
First-Void Urine Microbiome in Women with Chlamydia trachomatis Infection

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Study Population
2.2. CT Genotyping
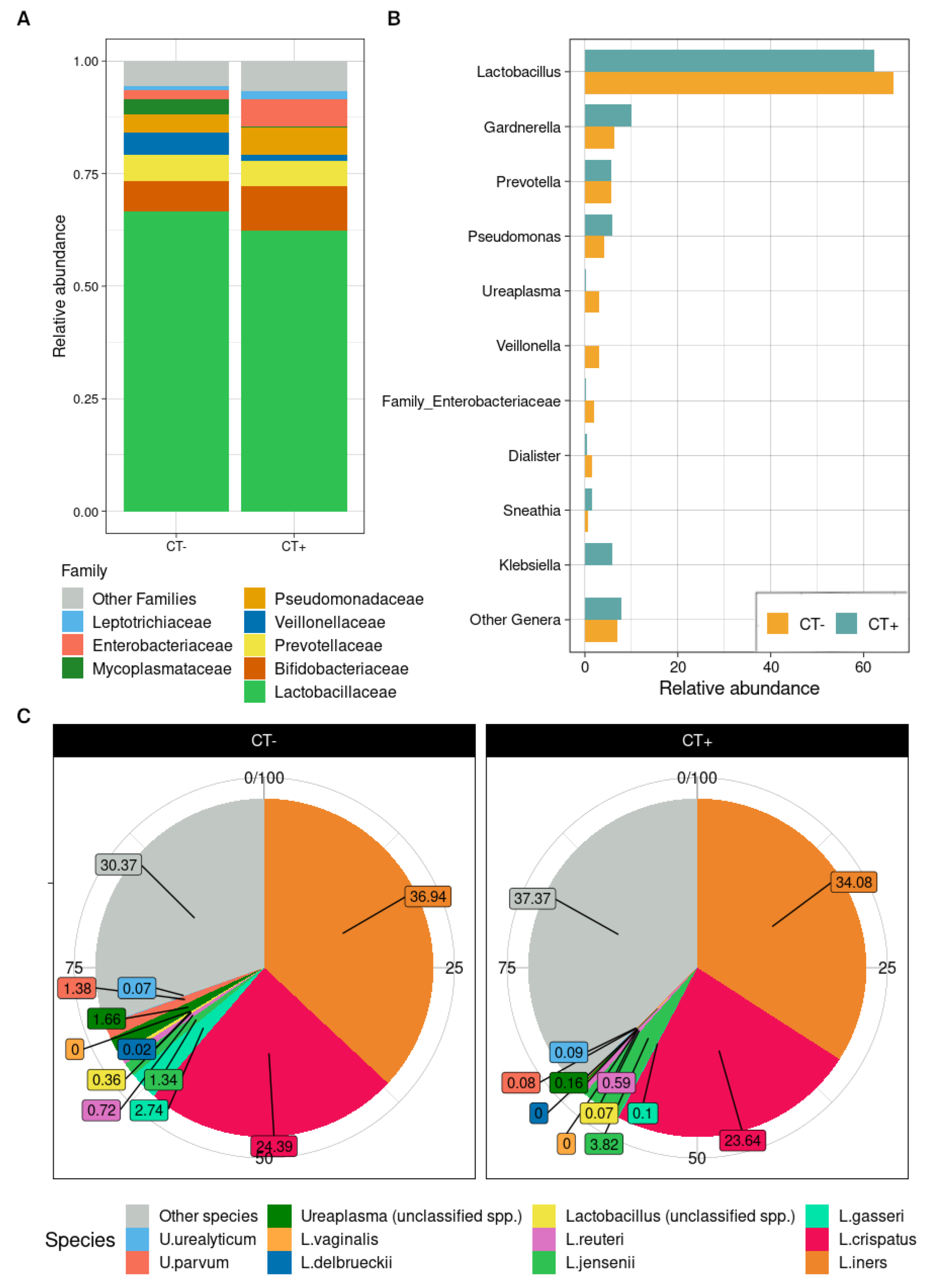
2.3. Taxonomic Composition of Urine Bacterial Communities
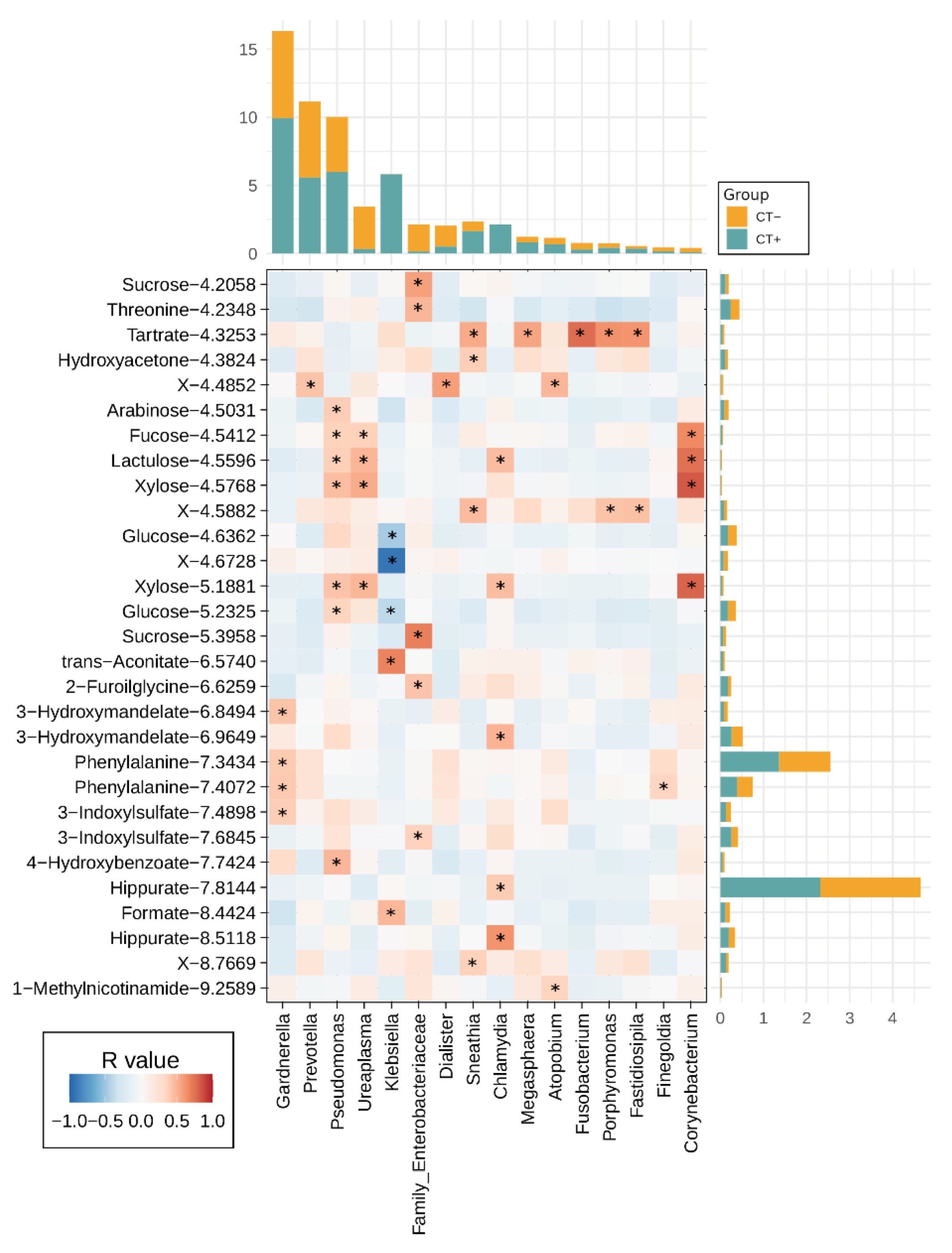
2.4. Urine Metabolites Composition and Metabolite Microbiome Correlation
3. Discussion
4. Materials and Methods
4.1. Study Population and Sample Collection
4.2. CT Genotyping
4.3. Urine Microbiome Analysis
4.4. Urine Metabolome Analysis by 1H-NMR
4.5. Data Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Senior, K. Chlamydia: A much underestimated STI. Lancet Infect. Dis. 2012, 12, 517–518. [Google Scholar] [CrossRef]
- Haggerty, C.L.; Gottlieb, S.L.; Taylor, B.D.; Low, N.; Xu, F.; Ness, R.B. Risk of Sequelae after Chlamydia trachomatis Genital Infection in Women. J. Infect. Dis. 2010, 201, S134–S155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foschi, C.; Nardini, P.; Banzola, N.; D’Antuono, A.; Compri, M.; Cevenini, R.; Marangoni, A. Chlamydia trachomatis infection prevalence and serovar distribution in a high-density urban area in the north of Italy. J. Med. Microbiol. 2016, 65, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Foschi, C.; Laghi, L.; D’Antuono, A.; Gaspari, V.; Zhu, C.; Dellarosa, N.; Salvo, M.; Marangoni, A. Urine metabolome in women with Chlamydia trachomatis infection. PLoS ONE 2018, 13, e0194827. [Google Scholar] [CrossRef] [PubMed]
- Elwell, C.; Mirrashidi, K.; Engel, J. Chlamydia cell biology and pathogenesis. Nat. Rev. Genet. 2016, 14, 385–400. [Google Scholar] [CrossRef]
- van der Veer, C.; Bruisten, S.M.; van der Helm, J.J.; de Vries, H.J.C.; van Houdt, R. The Cervicovaginal Microbiota in Women Notified for Chlamydia trachomatis Infection: A Case-Control Study at the Sexually Transmitted Infection Outpatient Clinic in Amsterdam, The Netherlands. Clin. Infect. Dis. 2017, 64, 24–31. [Google Scholar] [CrossRef] [Green Version]
- Tamarelle, J.; de Barbeyrac, B.; Le Hen, I.; Thiébaut, A.; Bébéar, C.; Ravel, J.; Delarocque-Astagneau, E. Vaginal microbiota composition and association with prevalent Chlamydia trachomatis infection: A cross-sectional study of young women attending a STI clinic in France. Sex. Transm. Infect. 2018, 94, 616–618. [Google Scholar] [CrossRef]
- Raimondi, S.; Candeliere, F.; Amaretti, A.; Foschi, C.; Morselli, S.; Gaspari, V.; Rossi, M.; Marangoni, A. Vaginal and Anal Microbiome during Chlamydia trachomatis Infections. Pathogens 2021, 10, 1347. [Google Scholar] [CrossRef]
- Chiu, S.-F.; Huang, P.-J.; Cheng, W.-H.; Huang, C.-Y.; Chu, L.J.; Lee, C.-C.; Lin, H.-C.; Chen, L.-C.; Lin, W.-N.; Tsao, C.-H.; et al. Vaginal Microbiota of the Sexually Transmitted Infections Caused by Chlamydia trachomatis and Trichomonas vaginalis in Women with Vaginitis in Taiwan. Microorganisms 2021, 9, 1864. [Google Scholar] [CrossRef]
- Cheong, H.C.; Yap, P.S.X.; Chong, C.W.; Cheok, Y.Y.; Lee, C.Y.Q.; Tan, G.M.Y.; Sulaiman, S.; Hassan, J.; Sabet, N.S.; Looi, C.Y.; et al. Diversity of endocervical microbiota associated with genital Chlamydia trachomatis infection and infertility among women visiting obstetrics and gynecology clinics in Malaysia. PLoS ONE 2019, 14, e0224658. [Google Scholar] [CrossRef]
- Filardo, S.; Di Pietro, M.; Tranquilli, G.; Latino, M.A.; Recine, N.; Porpora, M.G.; Sessa, R. Selected Immunological Mediators and Cervical Microbial Signatures in Women with Chlamydia trachomatis Infection. mSystems 2019, 4, e00094-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parolin, C.; Foschi, C.; Laghi, L.; Zhu, C.; Banzola, N.; Gaspari, V.; D’Antuono, A.; Giordani, B.; Severgnini, M.; Consolandi, C.; et al. Insights into Vaginal Bacterial Communities and Metabolic Profiles of Chlamydia trachomatis Infection: Positioning Between Eubiosis and Dysbiosis. Front. Microbiol. 2018, 9, 600. [Google Scholar] [CrossRef] [PubMed]
- Ceccarani, C.; Foschi, C.; Parolin, C.; D’Antuono, A.; Gaspari, V.; Consolandi, C.; Laghi, L.; Camboni, T.; Vitali, B.; Severgnini, M.; et al. Diversity of vaginal microbiome and metabolome during genital infections. Sci. Rep. 2019, 9, 14095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balle, C.; Lennard, K.; Dabee, S.; Barnabas, S.L.; Jaumdally, S.Z.; Gasper, M.A.; Maseko, V.; Mbulawa, Z.Z.A.; Williamson, A.-L.; Bekker, L.-G.; et al. Endocervical and vaginal microbiota in South African adolescents with asymptomatic Chlamydia trachomatis infection. Sci. Rep. 2018, 8, 11109. [Google Scholar] [CrossRef] [PubMed]
- Perez-Carrasco, V.; Soriano-Lerma, A.; Soriano, M.; Gutiérrez-Fernández, J.; Garcia-Salcedo, J.A. Urinary Microbiome: Yin and Yang of the Urinary Tract. Front. Cell. Infect. Microbiol. 2021, 11, 617002. [Google Scholar] [CrossRef]
- Brubaker, L.; Putonti, C.; Dong, Q.; Wolfe, A.J. The human urobiome. Mamm. Genome 2021, 32, 232–238. [Google Scholar] [CrossRef]
- Komesu, Y.M.; Dinwiddie, D.L.; Richter, H.E.; Lukacz, E.S.; Sung, V.W.; Siddiqui, N.Y.; Zyczynski, H.M.; Ridgeway, B.; Rogers, R.G.; Arya, L.A.; et al. Defining the relationship between vaginal and urinary microbiomes. Am. J. Obstet. Gynecol. 2020, 222, 154.e1–154.e10. [Google Scholar] [CrossRef]
- Novak, J.; Ravel, J.; Ma, B.; Ferreira, C.S.T.; Tristão, A.D.R.; Silva, M.G.; Marconi, C. Characteristics associated with Lactobacillus iners-dominated vaginal microbiota. Sex. Transm. Infect. 2021. [Google Scholar] [CrossRef]
- France, M.T.; Ma, B.; Gajer, P.; Brown, S.; Humphrys, M.S.; Holm, J.B.; Waetjen, L.E.; Brotman, R.M.; Ravel, J. VALENCIA: A nearest centroid classification method for vaginal microbial communities based on composition. Microbiome 2020, 8, 166. [Google Scholar] [CrossRef]
- Mueller, E.R.; Wolfe, A.J.; Brubaker, L. Female urinary microbiota. Curr. Opin. Urol. 2017, 27, 282–286. [Google Scholar] [CrossRef]
- Price, T.K.; Hilt, E.E.; Thomas-White, K.; Mueller, E.R.; Wolfe, A.J.; Brubaker, L. The urobiome of continent adult women: A cross-sectional study. BJOG Int. J. Obstet. Gynaecol. 2020, 127, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Saka, H.A.; Thompson, J.W.; Chen, Y.-S.; Kumar, Y.; Dubois, L.G.; Moseley, M.A.; Valdivia, R.H. Quantitative proteomics reveals metabolic and pathogenic properties of Chlamydia trachomatis developmental forms. Mol. Microbiol. 2011, 82, 1185–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtiss, N.; Balachandran, A.; Krska, L.; Peppiatt-Wildman, C.; Wildman, S.; Duckett, J. Age, menopausal status and the bladder microbiome. Eur. J. Obstet. Gynecol. Reprod. Biol. 2018, 228, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Foschi, C.; Salvo, M.; D’Antuono, A.; Gaspari, V.; Banzola, N.; Cevenini, R.; Marangoni, A. Distribution of genital Mollicutes in the vaginal ecosystem of women with different clinical conditions. New Microbiol. 2018, 41, 225–229. [Google Scholar]
- Rowlands, R.S.; Kragh, K.; Sahu, S.; Maddocks, S.E.; Bolhuis, A.; Spiller, O.B.; Beeton, M.L. A requirement for flow to enable the development of Ureaplasma parvum biofilms in vitro. J. Appl. Microbiol. 2021, 131, 2579–2585. [Google Scholar] [CrossRef]
- García-Castillo, M.; Morosini, M.-I.; Gálvez, M.; Baquero, F.; del Campo, R.; Meseguer, M.-A. Differences in biofilm development and antibiotic susceptibility among clinical Ureaplasma urealyticum and Ureaplasma parvum isolates. J. Antimicrob. Chemother. 2008, 62, 1027–1030. [Google Scholar] [CrossRef] [Green Version]
- Foschi, C.; Laghi, L.; Parolin, C.; Giordani, B.; Compri, M.; Cevenini, R.; Marangoni, A.; Vitali, B. Novel approaches for the taxonomic and metabolic characterization of lactobacilli: Integration of 16S rRNA gene sequencing with MALDI-TOF MS and 1H-NMR. PLoS ONE 2017, 12, e0172483. [Google Scholar] [CrossRef] [Green Version]
- Nicolò, S.; Tanturli, M.; Mattiuz, G.; Antonelli, A.; Baccani, I.; Bonaiuto, C.; Baldi, S.; Nannini, G.; Menicatti, M.; Bartolucci, G.; et al. Vaginal Lactobacilli and Vaginal Dysbiosis-Associated Bacteria Differently Affect Cervical Epithelial and Immune Homeostasis and Anti-Viral Defenses. Int. J. Mol. Sci. 2021, 22, 6487. [Google Scholar] [CrossRef]
- Sonthalia, S.; Aggarwal, P.; Das, S.; Sharma, P.; Sharma, R.; Singh, S. Aerobic vaginitis—An underdiagnosed cause of vaginal discharge—Narrative review. Int. J. STD AIDS 2020, 31, 1018–1027. [Google Scholar] [CrossRef]
- De Simone, G.; Balducci, C.; Forloni, G.; Pastorelli, R.; Brunelli, L. Hippuric acid: Could became a barometer for frailty and geriatric syndromes? Ageing Res. Rev. 2021, 72, 101466. [Google Scholar] [CrossRef]
- Piras, C.; Neri, I.; Pintus, R.; Noto, A.; Petrella, E.; Monari, F.; Dessì, A.; Fanos, V.; Atzori, L.; Facchinetti, F. First trimester metabolomics 1H-NMR study of the urinary profile predicts gestational diabetes mellitus development in obese women. J. Matern. Neonatal Med. 2021, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo-Garcia, J.L.; Comella-Del-Barrio, P.; Campos-Olivas, R.; Villar-Hernández, R.; Prat-Aymerich, C.; De Souza-Galvão, M.L.; Jiménez-Fuentes, M.A.; Ruiz-Manzano, J.; Stojanovic, Z.; González, A.; et al. Discovery and validation of an NMR-based metabolomic profile in urine as TB biomarker. Sci. Rep. 2020, 10, 122317. [Google Scholar] [CrossRef] [PubMed]
- Pallister, T.; Jackson, M.A.; Martin, T.; Zierer, J.; Jennings, A.; Mohney, R.P.; MacGregor, A.; Steves, C.; Cassidy, A.; Spector, T.D.; et al. Hippurate as a metabolomic marker of gut microbiome diversity: Modulation by diet and relationship to metabolic syndrome. Sci. Rep. 2017, 7, 13670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalska, K.; Wellington, S.; Maltseva, N.; Jedrzejczak, R.; Selem-Mojica, N.; Rosas-Becerra, L.R.; Barona-Gómez, F.; Hung, D.T.; Joachimiak, A. Catalytically impaired TrpA subunit of tryptophan synthase from Chlamydia trachomatis is an allosteric regulator of TrpB. Protein Sci. 2021, 30, 1904–1918. [Google Scholar] [CrossRef] [PubMed]
- Sherchand, S.P.; Aiyar, A. Ammonia generation by tryptophan synthase drives a key genetic difference between genital and ocular Chlamydia trachomatis isolates. Proc. Natl. Acad. Sci. USA 2019, 116, 12468–12477. [Google Scholar] [CrossRef] [Green Version]
- Nardini, P.; Palomino, R.A.N.; Parolin, C.; Laghi, L.; Foschi, C.; Cevenini, R.; Vitali, B.; Marangoni, A. Lactobacillus crispatus inhibits the infectivity of Chlamydia trachomatis elementary bodies, in vitro study. Sci. Rep. 2016, 6, 29024. [Google Scholar] [CrossRef]
- Marziali, G.; Marangoni, A.; Foschi, C.; Re, M.C.; Calonghi, N. Effect of Sugars on Chlamydia trachomatis Infectivity. Pathogens 2020, 9, 298. [Google Scholar] [CrossRef] [Green Version]
- Marangoni, A.; Foschi, C.; Nardini, P.; Compri, M.; Cevenini, R. Evaluation of the Versant CT/GC DNA 1.0 Assay (kPCR) for the Detection of Extra-Genital Chlamydia trachomatis and Neisseria gonorrhoeae Infections. PLoS ONE 2015, 10, e0120979. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SILVA 16S rRNA Database. Available online: https://www.arb-silva.de/documentation/release-138 (accessed on 1 March 2022).
- Foschi, C.; Salvo, M.; Laghi, L.; Zhu, C.; Ambretti, S.; Marangoni, A.; Re, M.C. Impact of meropenem on Klebsiella pneumoniae metabolism. PLoS ONE 2018, 13, e0207478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kneen, M.A.; Annegarn, H.J. Algorithm for fitting XRF, SEM and PIXE X-ray spectra backgrounds. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 1996, 109–110, 209–213. [Google Scholar] [CrossRef]
- Liland, K.H.; Almøy, T.; Mevik, B.-H. Optimal Choice of Baseline Correction for Multivariate Calibration of Spectra. Appl. Spectrosc. 2010, 64, 1007–1016. [Google Scholar] [CrossRef]
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S.; et al. HMDB: The Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–D526. [Google Scholar] [CrossRef]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic Quotient Normalization as Robust Method to Account for Dilution of Complex Biological Mixtures. Application in 1H NMR Metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H.; et al. Package “Vegan”. R Package Version 2.0–10. Available online: https://cran.r-project.org/src/contrib/Archive/vegan/vegan_2.0-10.tar.gz (accessed on 15 February 2022).



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaspari, V.; Ceccarani, C.; Severgnini, M.; Orioni, G.; Camboni, T.; Laghi, L.; Morselli, S.; Foschi, C.; Marangoni, A.; Consolandi, C.; et al. First-Void Urine Microbiome in Women with Chlamydia trachomatis Infection. Int. J. Mol. Sci. 2022, 23, 5625. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105625
Gaspari V, Ceccarani C, Severgnini M, Orioni G, Camboni T, Laghi L, Morselli S, Foschi C, Marangoni A, Consolandi C, et al. First-Void Urine Microbiome in Women with Chlamydia trachomatis Infection. International Journal of Molecular Sciences. 2022; 23(10):5625. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105625
Chicago/Turabian StyleGaspari, Valeria, Camilla Ceccarani, Marco Severgnini, Gionathan Orioni, Tania Camboni, Luca Laghi, Sara Morselli, Claudio Foschi, Antonella Marangoni, Clarissa Consolandi, and et al. 2022. "First-Void Urine Microbiome in Women with Chlamydia trachomatis Infection" International Journal of Molecular Sciences 23, no. 10: 5625. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105625