
The Amyotrophic Lateral Sclerosis M114T PFN1 Mutation Deregulates Alternative Autophagy Pathways and Mitochondrial Homeostasis

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. PFN1 Mutations Are Rare in French ALS Patients
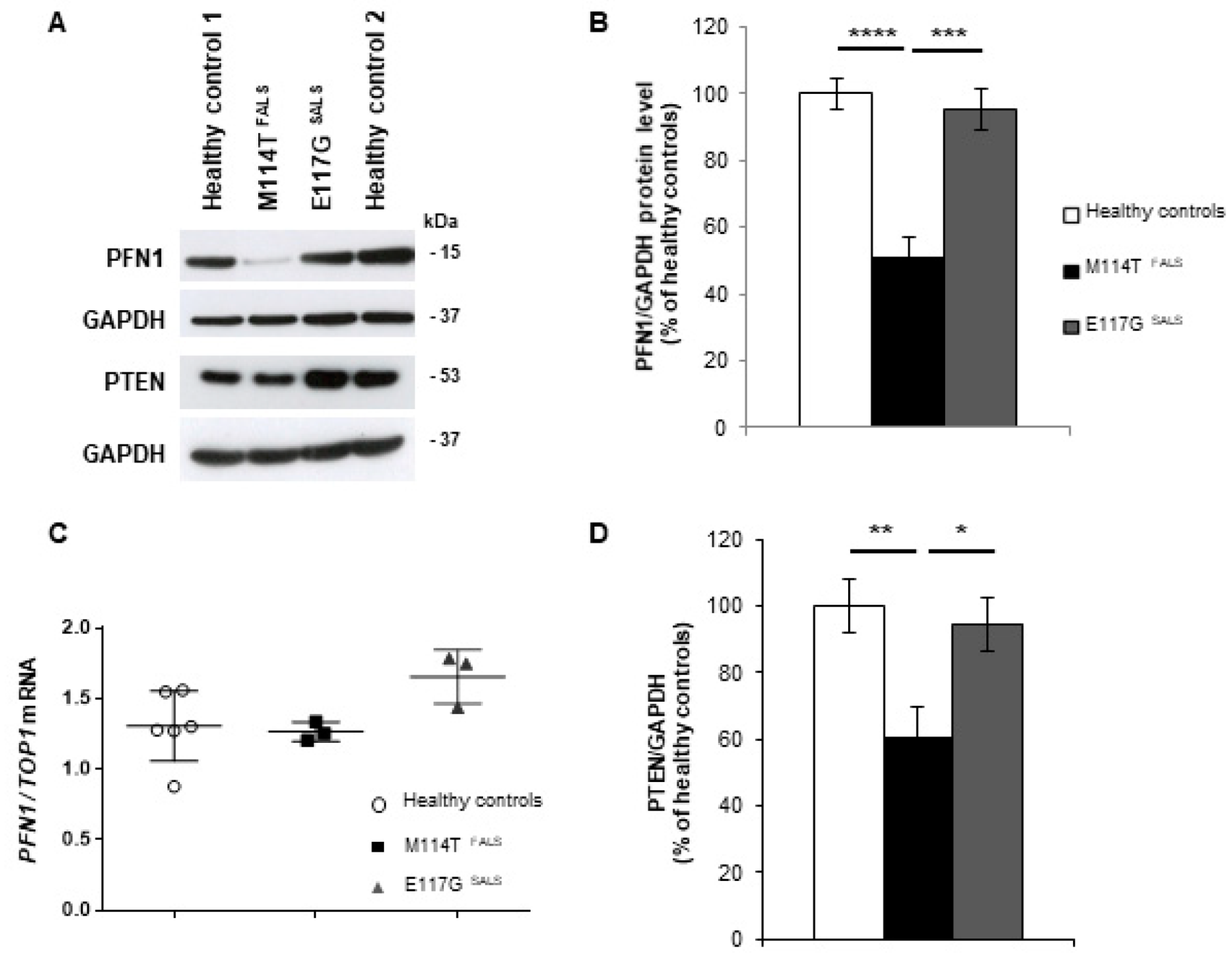
2.2. PFN1 Protein Levels Are Decreased in M114T Patient Lymphoblasts
2.3. Autophagy-Linked PTEN Shows Decreased Levels in M114T Patient Lymphoblasts
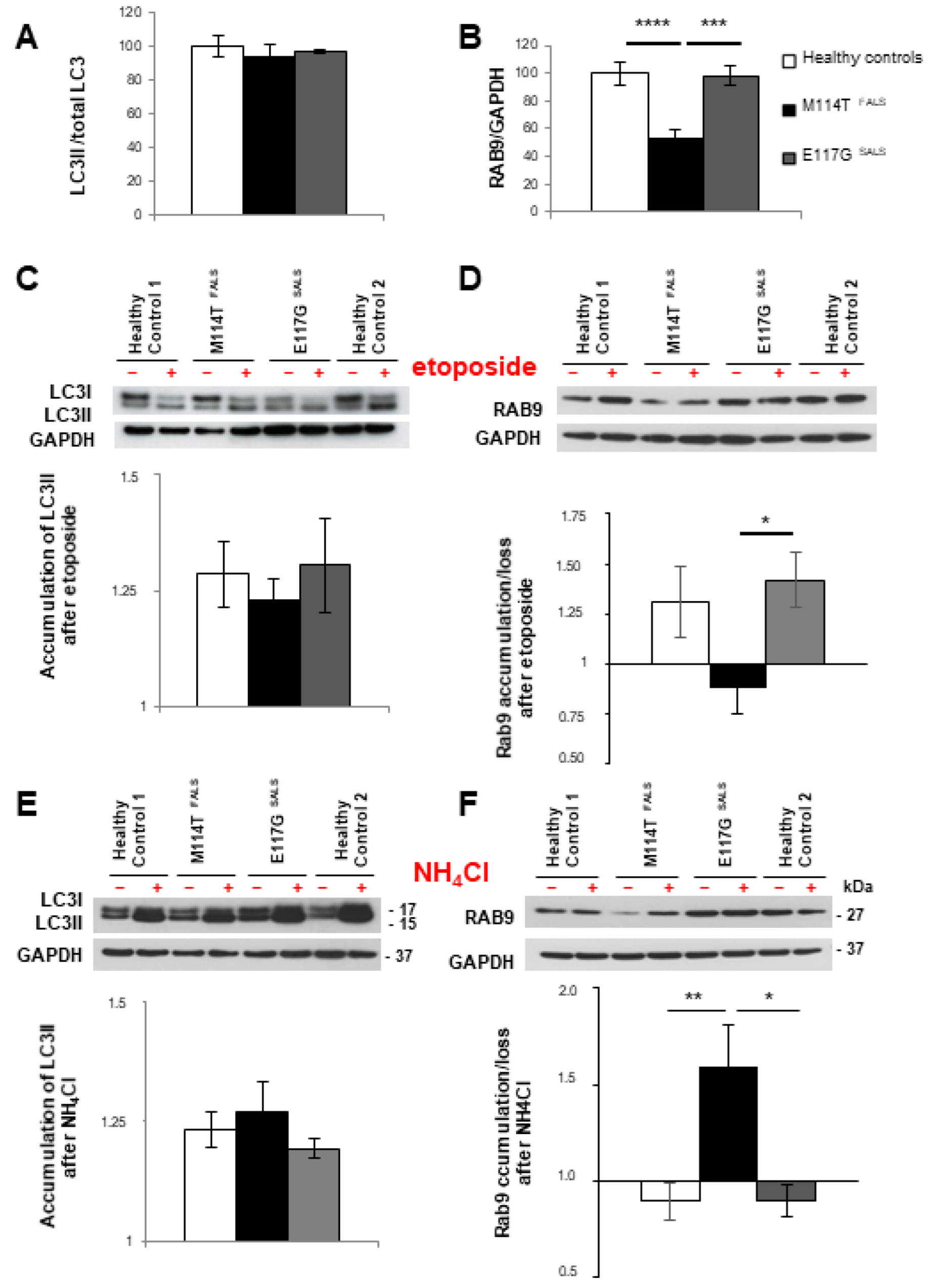
2.4. The Alternative Autophagy Pathway Is Deregulated in M114T Patient Lymphoblasts
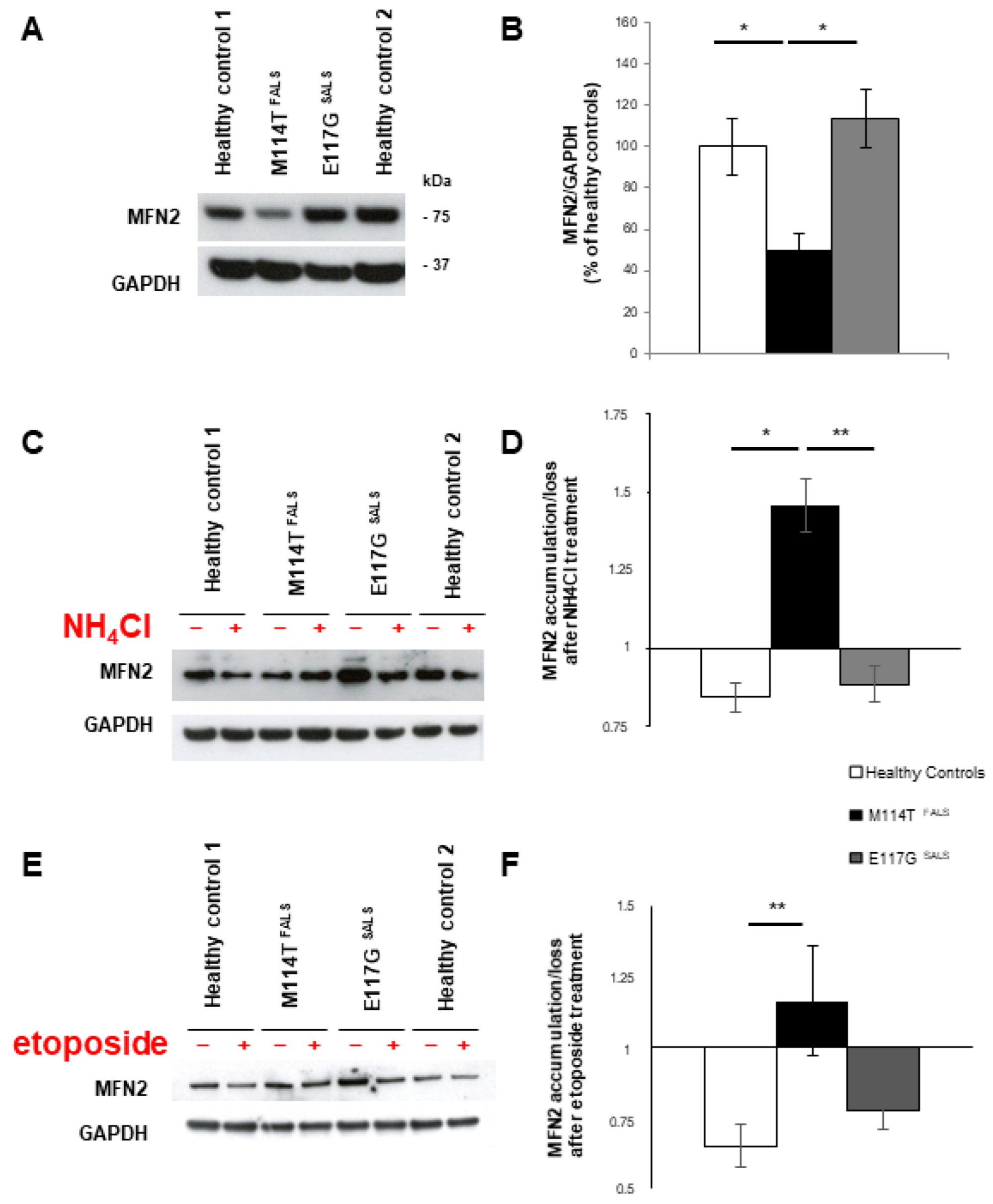
2.5. Mitochondrial Homeostasis Is Misregulated in M114T Lymphoblasts
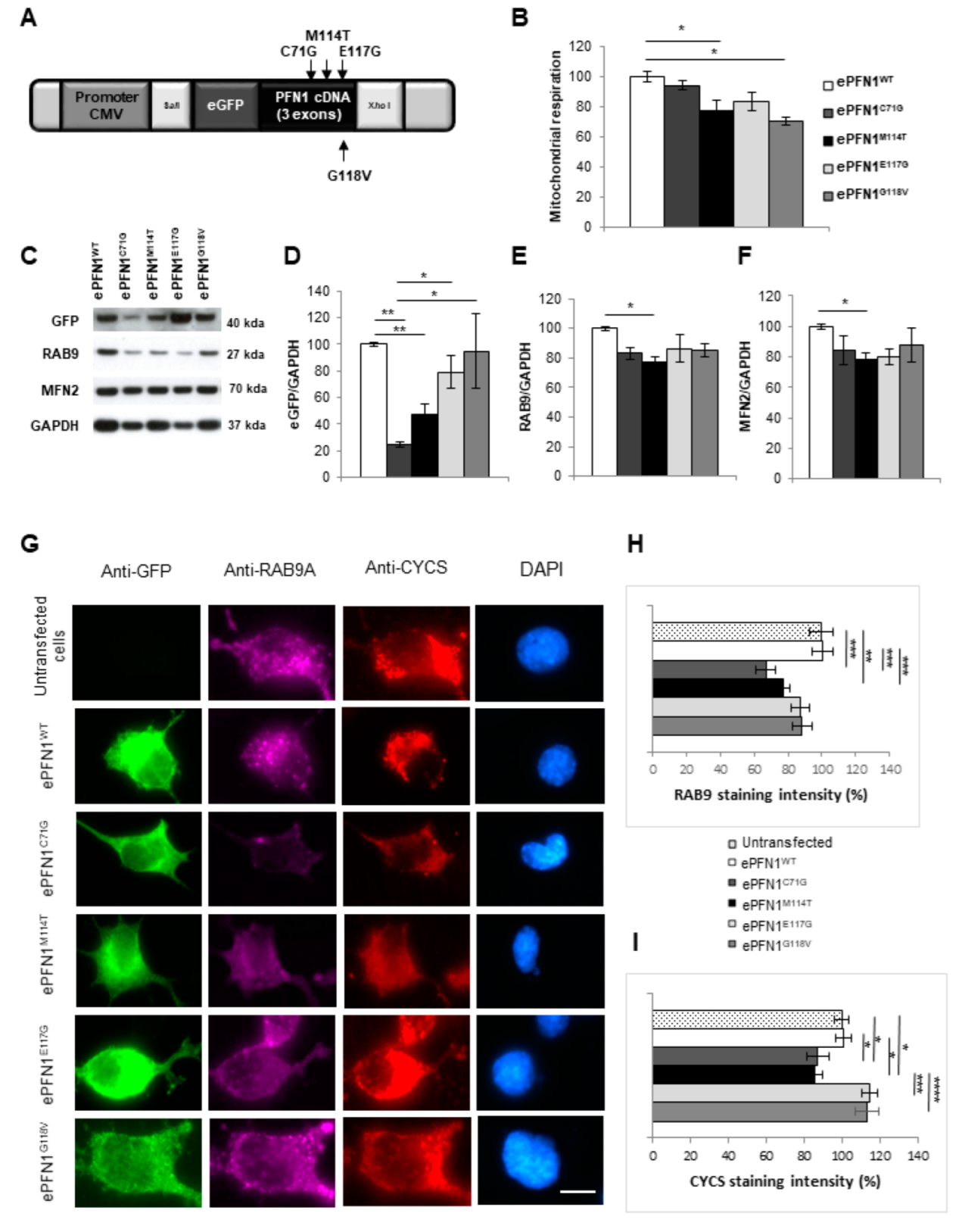
2.6. RAB9 and Mitochondria Levels Are Reduced in Cells Expressing a PFN1 M114T Transgene
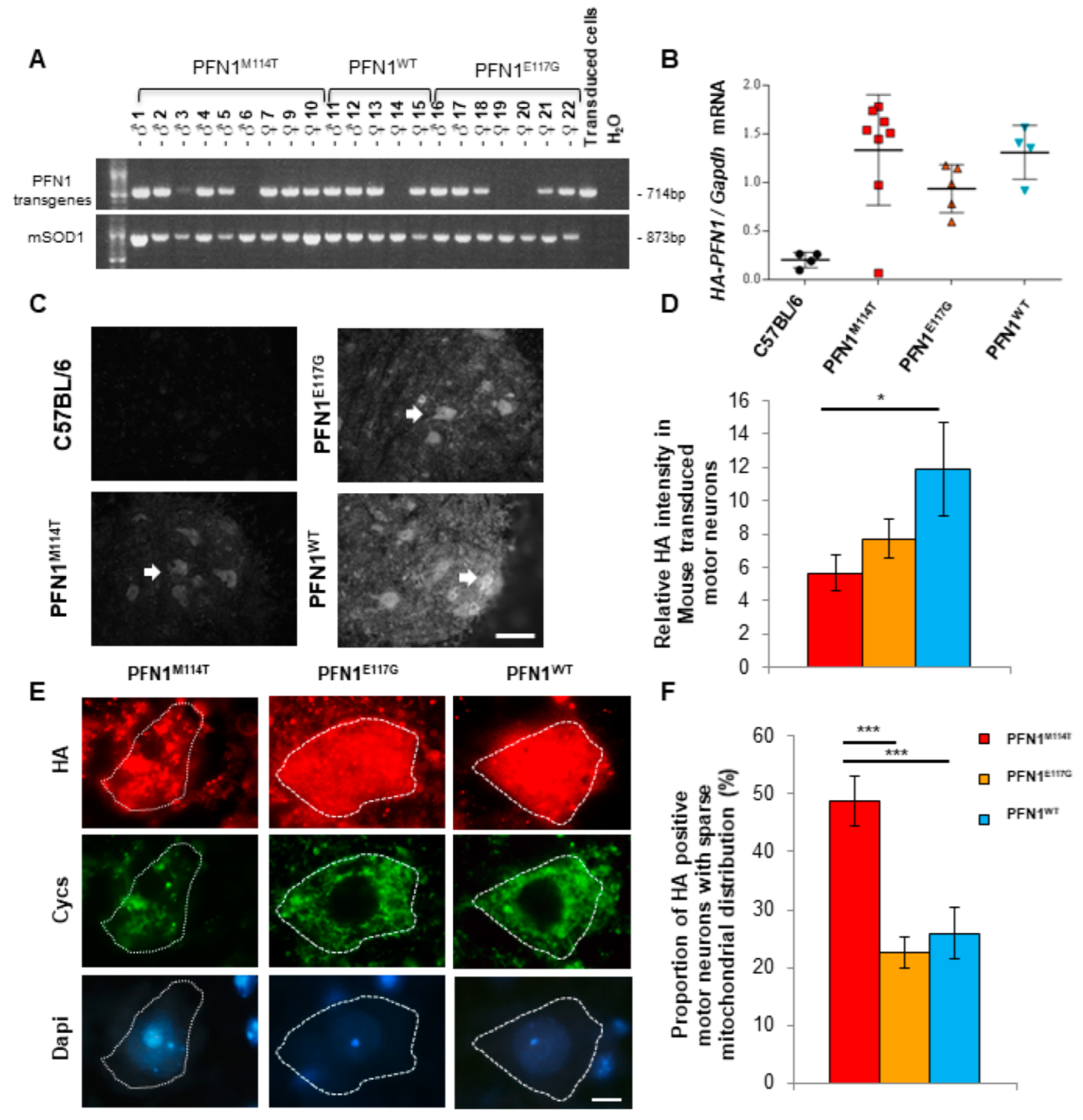
2.7. Expression of PFN1M114T Mutation in Motor Neurons In Vivo Leads to Abnormal Mitochondrial Distribution
3. Discussion
4. Materials and Methods
4.1. Genotyping
4.2. Patient Lymphoblasts
4.3. Semi Quantitative RT-PCR
4.4. Antibodies
4.5. Neuropathology
4.6. Immunoblotting
4.7. Plasmid and Lentiviral Constructs
4.8. Cell Transfection
4.9. Mitochondrial Activity Assays
4.10. Transgenic Mice Establishment and Housing
4.11. Behavioural Tests
4.12. Recordings of ElectroMyoGraphy and Compound Muscle Action Potential (CMAP)
4.13. Mouse Tissue Collection and Preparation
4.14. Immunohistological Analyses
4.15. Cell Counting and Signal Quantification
4.16. Electronic Microscopy Analysis
4.17. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weishaupt, J.H.; Hyman, T.; Dikic, I. Common Molecular Pathways in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Trends Mol. Med. 2016, 22, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Lowe, P.; Koppers, M.; McKenna-Yasek, D.; Baron, D.M.; et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012, 488, 499–503. [Google Scholar] [CrossRef] [PubMed]
- van Blitterswijk, M.; Baker, M.C.; Bieniek, K.F.; Knopman, D.S.; Josephs, K.A.; Boeve, B.; Caselli, R.; Wszolek, Z.K.; Petersen, R.; Graff-Radford, N.R.; et al. Profilin-1 mutations are rare in patients with amyotrophic lateral sclerosis and frontotemporal dementia. Amyotroph. Lateral Scler. Frontotemporal Degener. 2013, 14, 463–469. [Google Scholar] [PubMed] [Green Version]
- Kenna, K.P.; van Doormaal, P.T.; Dekker, A.M.; Ticozzi, N.; Kenna, B.J.; Diekstra, F.P.; van Rheenen, W.; van Eijk, K.R.; Jones, A.R.; Keagle, P.; et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1037–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingre, C.; Landers, J.E.; Rizik, N.; Volk, A.E.; Akimoto, C.; Birve, A.; Hubers, A.; Keagle, P.J.; Piotrowska, K.; Press, R.; et al. A novel phosphorylation site mutation in profilin 1 revealed in a large screen of US, Nordic, and German amyotrophic lateral sclerosis/frontotemporal dementia cohorts. Neurobiol. Aging 2013, 34, 1708.e1–1708.e6. [Google Scholar] [CrossRef]
- Chen, Y.; Zheng, Z.Z.; Huang, R.; Chen, K.; Song, W.; Zhao, B.; Chen, X.; Yang, Y.; Yuan, L.; Shang, H.F. PFN1 mutations are rare in Han Chinese populations with amyotrophic lateral sclerosis. Neurobiol. Aging 2013, 34, 1922.e1–1922.e5. [Google Scholar] [CrossRef]
- Smith, B.N.; Vance, C.; Scotter, E.L.; Troakes, C.; Wong, C.H.; Topp, S.; Maekawa, S.; King, A.; Mitchell, J.C.; Lund, K.; et al. Novel mutations support a role for Profilin 1 in the pathogenesis of ALS. Neurobiol. Aging 2015, 36, 1602.e17–1602.e27. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.Y.; Chen, S.D.; Feng, S.Y.; Liu, C.Y.; Cui, M.; Chen, S.F.; Feng, S.M.; Dong, Q.; Huang, H.; Yu, J.T. Familial flail leg ALS caused by PFN1 mutation. J. Neurol. Neurosurg. Psychiatry 2020, 91, 223–224. [Google Scholar] [CrossRef]
- Chi, J.; Chen, J.; Li, Y.; Huang, Z.; Wang, L.; Zhang, Y. A Familial Phenotypic and Genetic Study of Mutations in PFN1 Associated with Amyotrophic Lateral Sclerosis. Neurosci. Bull. 2020, 36, 174–178. [Google Scholar] [CrossRef]
- Schutt, C.E.; Myslik, J.C.; Rozycki, M.D.; Goonesekere, N.C.; Lindberg, U. The structure of crystalline profilin-beta-actin. Nature 1993, 365, 810–816. [Google Scholar] [CrossRef]
- Metzler, W.J.; Bell, A.J.; Ernst, E.; Lavoie, T.B.; Mueller, L. Identification of the poly-L-proline-binding site on human profilin. J. Biol. Chem. 1994, 269, 4620–4625. [Google Scholar] [CrossRef]
- Kovar, D.R.; Harris, E.S.; Mahaffy, R.; Higgs, H.N.; Pollard, T.D. Control of the assembly of ATP- and ADP-actin by formins and profilin. Cell 2006, 124, 423–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, E.J.; Funes, S.; McKeon, J.E.; Morgan, B.R.; Boopathy, S.; O’Connor, L.C.; Bilsel, O.; Massi, F.; Jegou, A.; Bosco, D.A. ALS-linked PFN1 variants exhibit loss and gain of functions in the context of formin-induced actin polymerization. Proc. Natl. Acad. Sci. USA 2021, 118, e2024605118. [Google Scholar] [CrossRef] [PubMed]
- Boopathy, S.; Silvas, T.V.; Tischbein, M.; Jansen, S.; Shandilya, S.M.; Zitzewitz, J.A.; Landers, J.E.; Goode, B.L.; Schiffer, C.A.; Bosco, D.A. Structural basis for mutation-induced destabilization of profilin 1 in ALS. Proc. Natl. Acad. Sci. USA 2015, 112, 7984–7989. [Google Scholar] [CrossRef] [Green Version]
- Freischmidt, A.; Schopflin, M.; Feiler, M.S.; Fleck, A.K.; Ludolph, A.C.; Weishaupt, J.H. Profilin 1 with the amyotrophic lateral sclerosis associated mutation T109M displays unaltered actin binding and does not affect the actin cytoskeleton. BMC Neurosci. 2015, 1677, 77. [Google Scholar] [CrossRef] [Green Version]
- Del Poggetto, E.; Gori, L.; Chiti, F. Biophysical analysis of three novel profilin-1 variants associated with amyotrophic lateral sclerosis indicates a correlation between their aggregation propensity and the structural features of their globular state. Biol. Chem. 2016, 397, 927–937. [Google Scholar] [CrossRef]
- Nekouei, M.; Ghezellou, P.; Aliahmadi, A.; Arjmand, S.; Kiaei, M.; Ghassempour, A. Changes in biophysical characteristics of PFN1 due to mutation causing amyotrophic lateral sclerosis. Metab. Brain Dis. 2018, 33, 1975–1984. [Google Scholar] [CrossRef]
- Henty-Ridilla, J.L.; Juanes, M.A.; Goode, B.L. Profilin Directly Promotes Microtubule Growth through Residues Mutated in Amyotrophic Lateral Sclerosis. Curr. Biol. 2017, 27, 3535–3543.e4. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nonaka, T.; Suzuki, G.; Kametani, F.; Hasegawa, M. Gain-of-function profilin 1 mutations linked to familial amyotrophic lateral sclerosis cause seed-dependent intracellular TDP-43 aggregation. Hum. Mol. Genet. 2016, 25, 1420–1433. [Google Scholar] [CrossRef] [Green Version]
- Giampetruzzi, A.; Danielson, E.W.; Gumina, V.; Jeon, M.; Boopathy, S.; Brown, R.H.; Ratti, A.; Landers, J.E.; Fallini, C. Modulation of actin polymerization affects nucleocytoplasmic transport in multiple forms of amyotrophic lateral sclerosis. Nat. Commun. 2019, 10, 3827. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.H.; Giampetruzzi, A.; Tran, H.; Fallini, C.; Gao, F.B.; Landers, J.E. A Drosophila model of ALS reveals a partial loss of function of causative human PFN1 mutants. Hum. Mol. Genet. 2017, 26, 2146–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fil, D.; DeLoach, A.; Yadav, S.; Alkam, D.; MacNicol, M.; Singh, A.; Compadre, C.M.; Goellner, J.J.; O’Brien, C.A.; Fahmi, T.; et al. Mutant Profilin1 transgenic mice recapitulate cardinal features of motor neuron disease. Hum. Mol. Genet. 2017, 26, 686–701. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Danielson, E.W.; Qiao, T.; Metterville, J.; Brown, R.H., Jr.; Landers, J.E.; Xu, Z. Mutant PFN1 causes ALS phenotypes and progressive motor neuron degeneration in mice by a gain of toxicity. Proc. Natl. Acad. Sci. USA 2016, 113, E6209–E6218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, G.; Cui, S.; Chen, X.; Song, H.; Huang, C.; Tong, J.; Yuan, Z.; Yu, L.; Xiong, X.; Zhao, J.; et al. Detergent-insoluble inclusion constitutes the first pathology in PFN1 transgenic rats. J. Neurochem. 2021, 157, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Brettle, M.; Stefen, H.; Djordjevic, A.; Fok, S.Y.Y.; Chan, J.W.; van Hummel, A.; van der Hoven, J.; Przybyla, M.; Volkerling, A.; Ke, Y.D.; et al. Developmental Expression of Mutant PFN1 in Motor Neurons Impacts Neuronal Growth and Motor Performance of Young and Adult Mice. Front. Mol. Neurosci. 2019, 12, 231. [Google Scholar] [CrossRef] [PubMed]
- Witke, W.; Sutherland, J.D.; Sharpe, A.; Arai, M.; Kwiatkowski, D.J. Profilin I is essential for cell survival and cell division in early mouse development. Proc. Natl. Acad. Sci. USA 2001, 98, 3832–3836. [Google Scholar] [CrossRef] [Green Version]
- Witke, W. The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol. 2004, 14, 461–469. [Google Scholar] [CrossRef]
- Figley, M.D.; Bieri, G.; Kolaitis, R.M.; Taylor, J.P.; Gitler, A.D. Profilin 1 associates with stress granules and ALS-linked mutations alter stress granule dynamics. J. Neurosci. 2014, 34, 8083–8097. [Google Scholar] [CrossRef] [Green Version]
- Elden, A.C.; Kim, H.J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Witke, W.; Podtelejnikov, A.V.; Di Nardo, A.; Sutherland, J.D.; Gurniak, C.B.; Dotti, C.; Mann, M. In mouse brain profilin I and profilin II associate with regulators of the endocytic pathway and actin assembly. EMBO J. 1998, 17, 967–976. [Google Scholar] [CrossRef] [Green Version]
- Ju, J.S.; Fuentealba, R.A.; Miller, S.E.; Jackson, E.; Piwnica-Worms, D.; Baloh, R.H.; Weihl, C.C. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 2009, 187, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tresse, E.; Salomons, F.A.; Vesa, J.; Bott, L.C.; Kimonis, V.; Yao, T.P.; Dantuma, N.P.; Taylor, J.P. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 2010, 6, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, A.H.; Manna, S.K. Profilin-PTEN interaction suppresses NF-kappaB activation via inhibition of IKK phosphorylation. Biochem. J. 2016, 473, 859–872. [Google Scholar] [CrossRef]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2001, 276, 35243–35246. [Google Scholar] [CrossRef] [Green Version]
- Seilhean, D.; Cazeneuve, C.; Thuries, V.; Russaouen, O.; Millecamps, S.; Salachas, F.; Meininger, V.; Leguern, E.; Duyckaerts, C. Accumulation of TDP-43 and alpha-actin in an amyotrophic lateral sclerosis patient with the K17I ANG mutation. Acta Neuropathol. 2009, 118, 561–573. [Google Scholar] [CrossRef] [Green Version]
- Seilhean, D.; Takahashi, J.; El Hachimi, K.H.; Fujigasaki, H.; Lebre, A.S.; Biancalana, V.; Durr, A.; Salachas, F.; Hogenhuis, J.; de The, H.; et al. Amyotrophic lateral sclerosis with neuronal intranuclear protein inclusions. Acta Neuropathol. 2004, 108, 81–87. [Google Scholar] [CrossRef]
- Das, T.; Bae, Y.H.; Wells, A.; Roy, P. Profilin-1 overexpression upregulates PTEN and suppresses AKT activation in breast cancer cells. J. Cell. Physiol. 2009, 218, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S. Biological Roles of Alternative Autophagy. Mol. Cells 2018, 41, 50–54. [Google Scholar]
- Shimizu, S.; Arakawa, S.; Nishida, Y. Autophagy takes an alternative pathway. Autophagy 2010, 6, 290–291. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Arakawa, S.; Fujitani, K.; Yamaguchi, H.; Mizuta, T.; Kanaseki, T.; Komatsu, M.; Otsu, K.; Tsujimoto, Y.; Shimizu, S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009, 461, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Chen, S.; Huang, K.X.; Le, W.D. Why should autophagic flux be assessed? Acta Pharmacol. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maskey, D.; Yousefi, S.; Schmid, I.; Zlobec, I.; Perren, A.; Friis, R.; Simon, H.U. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat. Commun. 2013, 4, 2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, A.H.; Hart, P.D.; Young, M.R. Ammonia inhibits phagosome-lysosome fusion in macrophages. Nature 1980, 286, 79–80. [Google Scholar] [CrossRef]
- Shimizu, S.; Honda, S.; Arakawa, S.; Yamaguchi, H. Alternative macroautophagy and mitophagy. Int. J. Biochem. Cell Biol. 2014, 50, 64–66. [Google Scholar] [CrossRef]
- Hirota, Y.; Yamashita, S.; Kurihara, Y.; Jin, X.; Aihara, M.; Saigusa, T.; Kang, D.; Kanki, T. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 2015, 11, 332–343. [Google Scholar] [CrossRef] [Green Version]
- Hansen, M.B.; Nielsen, S.E.; Berg, K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Methods 1989, 119, 203–210. [Google Scholar] [CrossRef]
- Dussaud, S.; Pardanaud-Glavieux, C.; Sauty-Colace, C.; Ravassard, P. Lentiviral Mediated Production of Transgenic Mice: A Simple and Highly Efficient Method for Direct Study of Founders. J. Vis. Exp. 2018, 7, e57609. [Google Scholar]
- Kennel, P.F.; Finiels, F.; Revah, F.; Mallet, J. Neuromuscular function impairment is not caused by motor neurone loss in FALS mice: An electromyographic study. Neuroreport 1996, 7, 1427–1431. [Google Scholar] [PubMed]
- Azzouz, M.; Leclerc, N.; Gurney, M.; Warter, J.M.; Poindron, P.; Borg, J. Progressive motor neuron impairment in an animal model of familial amyotrophic lateral sclerosis. Muscle Nerve 1997, 20, 45–51. [Google Scholar] [CrossRef]
- Fratta, P.; Charnock, J.; Collins, T.; Devoy, A.; Howard, R.; Malaspina, A.; Orrell, R.; Sidle, K.; Clarke, J.; Shoai, M.; et al. Profilin1 E117G is a moderate risk factor for amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2014, 85, 506–508. [Google Scholar] [CrossRef]
- Yang, S.; Fifita, J.A.; Williams, K.L.; Warraich, S.T.; Pamphlett, R.; Nicholson, G.A.; Blair, I.P. Mutation analysis and immunopathological studies of PFN1 in familial and sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2013, 34, 2235.e7–2235.e10. [Google Scholar] [CrossRef]
- Tiloca, C.; Ticozzi, N.; Pensato, V.; Corrado, L.; Del Bo, R.; Bertolin, C.; Fenoglio, C.; Gagliardi, S.; Calini, D.; Lauria, G.; et al. Screening of the PFN1 gene in sporadic amyotrophic lateral sclerosis and in frontotemporal dementia. Neurobiol. Aging 2013, 34, 1517.e9–1517.e10. [Google Scholar] [CrossRef] [Green Version]
- Greenway, M.J.; Andersen, P.M.; Russ, C.; Ennis, S.; Cashman, S.; Donaghy, C.; Patterson, V.; Swingler, R.; Kieran, D.; Prehn, J.; et al. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat. Genet. 2006, 38, 411–413. [Google Scholar] [CrossRef]
- Millecamps, S.; Boillee, S.; Le Ber, I.; Seilhean, D.; Teyssou, E.; Giraudeau, M.; Moigneu, C.; Vandenberghe, N.; Danel-Brunaud, V.; Corcia, P.; et al. Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J. Med. Genet. 2012, 49, 258–263. [Google Scholar] [CrossRef]
- Cady, J.; Allred, P.; Bali, T.; Pestronk, A.; Goate, A.; Miller, T.M.; Mitra, R.D.; Ravits, J.; Harms, M.B.; Baloh, R.H. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann. Neurol. 2015, 77, 100–113. [Google Scholar] [CrossRef] [Green Version]
- Lattante, S.; Ciura, S.; Rouleau, G.A.; Kabashi, E. Defining the genetic connection linking amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD). Trends Genet. 2015, 31, 263–273. [Google Scholar] [CrossRef]
- van Blitterswijk, M.; van Es, M.A.; Hennekam, E.A.; Dooijes, D.; van Rheenen, W.; Medic, J.; Bourque, P.R.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 3776–3784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Vande Velde, C.; McDonald, K.K.; Boukhedimi, Y.; McAlonis-Downes, M.; Lobsiger, C.S.; Bel Hadj, S.; Zandona, A.; Julien, J.P.; Shah, S.B.; Cleveland, D.W. Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS ONE 2011, 6, e22031. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Destroismaisons, L.; Peyrard, S.L.; Cadot, S.; Rouleau, G.A.; Brown, R.H., Jr.; Julien, J.P.; Arbour, N.; Vande Velde, C. Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Hum. Mol. Genet. 2013, 22, 3947–3959. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Wong, Y.C.; Simpson, C.L.; Holzbaur, E.L. Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat. Commun. 2016, 7, 12886. [Google Scholar] [CrossRef]
- Gautam, M.; Xie, E.F.; Kocak, N.; Ozdinler, P.H. Mitoautophagy: A Unique Self-Destructive Path Mitochondria of Upper Motor Neurons With TDP-43 Pathology Take, Very Early in ALS. Front. Cell. Neurosci. 2019, 13, 489. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, F.; Winkler, U.; Morawski, M.; Jager, C.; Reinecke, L.; Rossner, M.J.; Hirrlinger, P.G.; Hirrlinger, J. The human ubiquitin C promoter drives selective expression in principal neurons in the brain of a transgenic mouse line. Neurochem. Int. 2011, 59, 976–980. [Google Scholar] [CrossRef]
- de Carvalho, M.; Dengler, R.; Eisen, A.; England, J.D.; Kaji, R.; Kimura, J.; Mills, K.; Mitsumoto, H.; Nodera, H.; Shefner, J.; et al. Electrodiagnostic criteria for diagnosis of ALS. Clin. Neurophysiol. 2008, 119, 497–503. [Google Scholar] [CrossRef]
- Aguilera, M.O.; Beron, W.; Colombo, M.I. The actin cytoskeleton participates in the early events of autophagosome formation upon starvation induced autophagy. Autophagy 2012, 8, 1590–1603. [Google Scholar] [CrossRef] [Green Version]
- Izdebska, M.; Zielinska, W.; Halas-Wisniewska, M.; Grzanka, A. Involvement of Actin in Autophagy and Autophagy-Dependent Multidrug Resistance in Cancer. Cancers 2019, 11, 1209. [Google Scholar] [CrossRef] [Green Version]
- Vande Velde, C.; Miller, T.M.; Cashman, N.R.; Cleveland, D.W. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 4022–4027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiazzi, M.; D’Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Holzbaur, E.L. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granatiero, V.; Manfredi, G. Mitochondrial Transport and Turnover in the Pathogenesis of Amyotrophic Lateral Sclerosis. Biology 2019, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; van Swieten, J.C.; Seelaar, H.; Dopper, E.G.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134 Pt 9, 2456–2477. [Google Scholar] [CrossRef]
- Millecamps, S.; Salachas, F.; Cazeneuve, C.; Gordon, P.; Bricka, B.; Camuzat, A.; Guillot-Noel, L.; Russaouen, O.; Bruneteau, G.; Pradat, P.F.; et al. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: Genotype-phenotype correlations. J. Med. Genet. 2010, 47, 554–560. [Google Scholar] [CrossRef]
- Millecamps, S.; Boillee, S.; Chabrol, E.; Camu, W.; Cazeneuve, C.; Salachas, F.; Pradat, P.F.; Danel-Brunaud, V.; Vandenberghe, N.; Corcia, P.; et al. Screening of OPTN in French familial amyotrophic lateral sclerosis. Neurobiol. Aging 2011, 32, 557.e11–557.e13. [Google Scholar] [CrossRef]
- Millecamps, S.; Da Barroca, S.; Cazeneuve, C.; Salachas, F.; Pradat, P.F.; Danel-Brunaud, V.; Vandenberghe, N.; Lacomblez, L.; Le Forestier, N.; Bruneteau, G.; et al. Questioning on the role of D amino acid oxidase in familial amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2010, 107, E107. [Google Scholar] [CrossRef] [Green Version]
- Millecamps, S.; De Septenville, A.; Teyssou, E.; Daniau, M.; Camuzat, A.; Albert, M.; LeGuern, E.; Galimberti, D.; Brice, A.; Marie, Y.; et al. Genetic analysis of matrin 3 gene in French amyotrophic lateral sclerosis patients and frontotemporal lobar degeneration with amyotrophic lateral sclerosis patients. Neurobiol. Aging 2014, 35, 2882.e13–2882.e15. [Google Scholar] [CrossRef]
- Teyssou, E.; Takeda, T.; Lebon, V.; Boillee, S.; Doukoure, B.; Bataillon, G.; Sazdovitch, V.; Cazeneuve, C.; Meininger, V.; LeGuern, E.; et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: Genetics and neuropathology. Acta Neuropathol. 2013, 125, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Teyssou, E.; Vandenberghe, N.; Moigneu, C.; Boillee, S.; Couratier, P.; Meininger, V.; Pradat, P.F.; Salachas, F.; Leguern, E.; Millecamps, S. Genetic analysis of SS18L1 in French amyotrophic lateral sclerosis. Neurobiol. Aging 2014, 35, 1213.e9–1213.e12. [Google Scholar] [CrossRef] [PubMed]
- Schorpp, M.; Jager, R.; Schellander, K.; Schenkel, J.; Wagner, E.F.; Weiher, H.; Angel, P. The human ubiquitin C promoter directs high ubiquitous expression of transgenes in mice. Nucleic Acids Res. 1996, 24, 1787–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zennou, V.; Serguera, C.; Sarkis, C.; Colin, P.; Perret, E.; Mallet, J.; Charneau, P. The HIV-1 DNA flap stimulates HIV vector-mediated cell transduction in the brain. Nat. Biotechnol. 2001, 19, 446–450. [Google Scholar] [CrossRef]
- Cashman, N.R.; Durham, H.D.; Blusztajn, J.K.; Oda, K.; Tabira, T.; Shaw, I.T.; Dahrouge, S.; Antel, J.P. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 1992, 194, 209–221. [Google Scholar] [CrossRef]
- Pike Winer, L.S.; Wu, M. Rapid analysis of glycolytic and oxidative substrate flux of cancer cells in a microplate. PLoS ONE 2014, 9, e109916. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Barneoud, P.; Lolivier, J.; Sanger, D.J.; Scatton, B.; Moser, P. Quantitative motor assessment in FALS mice: A longitudinal study. Neuroreport 1997, 8, 2861–2865. [Google Scholar] [CrossRef]
- Evans, T.M.; Van Remmen, H.; Purkar, A.; Mahesula, S.; Gelfond, J.A.; Sabia, M.; Qi, W.; Lin, A.L.; Jaramillo, C.A.; Haskins, W.E. Microwave & Magnetic (M(2)) Proteomics of a Mouse Model of Mild Traumatic Brain Injury. Transl. Proteom. 2014, 3, 10–21. [Google Scholar]
- Reynolds, E.S. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 1963, 17, 208–212. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teyssou, E.; Chartier, L.; Roussel, D.; Perera, N.D.; Nemazanyy, I.; Langui, D.; Albert, M.; Larmonier, T.; Saker, S.; Salachas, F.; et al. The Amyotrophic Lateral Sclerosis M114T PFN1 Mutation Deregulates Alternative Autophagy Pathways and Mitochondrial Homeostasis. Int. J. Mol. Sci. 2022, 23, 5694. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105694
Teyssou E, Chartier L, Roussel D, Perera ND, Nemazanyy I, Langui D, Albert M, Larmonier T, Saker S, Salachas F, et al. The Amyotrophic Lateral Sclerosis M114T PFN1 Mutation Deregulates Alternative Autophagy Pathways and Mitochondrial Homeostasis. International Journal of Molecular Sciences. 2022; 23(10):5694. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105694
Chicago/Turabian StyleTeyssou, Elisa, Laura Chartier, Delphine Roussel, Nirma D. Perera, Ivan Nemazanyy, Dominique Langui, Mélanie Albert, Thierry Larmonier, Safaa Saker, François Salachas, and et al. 2022. "The Amyotrophic Lateral Sclerosis M114T PFN1 Mutation Deregulates Alternative Autophagy Pathways and Mitochondrial Homeostasis" International Journal of Molecular Sciences 23, no. 10: 5694. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105694