
Role of Mitochondrial Dynamics in Cocaine’s Neurotoxicity


Department of Forensic Medicine, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, 1-5-45, Yushima, Tokyo 113-8519, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(10), 5418; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105418
Submission received: 20 April 2022
/
Revised: 10 May 2022
/
Accepted: 10 May 2022
/
Published: 12 May 2022
(This article belongs to the Special Issue Mitochondrial Dysfunction in Neurodegenerative Diseases)
Abstract
:The dynamic balance of mitochondrial fission and fusion maintains mitochondrial homeostasis and optimal function. It is indispensable for cells such as neurons, which rely on the finely tuned mitochondria to carry out their normal physiological activities. The potent psychostimulant cocaine impairs mitochondria as one way it exerts its neurotoxicity, wherein the disturbances in mitochondrial dynamics have been suggested to play an essential role. In this review, we summarize the neurotoxicity of cocaine and the role of mitochondrial dynamics in cellular physiology. Subsequently, we introduce current findings that link disturbed neuronal mitochondrial dynamics with cocaine exposure. Finally, the possible role and potential therapeutic value of mitochondrial dynamics in cocaine neurotoxicity are discussed.
1. Introduction
Unity succeeds division, and division follows unity, such is the way with mitochondrial dynamics [1]. Though this famous double-membrane organelle is traditionally known for its bioenergetic function, mitochondria have profound influences on various physiological processes via morphological changes [2,3]. Mitochondrial dynamics participate in the regulation of mitochondrial quantity, quality, distribution, and metabolism [4]. When orchestrated in balance, this delicate mechanism maintains optimal mitochondrial output and cellular homeostasis, which allows the cell to resist intrinsic or extrinsic stress [5,6]. On the contrary, unbalanced mitochondrial dynamics leads to mitochondrial and cellular dysfunction, and is deeply involved in diverse pathological conditions [7]. It is particularly true for energy-intensive cells such as neurons, whose energy relies heavily on aerobic respiration [8]. Mitochondrial dynamics, by executing the bioenergetic function and participating in crucial cellular physiological processes, either independently or by interacting with other organelles, impact neurogenesis, neuronal plasticity, and functions including neurotransmission [9,10]. Disturbances in mitochondrial dynamics are a striking feature in the neurotoxicity of chemicals or drugs, including the potent psychostimulant cocaine [11]. Although indicating evidence suggested that altered neuronal mitochondrial dynamics may be closely associated with the appearance of clinical neurodegenerative symptoms [12], the causative role of defective mitochondrial dynamics in cocaine-induced neuronal pathogenesis remains to be illuminated. In this review article, we scrutinize the neurotoxicity of cocaine from the viewpoint of mitochondrial dynamics. We will describe the neurotoxicity of cocaine and the machinery of mitochondrial dynamics as well as its role in cellular physiology. We further introduce current findings that link disturbed mitochondrial dynamics with cocaine exposure on neurons. Finally, the possible role and potential therapeutic value of mitochondrial dynamics in the neurotoxicity of cocaine are discussed.
2. The Neurotoxicity of Cocaine
Cocaine (C17H21NO4) is a tropane alkaloid extracted from coca leaves. According to the latest report from the United Nations Office on Drugs and Crime [13], approximately 20,000,000 people used cocaine globally in 2019. This population represents a 22% increase in the last decade (Figure 1). The worldwide output of cocaine manufacture has also reached its highest level, with an especially sharp increase in Europe. Although the physiological effects of cocaine depend on the form, dose, administration route, and combination with other drugs [14], cocaine intake is associated with many mental or psychomotor disturbances [15,16] and detrimental brain consequences that can be life-threatening under some circumstances [17]. The detrimental effects of cocaine on the brain are comprehensively produced via multiple routes (Figure 2). Cocaine enters the central nervous system (CNS) either via direct crossing the BBB by passive diffusion and transport via proton-antiporter [18], or by destructing the blood–brain barrier (BBB) integrity through inducing acute hyperthermia and elevating the levels of brain serotonin [19,20]. The breakdown of the BBB leads to brain edema and neuronal damage in various brain regions [19], which contributes to the diverse brain consequences. The prolonged use of cocaine is also associated with accelerated brain aging, which may present as cognitive decline and brain atrophy [21,22]. A significant reduction in gray matter volume has been observed in chronic cocaine abusers [23]. By binding to monoamine transporters, cocaine blocks the reuptake of neurotransmitters (dopamine, serotonin, norepinephrine) and increases their release in synapses to carry out its strong stimulant effect [24,25]. The resultant constant and excessive synaptic concentration of dopamine leads to a dysregulated dopaminergic system and contributes to neurodegenerative processes [26]. Furthermore, the weak base property of cocaine enables its intracellular accumulation and direct effects on organelles such as mitochondria [27]. The neurotoxicity of cocaine results from the induction of subcellular stress in mitochondria and endoplasmic reticulum (ER), the following mitochondrial dysfunction, and the subsequent involvement of cell death pathways.
2.1. Overwhelmed Intracellular Oxidative Stress
Intracellular oxidative stress is a well-established feature of cocaine exposure. Mitochondria are not only the major organelle involved in the generation of reactive oxidative species (ROS) as a by-product of oxidative phosphorylation, but they are capable of scavenging ROS via their endogenous antioxidant enzymes including catalase, glutathione peroxide, and superoxide dismutase [5,28]. Overwhelming ROS generation that exceeds the endogenous antioxidant capacity contributes to oxidative stress and neuronal death [29]. Cocaine affects both the generation of stress and the antioxidant system. On one hand, during sustained excessive excitation of neurons, cocaine elevates ROS production within mitochondria by increasing calcium influx [30]. Its own degradation products, including norcocaine and norcocaine derivatives (nitroxide radical, N-hydroxy, nitrosonium, cocaine iminium, and formaldehyde), also have relatively higher oxidation potentials and result in stronger lipid peroxidation than cocaine induced itself [31]. Elevated oxidative stress has been well demonstrated in acute and chronic models of cocaine administration [32,33]. On the other hand, cocaine suppresses the expression and abolishes the activity of endogenous antioxidant enzymes and non-enzymatic antioxidants. The total antioxidant capacity was diminished as determined in blood samples of cocaine addicts [34]. Non-enzymatic antioxidants such as glutathione and vitamin E have also been found to be decreased after cocaine exposure [35,36]. The suppression of cocaine on antioxidant system has also been confirmed in in vivo and in vitro models involving single to chronic administration to withdrawal [35,37,38,39]. Aside from the direct effects, the aggregation of dopamine after cocaine exposure is considered another major route for intracellular oxidative stress formation. Dopamine results in the formation of ROS during its metabolism, either by monoamine oxidase-catalyzed intracellular oxidation or extracellular auto-oxidation [40]. Altogether, cocaine-induced oxidative stress leads to neuronal damage and is suggested to be connected to an increased risk of neurodegeneration. Aggregation of α-synuclein, an intrinsic disorder protein, results in various neurodegenerative changes related to oxidative stress and mitochondrial dysfunction; is the pathologic feature of neurodegenerative disorders known as synucleinopathies [41]. Increased expression of α-synuclein in the brain of cocaine abusers has been found and demonstrated to be a result of cocaine-induced dopamine accumulation and oxidative stress [42].
2.2. Mitochondrial Dysfunction
Mitochondrial dysfunction is considered to be a major event in cocaine neurotoxicity [43]. The canonical bioenergetic function of mitochondria relies on the steady operation of the electron transport chain (ETC) on the inner mitochondrial membrane. Electron transport during oxidative phosphorylation results in protons being pumped from the mitochondrial matrix to intermembrane spaces and the formation of an electrochemical gradient in the mitochondrial membrane [15]. Mitochondrial membrane potential is indispensable for bioenergetic functioning. Cocaine exposure directly impairs the enzyme activity of the ETC complexes via downregulating mitochondrial inner membrane and oxidative phosphorylation-related genes, as shown in different conditions including chronic administration and withdrawal [44,45]. Disruption of mitochondrial membrane potential and impaired respiration after cocaine administration has been illustrated in isolated mitochondria from rat brain, human and rat primary neurons [46,47,48].
Moreover, cocaine impacts the quality control mechanism of mitochondria. Mitochondrial biogenesis and mitophagy maintains the optimal quantity and quality of mitochondria by the coordination between the fresh formation of mitochondria and the degradation of unhealthy mitochondria via autophagosomes [49]. Under stress or injury conditions, both mitochondrial biogenesis and mitophagy are activated to preserve the normal mitochondrial function [50]. It is reasonable to be the same case with cocaine exposure. Brain region-specific increased expressions of mitochondrial biogenesis-related genes were found in a rat cocaine abuse model [51]. However, there is limited evidence to measure the exact performance of neuronal mitochondrial biogenesis after cocaine exposure. The machinery of mitochondrial biogenesis requires a well-organized collaboration between nuclear and mitochondrial genes [52], which can be difficult to accomplish under the damage of cocaine to mitochondrial genomes. Similarly, it remains unclear whether and how mitophagy participates against cocaine exposure on neurons. Supportive in vivo and in vitro findings in mouse microglial cells indicate an incomplete mitophagy in neurons under cocaine exposure with the deficient formation of autophagosomes, which results in the disordered mitochondrial quality control and leads to mitochondrial dysfunction [53].
Eventually, the overwhelmed oxidative stress and mitochondrial dysfunction lead to the activation of cell death pathways. By inducing oxidative stress and impairing oxidative phosphorylation, cocaine diminishes the mitochondrial membrane potential and causes the release of cytochrome c, resulting in neuronal apoptosis [35,46]. It has been found in neurons after cocaine exposure that acute or repeated administration triggers apoptosis via the upregulation of caspase-12 [54]. Cocaine-triggered apoptotic neuronal death has also been demonstrated in in vivo models in mouse, rat, and human [32,55,56]. Meanwhile, coordinating with mitochondria, ER stress induced by cocaine exposure via disrupting intracellular calcium homeostasis may also simultaneously induce apoptosis [57,58]. In addition, studies have suggested that autophagic cell death participates in the neurotoxicity of cocaine [59,60]. However, the role of cocaine-induced autophagy in neurotoxicity remains to be elucidated. Furthermore, mitochondria are also involved in other cell death pathways such as ferroptosis and necroptosis [61], yet whether these pathways play a role in cocaine-induced neurotoxicity needs further investigation.
As another key mechanism in mitochondrial quality control, mitochondrial dynamics have drawn increasing attention owing to the important and widely covered involvement in cellular physiology. With the progressing disclosure of the machinery of mitochondrial dynamics, growing evidence indicates the involvement and essential role of mitochondrial dynamics in the toxicity of cocaine, such as cardiotoxicity and neurotoxicity.
3. Machinery of Mitochondrial Dynamics
Mitochondria are bound by the outer and inner membranes. The inner membrane can be further divided into the inner boundary membrane, which runs parallel to the outer membrane and is distinguished by its intricate invaginated cristae where oxidative phosphorylation is conducted [62]. The changeable morphology of mitochondria, which varies from granules to unconnected or interconnected tubules, is governed by the continuous operating mechanism of fission and fusion, known as mitochondrial dynamics [8]. It is worth noting that the generalized concept of mitochondrial dynamics also encompasses mitochondrial trafficking, positioning, degradation, cristae biogenesis, and remodeling [8]. Herein, we will focus on the machinery of mitochondrial fission and fusion.
3.1. Mitochondrial Fission
Mitochondrial fission is an orchestration of multiple factors [63] (Figure 3). The major mediator is dynamin-related protein 1 (DRP1), a nucleotide guanosine triphosphate (GTP) hydrolyzing enzyme [64]. Most mitochondrial dynamics-related proteins, including DRP1 are encoded by nuclear genes [65]. At the beginning of fission, DRP1 is recruited from the cytosol to mitochondria by receptors in the outer membrane [66]. The cytosolic DRP1 exists in oligomeric states and organizes into higher-order spiral complexes after binding to its receptors [67]. Through its GTPase activity, the multimeric DRP1 winds around and constricts the mitochondria to scission [68].
The DRP1 receptors in the mitochondrial outer membrane consist mainly of mitochondrial fission factor (MFF) [69], mitochondrial protein of 51 kDa, (MiD51, also known as mitochondrial elongation factor 1) [70], mitochondrial protein of 49kDa (MiD49, also known as mitochondrial elongation factor 2) [71], and mitochondrial fission 1 protein (FIS1) [72]. It has been illustrated that these four receptors have distinct impacts on the fission process instead of functioning identically [73]. MFF, MiD51, and MiD49 are more prominent in the recruitment of DRP1 [74], and are largely associated with one another to form multimeric complexes [73]. On the other hand, FIS1 has been implied to interact with and suppress the fusion machinery, or mark fission sites, rather than collaborate with DRP1 in recruitment [75,76]. Although a MiDs level-dependent DRP1 regulation model has been proposed [77], the precise mechanism by which these four receptors coordinate in DRP1 recruitment still needs further clarification.
In order to modulate mitochondrial fission, DRP1 is regulated by multiple post-translational modifications to adapt its recruitment, polymerization, and activity to different cellular signals. These DRP1 modifications include phosphorylation or dephosphorylation, S-nitrosylation, SUMOylation [78], O-GlcNAcylation [79], and ubiquitination [80]. In particular, the different phosphorylation sites on DRP1, various kinases, and phosphates determine the activity of DRP1 based on cellular needs [81,82]. For instance, during processes such as entry to mitosis or tumor growth, the recruitment of DRP1 is strengthened by phosphorylation at Serine 616 [82,83]. Phosphorylation at Serine 637 suppresses DRP1 activity, whereas dephosphorylation at this site enhances it [84,85]. The ratio of phosphorylation at Serine 616 to that at Serine 637 has been suggested to reflect the exact activity of DRP1 under the added effects of diverse cues [86]. Apart from the classical machinery, complementary DRP1-independent mitochondrial fission mechanisms have been reported. In DRP1-deficient cells, FIS1 switches mitochondrial dynamics to favor fission by inducing the interaction between mitochondria and lysosomes or by inhibiting fusion mediators [75].
3.2. Mitochondrial Fusion
The machinery of mitochondrial fusion was well accepted over a decade ago. In the classical model, fusion starts with the tethering and fusing of the outer membranes from two apposing mitochondria by mitofusin1 and 2 (MFN1, 2) [87], and ends with the merging of the inner membranes from two sides mediated by optic atrophy 1 (OPA1) [88]. With progression in the understanding of proteins involved, the original impression of the fusion machinery has been challenged.
Similar to mitochondrial fission, three known major mediators of mitochondrial fusion belong to the dynamin superfamily with GTPase. The MFNs locate on the outer membrane and are associated with tethering and outer fusion [89,90]. To initiate mitochondrial fusion, two adjoining mitochondria tether together by binding the carboxyl-terminal of the opponent’s MFNs [87,91]. The mingled MFNs undergo conformational changes to create homogeneous or heterogeneous dimeric linkages that activate outer membrane fusion [92]. Although the two types of MFN have highly homologous structures [93], their functions are distinct [92]. MFN1 plays a central role in fusion machinery, while MFN2 seems to have more influence in other stages [94,95]. To adjust to a variety of cellular processes, the quantity and quality of MFNs are also modulated by post-translation modifications [96]. As of now, the mechanism for MFNs-mediated outer membrane fusion remains debated and awaits further elucidation.
OPA1, which is required for inner membrane fusion [97], anchors to the inner membrane and can be cleaved into a short form that aggregates in the mitochondrial matrix [98]. The exact mechanism by which the long and short forms of OPA1 mediate inner membrane fusion is controversial with differing interpretations. Some presume that the long form is adequate for promoting fusion while the short form may have other functions in inducing mitochondrial fission [99]. On the contrary, both isoforms have been indicated to possess independent fusion capacities [100]. Some argue that both forms of OPA1 are essential, and that an optimal balance is required for fusion depending on different conditions [101,102].
3.3. Mitochondrial Dynamics in Cellular Physiology
Although the relationship between mitochondrial fission and fusion literally appears to be biological opposite, it is more probable to be a mutual and cooperative partnership [103]. Certain mediators, such as DRP1 and MFNs, are even able to promote or compensate for a change in the other side to rebalance mitochondrial dynamics [104,105]. By working collectively, mitochondrial fission and fusion regulate bioenergetic metabolism [106], calcium homeostasis [107] and stress handling [108], and thereby contribute to cell proliferation [109], mitophagy, and cell death pathways [3,110].
Mitochondrial dynamics regulates bioenergetics of different paths. Fission is suggested to play an essential role in the assembly of ETC complexes [111]. Fusion mediators MFN2 and OPA1 also participate in ETC coupling, and OPA1 is further involved in oxidative phosphorylation by maintaining the cristae structure [112]. Consequently, disturbed mitochondrial dynamics affect the efficacy of mitochondrial respiration and the cellular energy supply [113]. In contrast, impaired mitochondrial respiration alters the dynamics and results in mitochondrial fragmentation [114].
Mitochondrial distribution is vital for the cellular energy supply because mitochondria should be trafficked and positioned within the cell so as to properly match the fluctuating energy demand in each region, especially in neurons with their extended axons and dendrites [115]. This is accomplished by the mitochondrial size regulation of dynamics, as well as the direct involvement of dynamics mediators in trafficking [116]. In neurons, mitochondrial distribution depends on the interactions between anchoring proteins in the mitochondrial outer membrane, the motor-adaptor proteins, and the motor proteins on microtubules, for example, the Miro–Milton-dynein interaction [117]. Owing to their tethering function, MFNs are essential effectors in interaction-mediated mitochondrial trafficking [118]. Mfn mutation-induced neurodegenerative disease has been attributed to the impairment in MFN2-related mitochondrial trafficking [119].
Furthermore, balanced mitochondrial dynamics are essential for the inheritance and integrity of mtDNA [120]. Despite most mitochondrial proteins being encoded by the nuclear genome, the mitochondrial DNA (mtDNA) genome encodes 13 proteins requisite for oxidative phosphorylation [8]. The mtDNAs are vulnerable and prone to mutations because of their relatively short half-life, sustained exposure to oxidative stress, and lack of protection or repair mechanisms [121,122]. Aggregating with aging, higher-level mtDNA mutations in the brain lead to impaired oxidative metabolism and mitochondrial dysfunction [123]. Mitochondrial fission enables segregation of mutant or damaged mtDNA for further degradation [124]. In addition, mtDNA determines the sites for mitochondrial fission so as to separate the replicated mitochondrial genome [125,126]. Fusion, on the other hand, provides toleration of mtDNA mutations by exchanging or diluting the detrimental effects among mitochondria [127]. Accordingly, disrupted mitochondrial fusion results in an elevated mutation rate and depletion of mtDNA [128]. In addition to mtDNA nucleoids, mitochondrial fusion allows the exchange of proteins, ETC complexes, and lipid membrane, so as to limit defective mitochondrial components to a tolerable level and maintain the homogenization of mitochondria [129,130]. This has been proposed as one of the foremost functions of mitochondrial fusion [131].
Moreover, mediators of mitochondrial dynamics are associated with mitophagy. When a mitochondrion is depolarized, PTEN-induced kinase 1 (PINK1) may induce the phosphorylation of MFN2 and the translocation of Parkin [96,132]. It has been suggested that, in the brain, MFN2 acts as a fusion component while a mitophagy effector or receptor of Parkin after phosphorylation induced by PINK1 [12]. The recruited Parkin ubiquitinates and induces the degeneration of MFN2 to inhibit further fusion of depolarized mitochondria and initiate mitophagy [133]. Meanwhile, DRP1-dependent fission separates the impaired mitochondria to allow further engulfment of autophagosomes to advance mitophagy [49].
Mitochondrial dynamics also participate in the regulation of apoptosis [134]. DRP1 has been proposed as a precursor of apoptosis, especially in neurodegenerative pathologies [124]. Overwhelming mitochondrial impairments, such as high-level DNA damage, outraged oxidative stress, or depolarization, trigger DRP1-mediated mitochondrial fragmentation and activate apoptosis [3]. Once apoptosis is initiated, FIS1 mediates the translocation of the cytosolic apoptosis regulators to the mitochondrial outer membrane [135]. The recruited apoptosis regulators colocalize with DRP1 or MFN2 and undergo oligomerization induced by DRP1 or MiD51 [136,137]. Simultaneously, OPA1 mediates the remodeling of mitochondrial cristae and the release of cytochrome c to the cytosol [138]. Collectively, these result in mitochondrial fragmentation and further apoptosis. Although fission is a basic event in apoptosis [135], the precise role of DRP1 in this intricate process remains to be elucidated. Apart from apoptosis, mitochondrial dynamics have been implied to be involved in other cell death pathways such as necroptosis and ferroptosis [61]. Altogether, the balance between mitochondrial fission and fusion cooperatively modulates diverse cellular physiological processes.
3.4. Mitochondrial Dynamics and Endoplasmic Reticulum
Mitochondria also interact with other organelles concomitantly instead of participating in cellular physiology independently. In particular, the crosstalk between mitochondria and ER has drawn increasing attention [139,140]. ER functions in protein folding and post-translational modifications, as well as in the maintenance of intracellular Ca2+ homeostasis [141]. The membranes of ER are capable of associating with the mitochondrial outer membrane by protein tethering [142] through what are known as mitochondria–ER contact (MERC) sites [143]. Approximately 5~20% of the mitochondrial outer membrane is connected to ER [144].
MERC is important for the exchange of content between the two organelles [145]. Intracellular calcium has a profound influence on a variety of cellular processes, and it is mainly centralized in mitochondria and ER. MERC provides a bidirectional lane for calcium influx, and enables mitochondria and ER to efficiently maintain cellular calcium homeostasis [146]. This calcium exchange is not only significant for mitochondrial bioenergetic function [147], but implicate the apoptotic pathway under pathological conditions [2]. The content exchange also includes lipids, which are associated with mitochondrial structural stability and functions [148]. Moreover, MERC may involve in protein import, mitochondrial distribution, and mitophagy [139]. Consequently, MERC can play a protective role under adverse conditions. When stress accumulates in ER, MERC sites are found to increase [149].
Intriguingly, many mitochondrial dynamics-related mediators are connected to MERC, in which MFN2 is suggested to play a key role. MFN2 is expressed in both the mitochondrial outer membrane and ER, and it constructs a bridge during their association by forming complexes with MFNs located on the mitochondrial outer membrane [150]. MFN2 is capable of promoting calcium uptake and further affect ER functions through MERC [151]. MFN2 ablation results in unstable MERC and deficits in mitochondrial calcium influx [95]. It should be noted that the topological determination of MFNs is still in progress, and the requisite role of MFNs in MERC has also been questioned [152,153]. Moreover, FIS1 has been implied to be involved in the regulation of this association [154].
Moreover, MERC participates directly in mitochondrial dynamics. This structure tethering seems to determine the position of mitochondrial fission. In a majority of the fission initiated stages, ER tubules have been found to wrap around the mitochondrion and constrict to reduce the diameter of the mitochondrion before the recruitment of DRP1 [155], after which fission mediators, including DRP1, MFF, MiD51, and MiD49, may colocalize at these sites with other effectors so as to advance the division process [156,157]. Meanwhile, MERC facilitates the mtDNA distribution during mitochondrial fission by coupling the replicated mtDNA nucleoids [126]. Disturbances in ER function are commonly observed in neurodegenerative diseases [158]. The possible implication of MERC in the pathogenesis of these neurodegenerative disorders has been proposed, in which calcium communication and oxidative stress are considered to play key roles [159,160]. The close interaction and coordination between mitochondria and ER in neurons should provide another appealing topic for further research concerning the neurodegenerative diseases and neurotoxicity of drugs.
To summarize, fine-tuned mitochondrial dynamics are closely connected with the appropriate operation of mitochondria and cellular fitness. Neurons rely heavily on mitochondrial oxidative phosphorylation for energy [53]. They have unique morphologies with extensive differentially extended arborizations and constantly remodeling synapses that demand sufficient energy supply and delicate calcium regulation [54,55]. All these characteristics make orchestrated mitochondrial dynamics pivotal for the normal functions of neurons. The balance of mitochondrial dynamics as a compensating mechanism is conversely governed by a variety of physiological signals and is sensitive to pathological or detrimental stimuli [161,162]. Disruption of this equilibrium leads to accumulated impaired mitochondrial fragments, and it is related to neurodegenerative pathologies [49,117]. Therefore, it is fascinating to explore the role of mitochondrial dynamics in the neurotoxicity of cocaine.
4. Mitochondrial Dynamics and the Neurotoxicity of Cocaine
Mitochondrial fragmentation resulting from disturbances of mitochondrial dynamics including enhanced DRP1-dependent mitochondrial fission and weakened mitochondrial fusion is a well-established feature that leads to mitochondrial dysfunction and neurodegeneration in common neurodegenerative diseases [163,164]. Indicative evidence has also been found in our former study on the cardiotoxicity of cocaine [165]. Before the occurrence of any structural changes in the myocardium, cocaine exposure to cardiomyocytes altered the mitochondrial dynamics with a similarly increased fission and decreased fusion as is found in neurodegenerative diseases. However, at this time, studies concentrated on the involvement of mitochondrial dynamics in the neurotoxicity of cocaine can be counted on the fingers of one hand. Sadakierska-Chudy et al. established a cocaine self-administration model (average of 100 mg/kg of cocaine intake within 3 days) in rats following short-term abstinence (3 days) to study the potential role of mitochondrial dynamics in the reward circuit and drug-seeking behaviors [166]. Using microarray and quantitative real-time PCR assays, they determined a significantly increased copy number of mtDNA and elevated expressions of nuclear-encoded mitochondrial genes associated with oxidative phosphorylation and mitochondrial dynamics. Increased fission was identified in the hippocampus whereas increased fusion was found in the prefrontal cortex (e.g., Mfn1, Opa1). Correspondingly, upregulated ER stress-induced genes were also revealed. Although the authors considered the increased mtDNA copy number as a compensative role against intracellular stress, it should be noted that the meaning of these findings can be indefinite, since neither change in relative protein levels nor the mitochondrial function were determined. Still, this study suggested the involvement of mitochondrial dynamics in early stage of the neurotoxicity of cocaine and provided indicative findings on possible alterations in the mitochondrial dynamics of different brain regions after short-term self-administered cocaine exposure.
In another study, Chandra R et al. associated the transcription factor early growth response 3 (Egr3) in nucleus accumbens (NAc) with cocaine reward and locomotor responses by applying an in vivo cocaine abuse model (20 mg/kg/day, intraperitoneal administration for 7 days) [167]. Erg3 is induced by acute cocaine exposure and has been found pivotal in cocaine-related signaling pathways. They found the overexpression of Egr3 in D1 type medium spiny neurons elevates the cocaine reward and locomotor responses while its overexpression in D2 type medium spiny neurons inhibits these responses. In the following research, they demonstrated the requisite role of Erg3 in facilitating mitochondrial fission in NAc after cocaine exposure utilizing intraperitoneal administration (20 mg/kg/day for 7 days), self-administration (1 mg/kg/infusion for 10 days) models, and postmortem brain samples of cocaine addicts. They found the binding of Erg3 to Drp1 was increased in NAc during cocaine exposure, whereas the Erg3 knockdown attenuated cocaine-induced mitochondrial fission [168]. These studies throw light on the potential involvement of mitochondrial dynamics in the behavioral modulation of cocaine abuse.
Subsequently, Chandra R et al. focused on the expressions of dynamics-related mediators in the NAc in mice and rats after cocaine exposure, and in the brains of cocaine addicts [169]. They found increased levels of NAc Drp1 mRNA in mice after 7 days of cocaine administration (intraperitoneal injection, 20 mg/kg) as well as in self-administered cocaine rats (1 mg/kg/infusion for 10 days). A similar increased expression of NAc Drp1 mRNA was also observed in the brains of cocaine addicts. Similarly, Drp1 has also been shown to be differentially regulated by cocaine in D1 type and D2 type medium spiny neurons, where Drp1 expression is upregulated in D1 type yet downregulated in D2 type medium spiny neurons upon cocaine exposure. Further, they found increased phosphorylation of DRP1 at serine 616 and a corresponding decrease in mitochondrial size, specifically in dopamine receptor 1-containing medium spiny neurons after cocaine self-administration. Moreover, by applying DRP1 inhibitor Mdivi-1 and Drp1 knockdown, they linked DRP1-dependent mitochondrial fission in dopamine receptor 1-containing medium spiny neurons to the development of cocaine-addictive behavior. Interestingly, there seems to be a threshold for DRP1-dependent mitochondrial fission in dopamine receptor 1-containing medium spiny neurons during cocaine administration. The enhancement of cocaine-seeking induced by promoted DRP1-dependent mitochondrial fission appeared to hit a limit at the end of the self-administration session, while the abstinent period restored the enhancement of DRP1-dependent mitochondrial fission on cocaine-seeking. This could reflect that the elevated mitochondrial fission during cocaine exposure has fallen back to the basal level. Due to its indispensable role in mitochondrial fission, DRP1 is important in neuronal maturation, synaptic neurotransmission, and neuronal plasticity [10,170]. Posttranslational modifications in DRP1 are related to neuronal injury under stress conditions [171]. Activated DRP1 causes an elevation in mitochondrial fission in affected neurons, leading not only to mitochondrial fragmentation, but also to impaired mitochondrial axonal distribution and synaptic degeneration resulting in neuronal damage [171,172]. This study provided solid evidence about the behavioral function of DRP1-dependent mitochondrial fission and its therapeutic value in cocaine abuse.
In another study, Funakoshi T. et, al. provided supporting evidence for the involvement of altered mitochondrial dynamics in the neurotoxicity of cocaine. They examined the effect of cocaine on mitochondrial dynamics in an in vitro model using Neuro2a neuroblastoma cells (600 mM, 2~3 times per week, 3 weeks) [11]. They found increased phosphorylation of DRP1 at serine 616 and the expression of Fis1 mRNA with a consequential decrease in mitochondrial size with the help of transmission electron microscopy. In addition, they also reported a decrease in mitochondrial membrane potential and activated autophagy as well as an elevation of Parkin after chronic cocaine exposure. Further clarification of the relationship between increased mitochondrial fission and mitochondrial dysfunction, as well as the potential involvement of autophagy after neuronal cocaine exposure should be worth pursuing. The cocaine’s effects on neuronal mitochondrial dynamics introduced in this section have been summarized in Table 1.
It has been suggested that mitochondrial fission might play a protective role against the detrimental effects of cocaine. Specific alterations of mitochondrial dynamics appear to be dependent on different brain regions or cell types, which indicates a potential role of mitochondrial dynamics in addiction and to a wider stage.
5. Conclusions
Insights into the causality and role of mitochondrial dynamics in neuronal degeneration should be beneficial for understanding pathogenesis and developing therapeutic strategies. Considering the implied role of DRP1-mediated fission and mitochondrial fragmentation in cocaine addiction [166], it would be enchanting to determine the participation of other key mitochondrial dynamics-related mediators and the potential regulating mechanisms in the neurotoxicity of cocaine under different experimental conditions.
Optimal mitochondrial morphologies and ultrastructure are correlated with multiple conditions including bioenergetic demands, cellular milieu, or occupied cell types [49,173]. Correspondingly, the profiles of balanced mitochondrial dynamics and mitochondrial morphology can differ greatly among brain regions [174]. Thus, the diagnosis of impaired dynamics should be based on comprehensive determinations including brain regions, dynamics profiles (including in-depth indexes such as the ratio of differently phosphorylated DRP1, or the ratio of short-form to total OPA1) as well as measurements of mitochondrial size. Deficits in mitochondrial dynamics appear to be systematic, as found in peripheral blood lymphocytes in neurodegenerative disorder patients [175,176]. It is reasonable to presume a similar consequence in cocaine addicts. Furthermore, since Midiv-1 has proved effective in attenuating cocaine addiction and its neurotoxicity in an in vivo model [169], it would be compelling to learn whether other agonists and antagonists of mitochondrial dynamics might have similar therapeutic effects in cocaine abuse models. However, it should be noted that both fission and fusion are essential elements for the normal functioning of neuronal mitochondria [124], so that a complete inhibition of either might have unexpected detrimental effects. As a result, no matter whether using drugs or gene strategies, therapeutic attempts should determine a suitable dose and target to achieve minute adjustments in mitochondrial dynamics and restore the balance between fission and fusion. In this review article, we have limited our focus to the involvement of mitochondrial dynamics in common cellular physiological processes and cocaine toxicity. It should be noted that mutations in dynamics-related genes also contribute to impaired neuronal development and neurodegenerative diseases, such as autosomal dominant optic atrophy and Charcot-Marie-Tooth neuropathy [177].
In conclusion, we introduced the neurotoxicity of cocaine and the machinery of mitochondrial dynamics. We further presented current findings that associate mitochondrial dynamics with neuronal exposure to cocaine. Balanced mitochondrial dynamics is indispensable for neuronal health. With the disclosure of cell signaling affecting dynamics, efforts to obtain a deeper understanding of mitochondrial dynamics are progressing. We hope this article provides beneficial information and outlooks for further studies on the role of mitochondrial dynamics in the neurotoxicity of cocaine.
Author Contributions
S.W. and T.A.; writing—original draft preparation, S.W. and T.A.; writing—review and editing, T.F., K.U. (Kana Unuma) and K.U. (Koichi Uemura). All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MEXT KAKENHI, grant number 20H03955 (to T.A.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
S.W. receives a scholarship funded by the Cooperation Program between Tokyo Medical and Dental University (TMDU), Sony Corporation, and Sony Group Corporation.
Conflicts of Interest
S.W. receives a scholarship funded by the Cooperation Program between Tokyo Medical and Dental University (TMDU), Sony Corporation, and Sony Group Corporation. The funders had no role in the design of the study, in the collection, analyses, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
References
- Lewis, M.R.; Lewis, W.H. Mitochondria in Tissue Culture. Science 1914, 39, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Simoni, A.M.; Chami, M.; Wieckowski, M.; Youle, R.J.; Rizzuto, R. Drp-1-Dependent Division of the Mitochondrial Network Blocks Intraorganellar Ca2+ Waves and Protects against Ca2+-Mediated Apoptosis. Mol. Cell 2004, 16, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Gaume, B.; Bergmann-Leitner, E.S.; Leitner, W.W.; Robert, E.G.; Catez, F.; Smith, C.L.; Youle, R.J. The Role of Dynamin-Related Protein 1, a Mediator of Mitochondrial Fission, in Apoptosis. Dev. Cell 2001, 1, 515–525. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W., 2nd; Kitsis, R.N. The Mitochondrial Dynamism-Mitophagy-Cell Death Interactome: Multiple Roles Performed by Members of a Mitochondrial Molecular Ensemble. Circ. Res. 2015, 116, 167–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More Than Just a Powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2014, 11, 11–24. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C. Mitochondrial Dynamics and Its Involvement in Disease. Annu. Rev. Pathol. 2020, 15, 235–259. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Okamoto, K.-I.; Hayashi, Y.; Sheng, M. The Importance of Dendritic Mitochondria in the Morphogenesis and Plasticity of Spines and Synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Verstreken, P.; Ly, C.V.; Venken, K.; Koh, T.-W.; Zhou, Y.; Bellen, H.J. Synaptic Mitochondria Are Critical for Mobilization of Reserve Pool Vesicles at Drosophila Neuromuscular Junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Funakoshi, T.; Furukawa, M.; Aki, T.; Uemura, K. Repeated exposure of cocaine alters mitochondrial dynamics in mouse neuroblastoma Neuro2a. NeuroToxicology 2019, 75, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Huang, C.-H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, J.W.; Youle, R.J. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- United Nations Office on Drugs and Crime. World Drug Report 2021; United Nations Office on Drugs and Crime: Vienna, Austria, 2022. [Google Scholar]
- Thornton, C.; Grad, E.; Yaka, R. The role of mitochondria in cocaine addiction. Biochem. J. 2021, 478, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Post, R.M.; Kopanda, R.T. Cocaine, Kindling, and Psychosis. Am. J. Psychiatry 1976, 133, 627–634. [Google Scholar] [PubMed]
- Dandekar, M.P.; Singru, P.S.; Kokare, D.M.; Subhedar, N.K. Cocaine- and Amphetamine-Regulated Transcript Peptide Plays a Role in the Manifestation of Depression: Social Isolation and Olfactory Bulbectomy Models Reveal Unifying Principles. Neuropsychopharmacology 2009, 34, 1288–1300. [Google Scholar] [CrossRef]
- Levine, S.R.; Welch, K.M. Cocaine and Stroke. Stroke 1988, 19, 779–783. [Google Scholar] [CrossRef] [Green Version]
- Chapy, H.; Smirnova, M.; André, P.; Schlatter, J.; Chiadmi, F.; Couraud, P.-O.; Scherrmann, J.-M.; Decleves, X.; Cisternino, S. Carrier-Mediated Cocaine Transport at the Blood-Brain Barrier as a Putative Mechanism in Addiction Liability. Int. J. Neuropsychopharmacol. 2014, 18, pyu001. [Google Scholar] [CrossRef] [Green Version]
- Sharma, H.S.; Muresanu, D.; Sharma, A.; Patnaik, R. Cocaine-Induced Breakdown of the Blood–Brain Barrier and Neurotoxicity. Int. Rev. Neurobiol. 2009, 88, 297–334. [Google Scholar] [CrossRef]
- Kousik, S.M.; Napier, T.C.; Carvey, P.M. The Effects of Psychostimulant Drugs on Blood Brain Barrier Function and Neuroinflammation. Front. Pharmacol. 2012, 3, 121. [Google Scholar] [CrossRef] [Green Version]
- Verdejo-Garcia, A.; Perez-Garcia, M. Profile of executive deficits in cocaine and heroin polysubstance users: Common and differential effects on separate executive components. Psychopharmacology 2006, 190, 517–530. [Google Scholar] [CrossRef]
- Siniscalchi, A.; Bonci, A.; Mercuri, N.B.; De Siena, A.; De Sarro, G.; Malferrari, G.; Diana, M.; Gallelli, L. Cocaine Dependence and Stroke: Pathogenesis and Management. Curr. Neurovasc. Res. 2015, 12, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Ersche, K.D.; Jones, P.S.; Williams, G.B.; Robbins, T.W.; Bullmore, E.T. Cocaine Dependence: A Fast-Track for Brain Ageing? Mol. Psychiatry 2013, 18, 134–135. [Google Scholar] [CrossRef] [PubMed]
- Howell, L.L.; Kimmel, H.L. Monoamine transporters and psychostimulant addiction. Biochem. Pharmacol. 2008, 75, 196–217. [Google Scholar] [CrossRef] [PubMed]
- White, S.M.; Lambe, C.J. The pathophysiology of cocaine abuse. J. Clin. Forensic Med. 2003, 10, 27–39. [Google Scholar] [CrossRef]
- Ravina, B.; Marek, K.; Eberly, S.; Oakes, D.; Kurlan, R.; Ascherio, A.; Beal, F.; Beck, J.; Flagg, E.; Galpern, W.R.; et al. Dopamine Transporter Imaging Is Associated with Long-Term Outcomes in Parkinson’s Disease. Mov. Disord. 2012, 27, 1392–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulzer, D.; Rayport, S. Amphetamine and other psychostimulants reduce pH gradients in midbrain dopaminergic neurons and chromaffin granules: A mechanism of action. Neuron 1990, 5, 797–808. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chong, Z.Z.; Li, F.; Maiese, K. Oxidative stress in the brain: Novel cellular targets that govern survival during neurodegenerative disease. Prog. Neurobiol. 2005, 75, 207–246. [Google Scholar] [CrossRef]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflug. Arch. 2010, 460, 525–542. [Google Scholar] [CrossRef]
- Kovacic, P. Role of oxidative metabolites of cocaine in toxicity and addiction: Oxidative stress and electron transfer. Med. Hypotheses 2005, 64, 350–356. [Google Scholar] [CrossRef]
- Dietrich, J.-B.; Mangeol, A.; Revel, M.-O.; Burgun, C.; Aunis, D.; Zwiller, J. Acute or repeated cocaine administration generates reactive oxygen species and induces antioxidant enzyme activity in dopaminergic rat brain structures. Neuropharmacology 2005, 48, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Beiser, T.; Numa, R.; Kohen, R.; Yaka, R. Chronic treatment with Tempol during acquisition or withdrawal from CPP abolishes the expression of cocaine reward and diminishes oxidative damage. Sci. Rep. 2017, 7, 11162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.; Winhusen, T.; Storkson, J.M.; Lewis, D.; Pariza, M.W.; Somoza, E.; Somoza, V. Total antioxidant capacity is significantly lower in cocaine-dependent and methamphetamine-dependent patients relative to normal controls: Results from a preliminary study. Hum. Psychopharmacol. Clin. Exp. 2014, 29, 537–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, H.F.; Abdullah, L.; Mullan, M.A.; Mullan, M.; Crawford, F.C. Cocaine-induced oxidative stress precedes cell death in human neuronal progenitor cells. Neurochem. Int. 2007, 50, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Lipton, J.W.; Gyawali, S.; Borys, E.D.; Koprich, J.B.; Ptaszny, M.; O McGuire, S. Prenatal cocaine administration increases glutathione and alpha-tocopherol oxidation in fetal rat brain. Dev. Brain Res. 2003, 147, 77–84. [Google Scholar] [CrossRef]
- Hahn, J.; Hopf, F.W.; Bonci, A. Chronic Cocaine Enhances Corticotropin-Releasing Factor-Dependent Potentiation of Excitatory Transmission in Ventral Tegmental Area Dopamine Neurons. J. Neurosci. 2009, 29, 6535–6544. [Google Scholar] [CrossRef]
- Muriach, M.; Lopez-Pedrajas, R.; Barcia, J.M.; Sanchez-Villarejo, M.V.; Almansa, I.; Romero, F.J. Cocaine Causes Memory and Learning Impairments in Rats: Involvement of Nuclear Factor Kappa B and Oxidative Stress, and Prevention by Topiramate. J. Neurochem. 2010, 114, 675–684. [Google Scholar] [CrossRef]
- Sordi, A.O.; Pechansky, F.; Kessler, F.; Kapczinski, F.; Pfaffenseller, B.; Gubert, C.; de Aguiar, B.W.; Narvaez, J.C.D.M.; Ornell, F.; von Diemen, L. Oxidative stress and BDNF as possible markers for the severity of crack cocaine use in early withdrawal. Psychopharmacology 2014, 231, 4031–4039. [Google Scholar] [CrossRef]
- Banerjee, K.; Munshi, S.; Sen, O.; Pramanik, V.; Roy Mukherjee, T.; Chakrabarti, S. Dopamine Cytotoxicity Involves Both Oxidative and Nonoxidative Pathways in Sh-Sy5y Cells: Potential Role of Alpha-Synuclein Overexpression and Proteasomal Inhibition in the Etiopathogenesis of Parkinson’s Disease. Parkinsons Dis. 2014, 2014, 878935. [Google Scholar] [CrossRef] [Green Version]
- Marvian, A.T.; Koss, D.J.; Aliakbari, F.; Morshedi, D.; Outeiro, T.F. In Vitro Models of Synucleinopathies: Informing on Molecular Mechanisms and Protective Strategies. J. Neurochem. 2019, 150, 535–565. [Google Scholar] [CrossRef] [Green Version]
- Mash, D.C.; Ouyang, Q.; Pablo, J.; Basile, M.; Izenwasser, S.; Lieberman, A.; Perrin, R.J. Cocaine Abusers Have an Overexpression of Alpha-Synuclein in Dopamine Neurons. J. Neurosci. 2003, 23, 2564–2571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, M.R.; Jardim, F.R. Cocaine and Mitochondria-Related Signaling in the Brain: A Mechanistic View and Future Directions. Neurochem. Int. 2016, 92, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Yuan, Q.; Mash, D.C.; Goldman, D. Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proc. Natl. Acad. Sci. USA 2011, 108, 6626–6631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacherjee, A.; Djekidel, M.N.; Chen, R.; Chen, W.; Tuesta, L.M.; Zhang, Y. Cell type-specific transcriptional programs in mouse prefrontal cortex during adolescence and addiction. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Cunha-Oliveira, T.; Rego, A.C.; Cardoso, S.M.; Borges, F.; Swerdlow, R.H.; Macedo, T.; de Oliveira, C.R. Mitochondrial dysfunction and caspase activation in rat cortical neurons treated with cocaine or amphetamine. Brain Res. 2006, 1089, 44–54. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Silva, L.; Silva, A.M.; Moreno, A.J.; Oliveira, C.R.; Santos, M.S. Mitochondrial complex I dysfunction induced by cocaine and cocaine plus morphine in brain and liver mitochondria. Toxicol. Lett. 2013, 219, 298–306. [Google Scholar] [CrossRef] [Green Version]
- De Simone, F.I.; Darbinian, N.; Amini, S.; Muniswamy, M.; White, M.K.; Elrod, J.; Datta, P.K.; Langford, D.; Khalili, K. HIV-1 Tat and Cocaine Impair Survival of Cultured Primary Neuronal Cells via a Mitochondrial Pathway. J. Neuroimmune Pharmacol. 2016, 11, 358–368. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Quiles, J.M.; Gustafsson, B. Mitochondrial Quality Control and Cellular Proteostasis: Two Sides of the Same Coin. Front. Physiol. 2020, 11, 515. [Google Scholar] [CrossRef]
- Pati, S.; Angel, P.; Drake, R.R.; Wagner, J.J.; Cummings, B.S. Lipidomic Changes in the Rat Hippocampus Following Cocaine Conditioning, Extinction, and Reinstatement of Drug-Seeking. Brain Behav. 2019, 9, e01451. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W., 2nd; Vega, R.B.; Kelly, D.P. Mitochondrial Biogenesis and Dynamics in the Developing and Diseased Heart. Genes Dev. 2015, 29, 1981–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thangaraj, A.; Periyasamy, P.; Guo, M.-L.; Chivero, E.T.; Callen, S.; Buch, S. Mitigation of cocaine-mediated mitochondrial damage, defective mitophagy and microglial activation by superoxide dismutase mimetics. Autophagy 2020, 16, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.M.; Kim, S.W.; Choe, E.S. Cocaine increases immunoglobulin heavy chain binding protein and caspase-12 expression in the rat dorsal striatum. Psychopharmacology 2007, 195, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Álvaro-Bartolomé, M.; La Harpe, R.; Callado, L.; Meana, J.; García-Sevilla, J. Molecular adaptations of apoptotic pathways and signaling partners in the cerebral cortex of human cocaine addicts and cocaine-treated rats. Neuroscience 2011, 196, 1–15. [Google Scholar] [CrossRef]
- Mai, H.N.; Sharma, N.; Jeong, J.H.; Shin, E.J.; Pham, D.T.; Trinh, Q.D.; Lee, Y.J.; Jang, C.G.; Nah, S.Y.; Bing, G.; et al. P53 Knockout Mice Are Protected from Cocaine-Induced Kindling Behaviors Via Inhibiting Mitochondrial Oxidative Burdens, Mitochondrial Dysfunction, and Proapoptotic Changes. Neurochem. Int. 2019, 124, 68–81. [Google Scholar] [CrossRef]
- Pereira, D.M.; Valentao, P.; Correia-da-Silva, G.; Teixeira, N.; Andrade, P.B. Translating Endoplasmic Reticulum Biology into the Clinic: A Role for Er-Targeted Natural Products? Nat. Prod. Rep. 2015, 32, 705–722. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Guha, P.; Harraz, M.M.; Snyder, S.H. Cocaine elicits autophagic cytotoxicity via a nitric oxide-GAPDH signaling cascade. Proc. Natl. Acad. Sci. USA 2016, 113, 1417–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Xiao, W.; Deng, S.; Cheng, X.; Zheng, H.; Chen, J.; Wang, F. Activation of AMPK-dependent autophagy in the nucleus accumbens opposes cocaine-induced behaviors of mice. Addict. Biol. 2020, 25, e12736. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Kraus, F.; Ryan, M.T. The constriction and scission machineries involved in mitochondrial fission. J. Cell Sci. 2017, 130, 2953–2960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnova, E.; Griparic, L.; Shurland, D.-L.; van der Bliek, A.M. Dynamin-related Protein Drp1 Is Required for Mitochondrial Division in Mammalian Cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koopman, W.J.; Willems, P.H.; Smeitink, J.A. Monogenic Mitochondrial Disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Macdonald, P.J.; Stepanyants, N.; Mehrotra, N.; Mears, J.A.; Qi, X.; Sesaki, H.; Ramachandran, R. A dimeric equilibrium intermediate nucleates Drp1 reassembly on mitochondrial membranes for fission. Mol. Biol. Cell 2014, 25, 1905–1915. [Google Scholar] [CrossRef]
- Kalia, R.; Wang, R.Y.-R.; Yusuf, A.; Thomas, P.V.; Agard, D.A.; Shaw, J.M.; Frost, A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018, 558, 401–405. [Google Scholar] [CrossRef]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Liu, T.; Jin, S.; Wang, X.; Qu, M.; Uhlén, P.; Tomilin, N.; Shupliakov, O.; Lendahl, U.; Nistér, M. Human MIEF1 recruits Drp1 to mitochondrial outer membranes and promotes mitochondrial fusion rather than fission. EMBO J. 2011, 30, 2762–2778. [Google Scholar] [CrossRef]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- James, D.I.; Parone, P.A.; Mattenberger, Y.; Martinou, J.-C. hFis1, a Novel Component of the Mammalian Mitochondrial Fission Machinery. J. Biol. Chem. 2003, 278, 36373–36379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osellame, L.D.; Singh, A.P.; Stroud, D.A.; Palmer, C.S.; Stojanovski, D.; Ramachandran, R.; Ryan, M.T. Cooperative and Independent Roles of the Drp1 Adaptors Mff, Mid49 and Mid51 in Mitochondrial Fission. J. Cell Sci. 2016, 129, 2170–2181. [Google Scholar] [PubMed] [Green Version]
- Otera, H.; Miyata, N.; Kuge, O.; Mihara, K. Drp1-Dependent Mitochondrial Fission Via Mid49/51 Is Essential for Apoptotic Cristae Remodeling. J. Cell Biol. 2016, 212, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria–lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Yu, R.; Jin, S.; Lendahl, U.; Nistér, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38, e99748. [Google Scholar] [CrossRef]
- Yu, R.; Liu, T.; Jin, S.B.; Ning, C.; Lendahl, U.; Nister, M.; Zhao, J. Mief1/2 Function as Adaptors to Recruit Drp1 to Mitochondria and Regulate the Association of Drp1 with Mff. Sci. Rep. 2017, 7, 880. [Google Scholar] [CrossRef] [Green Version]
- Braschi, E.; Zunino, R.; McBride, H.M. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009, 10, 748–754. [Google Scholar] [CrossRef]
- Chang, C.-R.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N.Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Song, P.; Du, L.; Tian, W.; Yue, W.; Liu, M.; Li, D.; Wang, B.; Zhu, Y.; Cao, C.; et al. Parkin Ubiquitinates Drp1 for Proteasome-Dependent Degradation: Implication of Dysregulated Mitochondrial Dynamics in Parkinson Disease. J. Biol. Chem. 2011, 286, 11649–11658. [Google Scholar] [CrossRef] [Green Version]
- Han, X.J.; Lu, Y.F.; Li, S.A.; Kaitsuka, T.; Sato, Y.; Tomizawa, K.; Nairn, A.C.; Takei, K.; Matsui, H.; Matsushita, M. Cam Kinase I Alpha-Induced Phosphorylation of Drp1 Regulates Mitochondrial Morphology. J. Cell Biol. 2008, 182, 573–585. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic Phosphorylation of Dynamin-related GTPase Drp1 Participates in Mitochondrial Fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.; Hoehn, K.; Counter, C.M.; Kashatus, D. Erk2 Phosphorylation of Drp1 Promotes Mitochondrial Fission and MAPK-Driven Tumor Growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-R.; Blackstone, C. Cyclic AMP-dependent Protein Kinase Phosphorylation of Drp1 Regulates Its GTPase Activity and Mitochondrial Morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cereghetti, G.M.; Stangherlin, A.; de Brito, O.M.; Chang, C.-R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [Green Version]
- Archer, S.L. Mitochondrial Dynamics — Mitochondrial Fission and Fusion in Human Diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef] [Green Version]
- Koshiba, T.; Detmer, S.A.; Kaiser, J.T.; Chen, H.; McCaffery, J.M.; Chan, D.C. Structural Basis of Mitochondrial Tethering by Mitofusin Complexes. Science 2004, 305, 858–862. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 Mediate Sequential Steps in Mitochondrial Membrane Fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef]
- Qi, Y.; Yan, L.; Yu, C.; Guo, X.; Zhou, X.; Hu, X.; Huang, X.; Rao, Z.; Lou, Z.; Hu, J. Structures of human mitofusin 1 provide insight into mitochondrial tethering. J. Cell Biol. 2016, 215, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.-L.; Meng, S.; Chen, Y.; Feng, J.-X.; Gu, D.-D.; Yu, B.; Li, Y.-J.; Yang, J.-Y.; Liao, S.; Chan, D.C.; et al. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 2017, 542, 372–376. [Google Scholar] [CrossRef]
- Franco, A.; Kitsis, R.N.; Fleischer, J.A.; Gavathiotis, E.; Kornfeld, O.S.; Gong, G.; Biris, N.; Benz, A.; Qvit, N.; Donnelly, S.; et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 2016, 540, 74–79. [Google Scholar] [CrossRef]
- Chen, H.C.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Rojo, M.; Legros, F.; Chateau, D.; Lombès, A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J. Cell Sci. 2002, 115, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 Tethers Endoplasmic Reticulum to Mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Dorn, G.W. PINK1-Phosphorylated Mitofusin 2 Is a Parkin Receptor for Culling Damaged Mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Cipolat, S.; De Brito, O.M.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [Green Version]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; Van Der Bliek, A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009, 187, 959–966. [Google Scholar] [CrossRef]
- Anand, R.; Wai, T.; Baker, M.J.; Kladt, N.; Schauss, A.C.; Rugarli, E.; Langer, T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014, 204, 919–929. [Google Scholar] [CrossRef]
- Ban, T.; Kohno, H.; Ishihara, T.; Ishihara, N. Relationship between OPA1 and cardiolipin in mitochondrial inner-membrane fusion. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 951–957. [Google Scholar] [CrossRef]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. Opa1 Processing Controls Mitochondrial Fusion and Is Regulated by Mrna Splicing, Membrane Potential, and Yme1l. J. Cell Biol 2007, 178, 749–755. [Google Scholar] [CrossRef]
- Ge, Y.; Shi, X.; Boopathy, S.; McDonald, J.; Smith, A.W.; Chao, L.H. Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. eLife 2020, 9, e50973. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W. Evolving Concepts of Mitochondrial Dynamics. Annu. Rev. Physiol. 2019, 81, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Galloway, C.A.; Yoon, Y. Control of Mitochondrial Morphology Through Differential Interactions of Mitochondrial Fusion and Fission Proteins. PLoS ONE 2011, 6, e20655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120, 838–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frieden, M.; James, D.; Castelbou, C.; Danckaert, A.; Martinou, J.-C.; Demaurex, N. Ca2+ Homeostasis during Mitochondrial Fragmentation and Perinuclear Clustering Induced by hFis1. J. Biol. Chem. 2004, 279, 22704–22714. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Robotham, J.L.; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 2653–2658. [Google Scholar] [CrossRef] [Green Version]
- Mitra, K. Mitochondrial fission-fusion as an emerging key regulator of cell proliferation and differentiation. BioEssays 2013, 35, 955–964. [Google Scholar] [CrossRef]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of Opa1 Perturbates the Mitochondrial Inner Membrane Structure and Integrity, Leading to Cytochrome C Release and Apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Acín-Peréz, R.; Geghman, K.D.; Manfredi, G.; Lu, B.; Li, C. Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc. Natl. Acad. Sci. USA 2011, 108, 12920–12924. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic Cleavage of Opa1 Stimulates Mitochondrial Inner Membrane Fusion and Couples Fusion to Oxidative Phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griparic, L.; van der Wel, N.N.; Orozco, I.J.; Peters, P.J.; van der Bliek, A.M. Loss of the Intermembrane Space Protein Mgm1/Opa1 Induces Swelling and Localized Constrictions Along the Lengths of Mitochondria. J. Biol. Chem. 2004, 279, 18792–18798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knott, A.B.; Bossy-Wetzel, E. Impairing the Mitochondrial Fission and Fusion Balance: A New Mechanism of Neurodegeneration. Ann. N.Y. Acad. Sci. 2008, 1147, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, T.J.; Berridge, M.J.; Lipp, P.; Bootman, M. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002, 21, 1616–1627. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics—Fusion, Fission, Movement, and Mitophagy—In Neurodegenerative Diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef] [PubMed]
- Bertholet, A.; Delerue, T.; Millet, A.; Moulis, M.; David, C.; Daloyau, M.; Arnauné-Pelloquin, L.; Davezac, N.; Mils, V.; Miquel, M.-C.; et al. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol. Dis. 2016, 90, 3–19. [Google Scholar] [CrossRef]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 Is Necessary for Transport of Axonal Mitochondria and Interacts with the Miro/Milton Complex. J. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [Green Version]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Parone, P.A.; Da Cruz, S.; Tondera, D.; Mattenberger, Y.; James, D.I.; Maechler, P.; Barja, F.; Martinou, J.-C. Preventing Mitochondrial Fission Impairs Mitochondrial Function and Leads to Loss of Mitochondrial DNA. PLoS ONE 2008, 3, e3257. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondrial Genetics: A Paradigm for Aging and Degenerative Diseases? Science 1992, 256, 628–632. [Google Scholar] [CrossRef]
- Wang, D.; Kreutzer, D.A.; Essigmann, J.M. Mutagenicity and repair of oxidative DNA damage: Insights from studies using defined lesions. Mutat. Res. Mol. Mech. Mutagen. 1998, 400, 99–115. [Google Scholar] [CrossRef]
- Carelli, V.; Chan, D.C. Mitochondrial DNA: Impacting Central and Peripheral Nervous Systems. Neuron 2014, 84, 1126–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knott, A.B.; Perkins, G.; Schwarzenbacher, R.; Bossy-Wetzel, E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 505–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ban-Ishihara, R.; Ishihara, T.; Sasaki, N.; Mihara, K.; Ishihara, N. Dynamics of nucleoid structure regulated by mitochondrial fission contributes to cristae reformation and release of cytochrome c. Proc. Natl. Acad. Sci. USA 2013, 110, 11863–11868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Long, Q.; Liu, J.; Tang, H.; Li, Y.; Bao, F.; Qin, D.; Pei, D.; Liu, X. Mitochondrial fusion provides an ‘initial metabolic complementation’ controlled by mtDNA. Cell Mol. Life Sci. 2015, 72, 2585–2598. [Google Scholar] [CrossRef]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of Fusion Results in Mitochondrial Heterogeneity and Dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [Green Version]
- Wilkens, V.; Kohl, W.; Busch, K. Restricted diffusion of OXPHOS complexes in dynamic mitochondria delays their exchange between cristae and engenders a transitory mosaic distribution. J. Cell Sci. 2013, 126, 103–116. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; McCaffery, J.M.; Chan, D.C. Mitochondrial Fusion Protects against Neurodegeneration in the Cerebellum. Cell 2007, 130, 548–562. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Weaver, D.; Shirihai, O.; Hajnoczky, G. Mitochondrial ’Kiss-and-Run’: Interplay between Mitochondrial Motility and Fusion-Fission Dynamics. EMBO J. 2009, 28, 3074–3089. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gegg, M.E.; Cooper, J.M.; Chau, K.Y.; Rojo, M.; Schapira, A.H.; Taanman, J.W. Mitofusin 1 and Mitofusin 2 Are Ubiquitinated in a Pink1/Parkin-Dependent Manner Upon Induction of Mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.-Y.; Seol, D.-W. The role of mitochondria in apoptosis. BMB Rep. 2008, 41, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Jeong, S.Y.; Karbowski, M.; Smith, C.L.; Youle, R.J. Roles of the Mammalian Mitochondrial Fission and Fusion Mediators Fis1, Drp1, and Opa1 in Apoptosis. Mol. Biol. Cell 2004, 15, 5001–5011. [Google Scholar] [CrossRef] [Green Version]
- Montessuit, S.; Somasekharan, S.P.; Terrones, O.; Lucken-Ardjomande, S.; Herzig, S.; Schwarzenbacher, R.; Manstein, D.J.; Bossy-Wetzel, E.; Basañez, G.; Meda, P.; et al. Membrane Remodeling Induced by the Dynamin-Related Protein Drp1 Stimulates Bax Oligomerization. Cell 2010, 142, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Xian, H.; Liou, Y.C. Loss of Mief1/Mid51 Confers Susceptibility to Bax-Mediated Cell Death and Pink1-Prkn-Dependent Mitophagy. Autophagy 2019, 15, 2107–2125. [Google Scholar] [CrossRef]
- Olichon, A.; ElAchouri, G.; Baricault, L.; Delettre, C.; Belenguer, P.; Lenaers, G. OPA1 alternate splicing uncouples an evolutionary conserved function in mitochondrial fusion from a vertebrate restricted function in apoptosis. Cell Death Differ. 2007, 14, 682–692. [Google Scholar] [CrossRef] [Green Version]
- de Brito, O.M.; Scorrano, L. An Intimate Liaison: Spatial Organization of the Endoplasmic Reticulum-Mitochondria Relationship. EMBO J. 2010, 29, 2715–2723. [Google Scholar] [CrossRef] [Green Version]
- Area-Gomez, E.; Del Carmen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; De Groof, A.J.C.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.L.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef] [Green Version]
- E Rusiñol, A.; Cui, Z.; Chen, M.H.; E Vance, J. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 1994, 269, 27494–27502. [Google Scholar] [CrossRef]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria–ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-Mitochondria Tethering Complex Revealed by a Synthetic Biology Screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef] [Green Version]
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal. 2011, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Janer, A.; Prudent, J.; Paupe, V.; Fahiminiya, S.; Majewski, J.; Sgarioto, N.; Des Rosiers, C.; Forest, A.; Lin, Z.Y.; Gingras, A.C.; et al. Slc25a46 Is Required for Mitochondrial Lipid Homeostasis and Cristae Maintenance and Is Responsible for Leigh Syndrome. EMBO Mol. Med. 2016, 8, 1019–1038. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased Er-Mitochondrial Coupling Promotes Mitochondrial Respiration and Bioenergetics During Early Phases of Er Stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [Green Version]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernandez-Alvarez, M.I.; et al. Critical Reappraisal Confirms That Mitofusin 2 Is an Endoplasmic Reticulum-Mitochondria Tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, J.P.; Ivanova, S.; Sánchez-Wandelmer, J.; Martínez-Cristóbal, P.; Noguera, E.; Sancho, A.; Díaz-Ramos, A.; Hernández-Alvarez, M.I.; Sebastián, D.; Mauvezin, C.; et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013, 32, 2348–2361. [Google Scholar] [CrossRef] [Green Version]
- Cosson, P.; Marchetti, A.; Ravazzola-Schreyer, M.; Orci, L. Mitofusin-2 Independent Juxtaposition of Endoplasmic Reticulum and Mitochondria: An Ultrastructural Study. PLoS ONE 2012, 7, e46293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum–mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasawa, R.; Mahul-Mellier, A.-L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2010, 30, 556–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korobova, F.; Ramabhadran, V.; Higgs, H.N. An Actin-Dependent Step in Mitochondrial Fission Mediated by the ER-Associated Formin INF2. Science 2013, 339, 464–467. [Google Scholar] [CrossRef] [Green Version]
- Manor, U.; Bartholomew, S.; Golani, G.; Christenson, E.; Kozlov, M.; Higgs, H.; Spudich, J.; Lippincott-Schwartz, J. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. eLife 2015, 4, e08828. [Google Scholar] [CrossRef]
- Matus, S.; Glimcher, L.H.; Hetz, C. Protein folding stress in neurodegenerative diseases: A glimpse into the ER. Curr. Opin. Cell Biol. 2011, 23, 239–252. [Google Scholar] [CrossRef]
- Area-Gomez, E.; De Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic Reticulum–Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Jendrach, M.; Mai, S.; Pohl, S.; Voth, M.; Bereiter-Hahn, J. Short- and Long-Term Alterations of Mitochondrial Morphology, Dynamics and Mtdna after Transient Oxidative Stress. Mitochondrion 2008, 8, 293–304. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of Mitochondrial Fusion by Alpha-Synuclein Is Rescued by Pink1, Parkin and Dj-1. EMBO J. 2010, 29, 3571–3589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Lim, P.J.; Karbowski, M.; Monteiro, M.J. Effects of overexpression of Huntingtin proteins on mitochondrial integrity. Hum. Mol. Genet. 2008, 18, 737–752. [Google Scholar] [CrossRef] [Green Version]
- Wen, S.; Unuma, K.; Funakoshi, T.; Aki, T.; Uemura, K. Altered cardiac mitochondrial dynamics and biogenesis in rat after short-term cocaine administration. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Sadakierska-Chudy, A.; Kotarska, A.; Frankowska, M.; Jastrzębska, J.; Wydra, K.; Miszkiel, J.; Przegaliński, E.; Filip, M. The Alterations in Mitochondrial DNA Copy Number and Nuclear-Encoded Mitochondrial Genes in Rat Brain Structures after Cocaine Self-Administration. Mol. Neurobiol. 2017, 54, 7460–7470. [Google Scholar] [CrossRef] [Green Version]
- Chandra, R.; Francis, T.; Konkalmatt, P.; Amgalan, A.; Gancarz, A.M.; Dietz, D.; Lobo, M.K. Opposing Role for Egr3 in Nucleus Accumbens Cell Subtypes in Cocaine Action. J. Neurosci. 2015, 35, 7927–7937. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.L.; Chandra, R.; Harris, M.; Patel, I.; Wang, T.; Kim, H.; Jensen, L.; Russo, S.J.; Turecki, G.; Gancarz-Kausch, A.M.; et al. Cocaine-induced neuron subtype mitochondrial dynamics through Egr3 transcriptional regulation. Mol. Brain 2021, 14, 1–14. [Google Scholar] [CrossRef]
- Chandra, R.; Engeln, M.; Schiefer, C.; Patton, M.; Martin, J.A.; Werner, C.; Riggs, L.M.; Francis, T.C.; McGlincy, M.; Evans, B.; et al. Drp1 Mitochondrial Fission in D1 Neurons Mediates Behavioral and Cellular Plasticity during Early Cocaine Abstinence. Neuron 2017, 96, 1327–1341.e6. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.-I.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-Nitrosylation of Drp1 Mediates Beta-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [Green Version]
- Calkins, M.J.; Manczak, M.; Mao, P.; Shirendeb, U.; Reddy, P.H. Impaired Mitochondrial Biogenesis, Defective Axonal Transport of Mitochondria, Abnormal Mitochondrial Dynamics and Synaptic Degeneration in a Mouse Model of Alzheimer’s Disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.R.; Wardell, S.E.; Cakir, M.; Yip, C.; Ahn, Y.-R.; Ali, M.; Yllanes, A.P.; Chao, C.A.; McDonnell, D.P.; Wood, K.C. Dysregulation of mitochondrial dynamics proteins are a targetable feature of human tumors. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsov, A.V.; Hermann, M.; Saks, V.; Hengster, P.; Margreiter, R. The cell-type specificity of mitochondrial dynamics. Int. J. Biochem. Cell Biol. 2009, 41, 1928–1939. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, J.; Tan, M.; Albers, K.M.; Jia, J. Mitochondrial fission proteins in peripheral blood lymphocytes are potential biomarkers for Alzheimer’s disease. Eur. J. Neurol. 2012, 19, 1015–1022. [Google Scholar] [CrossRef]
- Annesley, S.J.; Fisher, P.R. Lymphoblastoid Cell Lines as Models to Study Mitochondrial Function in Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 4536. [Google Scholar] [CrossRef]
- Itoh, K.; Nakamura, K.; Iijima, M.; Sesaki, H. Mitochondrial dynamics in neurodegeneration. Trends Cell Biol. 2013, 23, 64–71. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Chemical structure of cocaine and its action on dopaminergic neurons. Cocaine antagonizes dopamine reuptake from the synaptic cleft through dopamine transporters. This leads to an increase in the levels of dopamine in the cleft, boosting the effects of dopamine in the central nervous system.
Figure 1.
Chemical structure of cocaine and its action on dopaminergic neurons. Cocaine antagonizes dopamine reuptake from the synaptic cleft through dopamine transporters. This leads to an increase in the levels of dopamine in the cleft, boosting the effects of dopamine in the central nervous system.

Figure 2.
Effects of cocaine on neuronal cells. Cocaine enters the central nervous system by crossing and/or disrupting the blood–brain barrier (BBB). After entering neuronal cells, cocaine and its metabolites induce subcellular stress within mitochondria and the endoplasmic reticulum (ER). The aggregation of dopamine induced by cocaine also results in oxidative stress during the degradation of dopamine. Overwhelmed stress and direct damage from cocaine lead to mitochondrial dysfunction which results in cell death.
Figure 2.
Effects of cocaine on neuronal cells. Cocaine enters the central nervous system by crossing and/or disrupting the blood–brain barrier (BBB). After entering neuronal cells, cocaine and its metabolites induce subcellular stress within mitochondria and the endoplasmic reticulum (ER). The aggregation of dopamine induced by cocaine also results in oxidative stress during the degradation of dopamine. Overwhelmed stress and direct damage from cocaine lead to mitochondrial dysfunction which results in cell death.

Figure 3.
General aspects of mitochondrial fission and fusion mediated by a panel of regulating molecules. DRP1, which ordinarily resides in cytosol, is recruited to mitochondrial outer membranes upon activation through, e.g., phosphorylation at Ser-616. DRP1 assembles into a ring-like structure and act as scissors to promote mitochondrial division. Mitochondrial fusion consists of two consecutive processes: outer and inner membrane fusions. The former process is mediated by MFN1/2, while the latter is mainly executed through OPA1.
Figure 3.
General aspects of mitochondrial fission and fusion mediated by a panel of regulating molecules. DRP1, which ordinarily resides in cytosol, is recruited to mitochondrial outer membranes upon activation through, e.g., phosphorylation at Ser-616. DRP1 assembles into a ring-like structure and act as scissors to promote mitochondrial division. Mitochondrial fusion consists of two consecutive processes: outer and inner membrane fusions. The former process is mediated by MFN1/2, while the latter is mainly executed through OPA1.


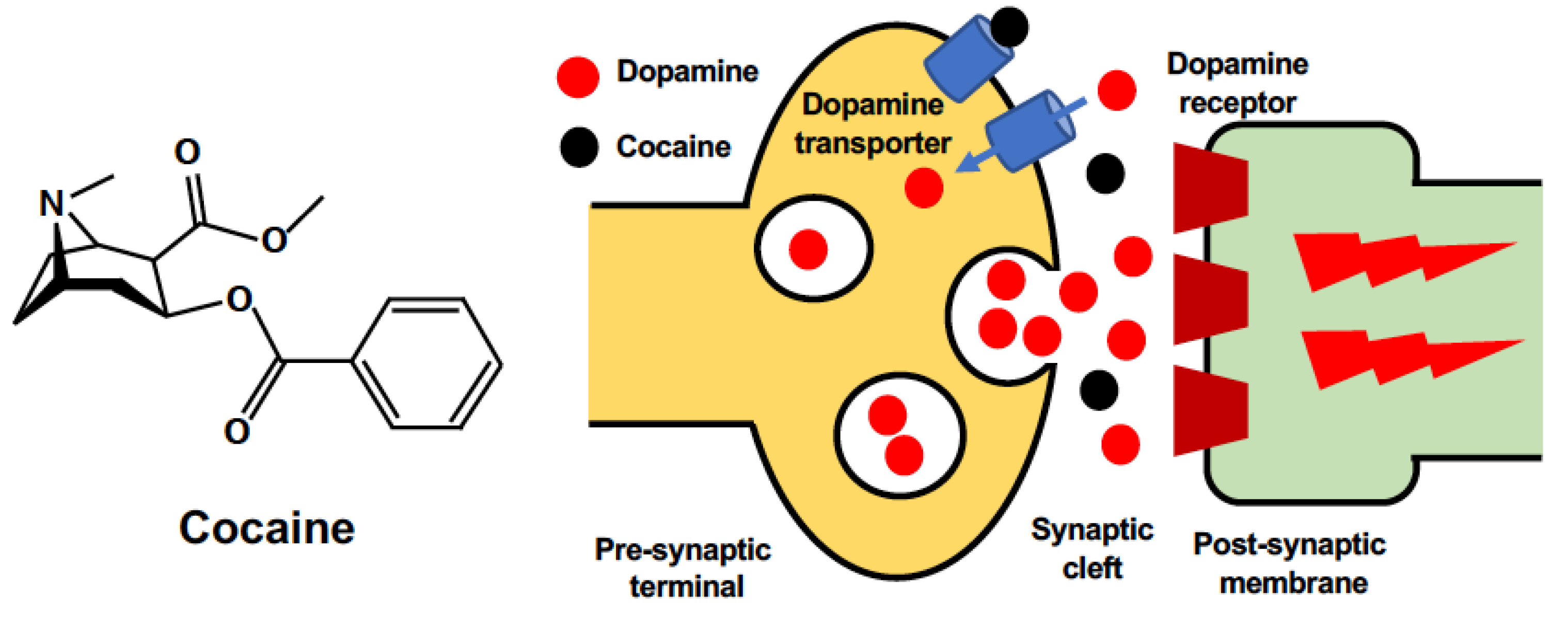{kind=link}
{kind=link}
{kind=link}
Table 1.
Current findings of how cocaine affects neuronal mitochondrial dynamics.
| Mitochondrial Dynamics | Altered Markers | Tested Brain Region/Cell Type | Model Details |
|---|---|---|---|
| Fission related | Mtfr1 (↑) | Prefrontal cortex | Rat, self-administration (average 100 mg/kg of cocaine intake, 3 days) and abstinence (3 days) [166] |
| Opa3 (↑) | Prefrontal cortex | ||
| Drp1 (↑) | Nucleus accumbens | Mice, repeated cocaine (7 days, 20 mg/kg, ip) [169] | |
| Nucleus accumbens | Rat, self-administration (1 mg/kg/infusion, 10 days) [169] | ||
| Nucleus accumbens | Human samples [169] | ||
| D1 type medium spiny neurons | Mice, repeated cocaine (7 days, 20 mg/kg, ip) [169] | ||
| Drp1 (↓) | D2 type medium spiny neurons | ||
| pDRP1 at ser616(↑) | D1 type medium spiny neurons | ||
| Neuro2a neuroblastoma cells | 600 mM, 2~3 times per week, 3 weeks [11] | ||
| pDRP1 at ser616 (↓) | D2 type medium spiny neurons | Mice, repeated cocaine (7 days, 20 mg/kg, ip) [169] | |
| Mitochondrial size (↓) | D1 type medium spiny neurons | Rat, self-administration (1 mg/kg/infusion, 10 days) [169] | |
| Neuro2a neuroblastoma cells | 600 mM, 2~3 times per week, 3 weeks [11] | ||
| Fusion related | Mfn1 (↑) | prefrontal cortex | Rat, self-administration (average 100 mg/kg of cocaine intake) and abstinence (3 days) [166] |
| Opa1 (↑) | prefrontal cortex |
Mtfr1, mitochondrial fission regulator 1, Opa3, optic atrophy 3, Drp1, dynamin-related protein 1, pDRP1 at ser616, phosphorylation of DRP1 at serine 616, Mfn1, mitofusion 1, Opa1, optic atrophy 1, ip, intraperitoneal injection.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wen, S.; Aki, T.; Funakoshi, T.; Unuma, K.; Uemura, K. Role of Mitochondrial Dynamics in Cocaine’s Neurotoxicity. Int. J. Mol. Sci. 2022, 23, 5418. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105418
AMA Style
Wen S, Aki T, Funakoshi T, Unuma K, Uemura K. Role of Mitochondrial Dynamics in Cocaine’s Neurotoxicity. International Journal of Molecular Sciences. 2022; 23(10):5418. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105418
Chicago/Turabian StyleWen, Shuheng, Toshihiko Aki, Takeshi Funakoshi, Kana Unuma, and Koichi Uemura. 2022. "Role of Mitochondrial Dynamics in Cocaine’s Neurotoxicity" International Journal of Molecular Sciences 23, no. 10: 5418. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105418
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.