novel insights into their mechanisms of action. Chem. Commun. 2020, 56, 6420–6423. [Google Scholar] [CrossRef]
- Clyne, M.; May, F.E.B. The Interaction of Helicobacter pylori with TFF1 and Its Role in Mediating the Tropism of the Bacteria Within the Stomach. Int. J. Mol. Sci. 2019, 20, 4400. [Google Scholar] [CrossRef] [Green Version]
- Bossenmeyer-Pourié, C.; Kannan, R.; Ribieras, S.; Wendling, C.; Stoll, I.; Thim, L.; Tomasetto, C.; Rio, M.C. The trefoil factor 1 participates in gastrointestinal cell differentiation by delaying G1-S phase transition and reducing apoptosis. J. Cell Biol. 2002, 157, 761–770. [Google Scholar] [CrossRef]
- Fu, T.; Kalbacher, H.; Hoffmann, W. TFF1 is differentially expressed in stationary and migratory rat gastric epithelial cells (RGM-1) after in vitro wounding: Influence of TFF1 RNA interference on cell migration. Cell. Physiol. Biochem. 2013, 32, 997–1010. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Klemke, R.L. Extracellular-regulated kinase activation and CAS/Crk coupling regulate cell migration and suppress apoptosis during invasion of the extracellular matrix. J. Cell Biol. 2000, 149, 223–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rio, M.C.; Chenard, M.P.; Wolf, C.; Marcellin, L.; Tomasetto, C.; Lathe, R.; Bellocq, J.P.; Chambon, P. Induction of pS2 and hSP genes as markers of mucosal ulceration of the digestive tract. Gastroenterology 1991, 100, 375–379. [Google Scholar] [CrossRef]
- Znalesniak, E.B.; Fu, T.; Guttek, K.; Händel, U.; Reinhold, D.; Hoffmann, W. Increased Cerebral Tff1 Expression in Two Murine Models of Neuroinflammation. Cell. Physiol. Biochem. 2016, 39, 2287–2296. [Google Scholar] [CrossRef] [PubMed]
- Hertel, S.C.; Chwieralski, C.E.; Hinz, M.; Rio, M.C.; Tomasetto, C.; Hoffmann, W. Profiling trefoil factor family (TFF) expression in the mouse: Identification of an antisense TFF1-related transcript in the kidney and liver. Peptides 2004, 25, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Karam, S.M.; Tomasetto, C.; Rio, M.C. Amplification and invasiveness of epithelial progenitors during gastric carcinogenesis in trefoil factor 1 knockout mice. Cell Prolif. 2008, 41, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Soutto, M.; Belkhiri, A.; Piazuelo, M.B.; Schneider, B.G.; Peng, D.; Jiang, A.; Washington, M.K.; Kokoye, Y.; Crowe, S.E.; Zaika, A.; et al. Loss of TFF1 is associated with activation of NF-κB-mediated inflammation and gastric neoplasia in mice and humans. J. Clin. Investig. 2011, 121, 1753–1767. [Google Scholar] [CrossRef] [PubMed]
- Soutto, M.; Saleh, M.; Arredouani, M.S.; Piazuelo, B.; Belkhiri, A.; El-Rifai, W. Loss of Tff1 Promotes Pro-Inflammatory Phenotype with Increase in the Levels of RORγt+ T Lymphocytes and Il-17 in Mouse Gastric Neoplasia. J. Cancer 2017, 8, 2424–2435. [Google Scholar] [CrossRef] [Green Version]
- Saukkonen, K.; Tomasetto, C.; Narko, K.; Rio, M.C.; Ristimäki, A. Cyclooxygenase-2 expression and effect of celecoxib in gastric adenomas of trefoil factor 1-deficient mice. Cancer Res. 2003, 63, 3032–3036. [Google Scholar]
- Thiel, A.; Narko, K.; Heinonen, M.; Hemmes, A.; Tomasetto, C.; Rio, M.C.; Haglund, C.; Mäkelä, T.P.; Ristimäki, A. Inhibition of cyclooxygenase-2 causes regression of gastric adenomas in trefoil factor 1 deficient mice. Int. J. Cancer 2012, 131, 1032–1041. [Google Scholar] [CrossRef]
- Soutto, M.; Peng, D.; Katsha, A.; Chen, Z.; Piazuelo, M.B.; Washington, M.K.; Belkhiri, A.; Correa, P.; El-Rifai, W. Activation of β-catenin signalling by TFF1 loss promotes cell proliferation and gastric tumorigenesis. Gut 2015, 64, 1028–1039. [Google Scholar] [CrossRef] [Green Version]
- Tomita, H.; Takaishi, S.; Menheniott, T.R.; Yang, X.; Shirata, W.; Jin, G.; Betz, K.S.; Kawakami, K.; Minamoto, T.; Tomasetto, C.; et al. Inhibition of Gastric Carcinogenesis by the Hormone Gastrin is Mediated by Suppression of TFF1 Epigenetic Silencing. Gastroenterology 2011, 140, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Thiem, S.; Eissmann, M.F.; Elzer, J.; Jonas, A.; Putoczki, T.L.; Poh, A.; Nguyen, P.; Preaudet, A.; Flanagan, D.; Vincan, E.; et al. Stomach-Specific Activation of Oncogenic KRAS and STAT3-Dependent Inflammation Cooperatively Promote Gastric Tumorigenesis in a Preclinical Model. Cancer Res. 2016, 76, 2277–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, H.; Hayakawa, Y.; Konishi, M.; Hata, M.; Tsuboi, M.; Hayata, Y.; Hikiba, Y.; Ihara, S.; Nakagawa, H.; Ikenoue, T.; et al. Three types of metaplasia model through Kras activation, Pten deletion, or Cdh1 deletion in the gastric epithelium. J. Pathol. 2019, 247, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Kirikoshi, H.; Katoh, M. Expression of TFF1, TFF2 and TFF3 in gastric cancer. Int. J. Oncol. 2002, 21, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Beckler, A.D.; Roche, J.K.; Harper, J.C.; Petroni, G.; Frierson, H.F., Jr.; Moskaluk, C.A.; El-Rifai, W.; Powell, S.M. Decreased abundance of trefoil factor 1 transcript in the majority of gastric carcinomas. Cancer 2003, 98, 2184–2191. [Google Scholar] [CrossRef]
- Park, W.S.; Oh, R.R.; Park, J.Y.; Lee, J.H.; Shin, M.S.; Kim, H.S.; Lee, H.K.; Kim, Y.S.; Kim, S.Y.; Lee, S.H.; et al. Somatic mutations of the trefoil factor family 1 gene in gastric cancer. Gastroenterology 2000, 119, 691–698. [Google Scholar] [CrossRef]
- Yio, X.; Diamond, M.; Zhang, J.Y.; Weinstein, H.; Wang, L.H.; Werther, L.; Itzkowitz, S. Trefoil factor family-1 mutations enhance gastric cancer cell invasion through distinct signaling pathways. Gastroenterology 2006, 130, 1696–1706. [Google Scholar] [CrossRef]
- Fujimoto, J.; Yasui., W.; Tahara, H.; Tahara, E.; Kudo, Y.; Yokozaki, H. DNA hypermethylation at the pS2 promoter region is associated with early stage of stomach carcinogenesis. Cancer Lett. 2000, 149, 125–134. [Google Scholar] [CrossRef]
- Carvalho, R.; Kayademir, T.; Soares, P.; Canedo, P.; Sousa, S.; Oliveira, C.; Leistenschneider, P.; Seruca, R.; Gött, P.; Blin, N.; et al. Loss of Heterozygosity and Promoter Methylation, but not Mutation, May Underlie Loss of TFF1 in Gastric Carcinoma. Lab. Investig. 2002, 82, 1319–1326. [Google Scholar] [CrossRef] [Green Version]
- Torres, L.F.; Karam, S.M.; Wendling, C.; Chenard, M.P.; Kershenobich, D.; Tomasetto, C.; Rio, M.C. Trefoil factor 1 (TFF1/pS2) deficiency activates the unfolded protein response. Mol. Med. 2002, 8, 273–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heazlewood, C.K.; Cook, M.C.; Eri, R.; Price, G.R.; Tauro, S.B.; Taupin, D.; Thornton, D.J.; Png, C.W.; Crockford, T.L.; Cornall, R.J.; et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 2008, 5, e54. [Google Scholar] [CrossRef] [Green Version]
- Muthupalani, S.; Ge, Z.; Joy, J.; Feng, Y.; Dobey, C.; Cho, H.Y.; Langenbach, R.; Wang, T.C.; Hagen, S.J.; Fox, J.G. Muc5ac null mice are predisposed to spontaneous gastric antro-pyloric hyperplasia and adenomas coupled with attenuated H. pylori-induced corpus mucous metaplasia. Lab. Investig. 2019, 99, 1887–1905. [Google Scholar] [CrossRef]
- Kouznetsova, I.; Laubinger, W.; Kalbacher, H.; Kalinski, T.; Meyer, F.; Roessner, F.; Hoffmann, W. Biosynthesis of gastrokine-2 in the human gastric mucosa: Restricted spatial expression along the antral gland axis and differential interactions with TFF1, TFF2, and mucins. Cell. Physiol. Biochem. 2007, 20, 899–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulsen, S.S.; Thulesen, J.; Hartmann, B.; Kissow, H.L.; Nexø, E.; Thim, L. Injected TFF1 and TFF3 bind to TFF2-immunoreactive cells in the gastrointestinal tract in rats. Regul. Pept. 2003, 115, 91–99. [Google Scholar] [CrossRef]
- Poulsen, S.S.; Thulesen, J.; Nexø, E.; Thim, L. Distribution and metabolism of intravenously administered trefoil factor 2/porcine spasmolytic polypeptide in the rat. Gut 1998, 43, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Kjellev, S.; Nexø, E.; Thim, L.; Poulsen, S.S. Systemically administered trefoil factors are secreted into the gastric lumen and increase the viscosity of gastric contents. Br. J. Pharmacol. 2006, 149, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Braga Emidio, N.; Hoffmann, W.; Brierley, S.M.; Muttenthaler, M. Trefoil Factor Family: Unresolved Questions and Clinical Perspectives. Trends Biochem. Sci. 2019, 44, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Kurt-Jones, E.A.; Cao, L.; Sandor, F.; Rogers, A.B.; Whary, M.T.; Nambiar, P.R.; Cerny, A.; Bowen, G.; Yan, J.; Takaishi, S.; et al. Trefoil family factor 2 is expressed in murine gastric and immune cells and controls both gastrointestinal inflammation and systemic immune responses. Infect. Immun. 2007, 75, 471–480. [Google Scholar] [CrossRef] [Green Version]
- McBerry, C.; Egan, C.E.; Rani, R.; Yang, Y.; Wu, D.; Boespflug, N.; Boon, L.; Butcher, B.; Mirpuri, J.; Hogan, S.P.; et al. Trefoil factor 2 negatively regulates type 1 immunity against Toxoplasma gondii. J. Immunol. 2012, 189, 3078–3084. [Google Scholar] [CrossRef] [Green Version]
- Omar, O.M.; Soutto, M.; Bhat, N.S.; Bhat, A.A.; Lu, H.; Chen, Z.; El-Rifai, W. TFF1 antagonizes TIMP-1 mediated proliferative functions in gastric cancer. Mol. Carcinog. 2018, 57, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Soutto, M.; Chen, Z.; Bhat, A.A.; Wang, L.; Zhu, S.; Gomaa, A.; Bates, A.; Bhat, N.S.; Peng, D.; Belkhiri, A.; et al. Activation of STAT3 signaling is mediated by TFF1 silencing in gastric neoplasia. Nat. Commun. 2019, 10, 3039. [Google Scholar] [CrossRef] [PubMed]
- Limaye, S.A.; Haddad, R.I.; Cilli, F.; Sonis, S.T.; Colevas, A.D.; Brennan, M.T.; Hu, K.S.; Murphy, B.A. Phase 1b, multicenter, single blinded, placebo-controlled, sequential dose escalation study to assess the safety and tolerability of topically applied AG013 in subjects with locally advanced head and neck cancer receiving induction chemotherapy. Cancer 2013, 119, 4268–4276. [Google Scholar] [CrossRef] [PubMed]
- Vilchez-Vargas, R.; Salm, F.; Znalesniak, E.B.; Haupenthal, K.; Schanze, D.; Zenker, M.; Link, A.; Hoffmann, W. Profiling of the Bacterial Microbiota along the Murine Alimentary Tract. Int. J. Mol. Sci. 2022, 23, 1783. [Google Scholar] [CrossRef]
- Idowu, S.; Bertrand, P.P.; Walduck, A.K. Homeostasis and cancer initiation: Organoids as models to study the initiation of gastric cancer. Int. J. Mol. Med. 2022, 23, 2790. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoffmann, W. Self-Renewal and Cancers of the Gastric Epithelium: An Update and the Role of the Lectin TFF1 as an Antral Tumor Suppressor. Int. J. Mol. Sci. 2022, 23, 5377. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105377
Hoffmann W. Self-Renewal and Cancers of the Gastric Epithelium: An Update and the Role of the Lectin TFF1 as an Antral Tumor Suppressor. International Journal of Molecular Sciences. 2022; 23(10):5377. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105377
Chicago/Turabian StyleHoffmann, Werner. 2022. "Self-Renewal and Cancers of the Gastric Epithelium: An Update and the Role of the Lectin TFF1 as an Antral Tumor Suppressor" International Journal of Molecular Sciences 23, no. 10: 5377. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105377