
Delayed Ventricular Repolarization and Sodium Channel Current Modification in a Mouse Model of Rett Syndrome


Abstract
:1. Introduction
2. Results
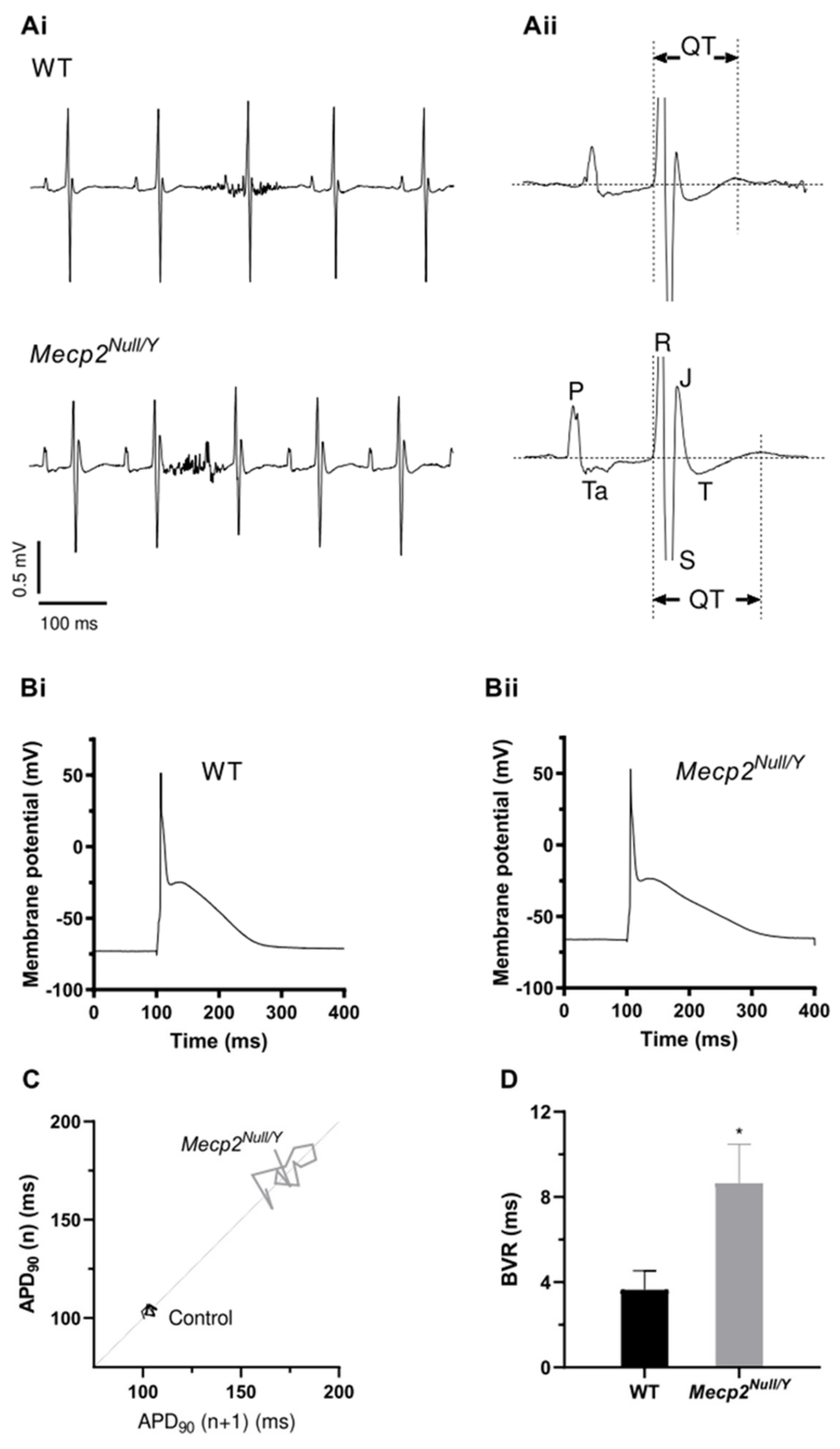
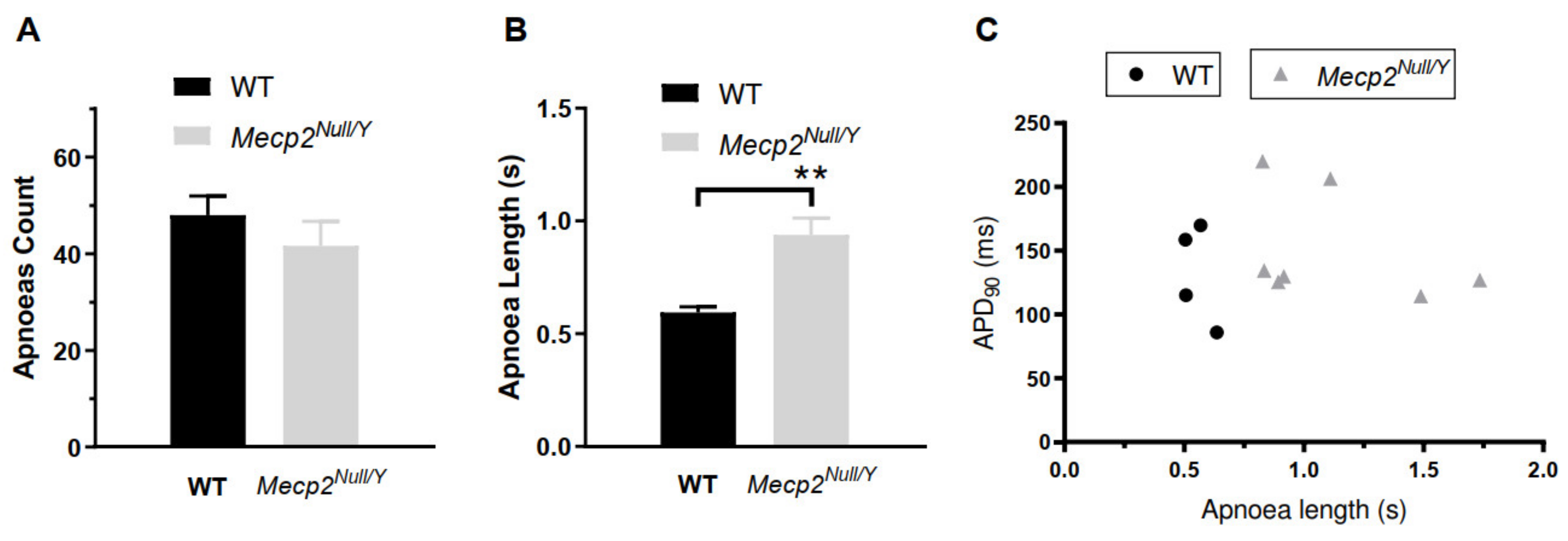
2.1. Delayed Ventricular Repolarization in Mecp2Null/Y Mice
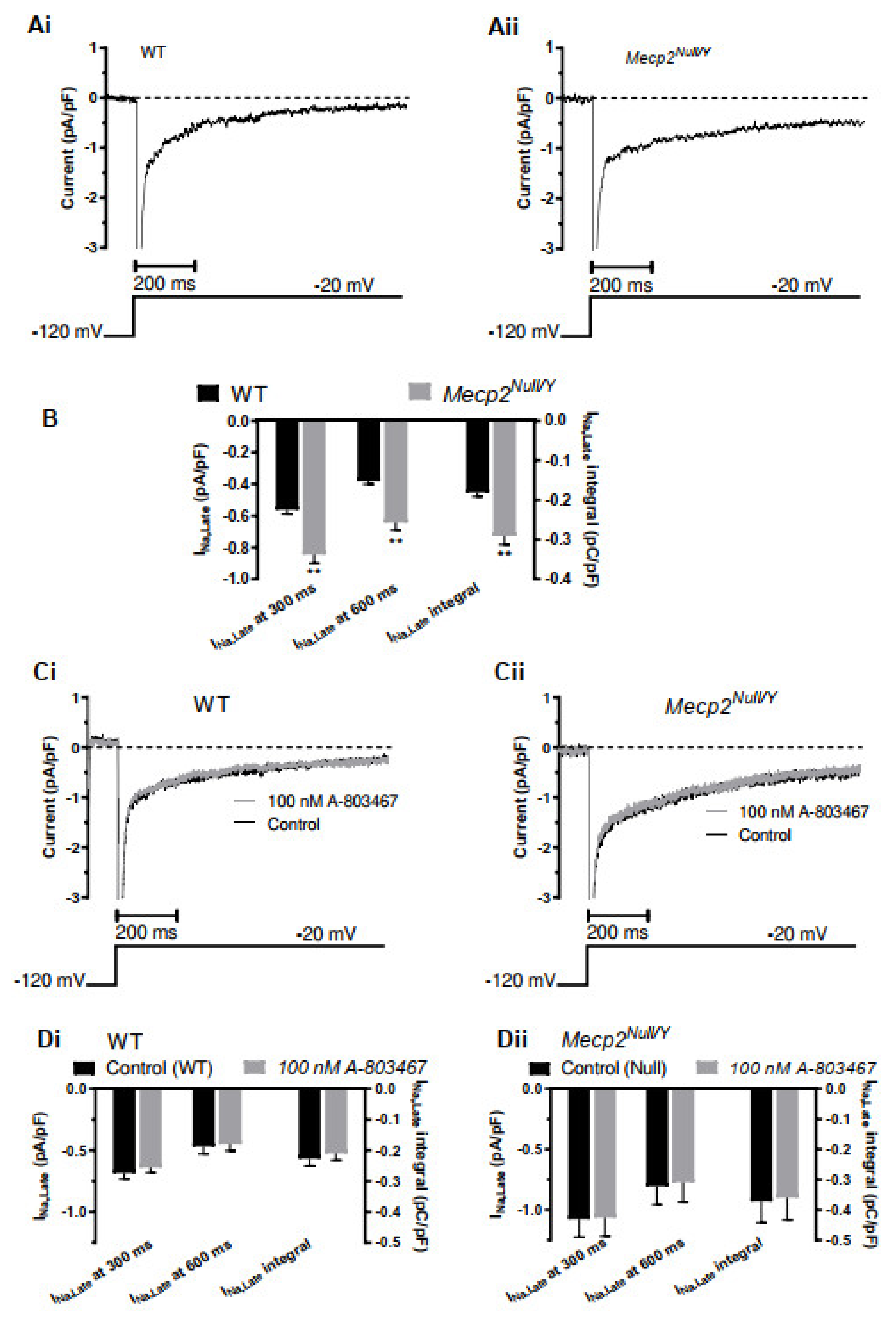
2.2. Late Sodium Current, INa,Late, Enhancement in Mecp2Null/Y Mice
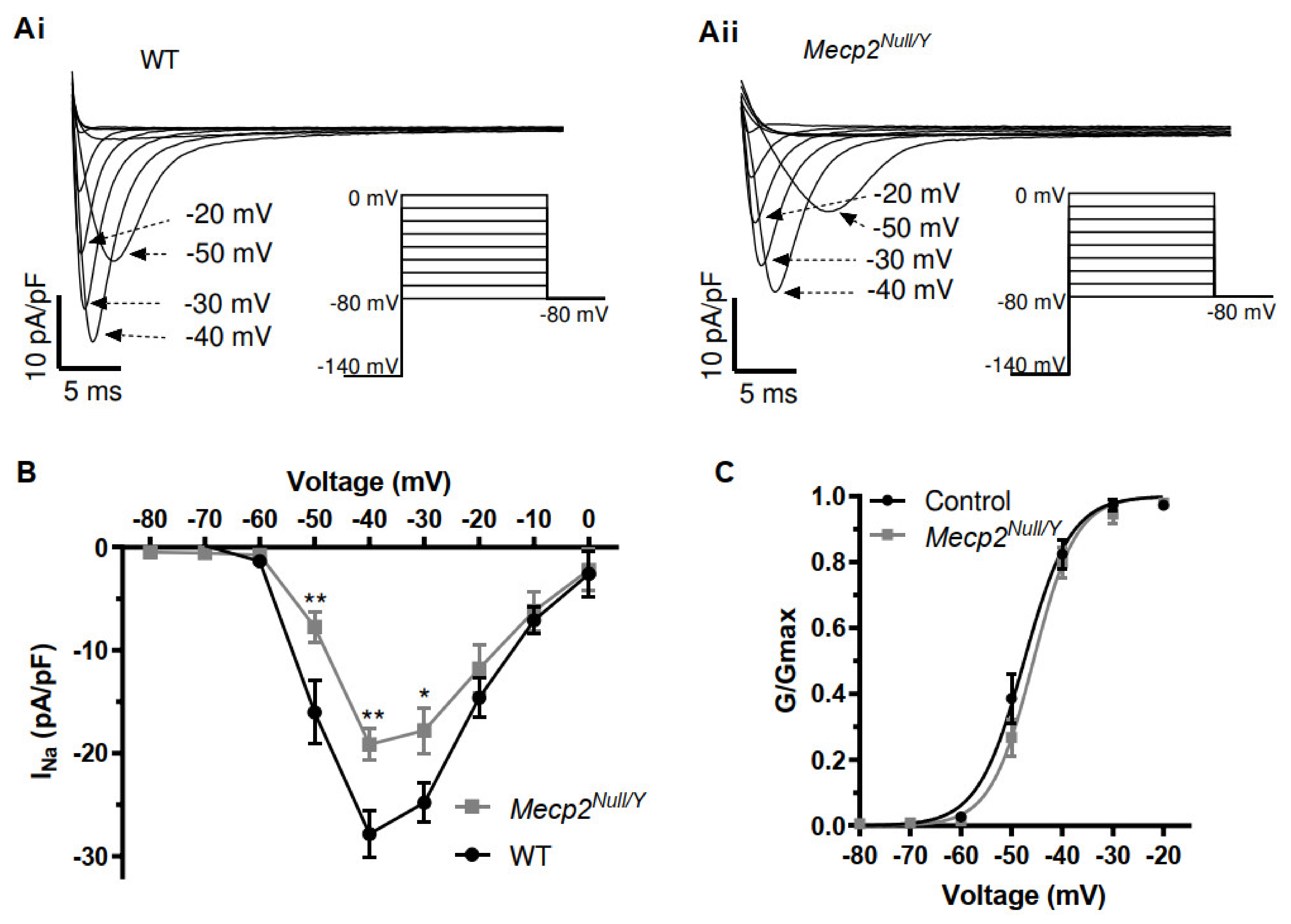
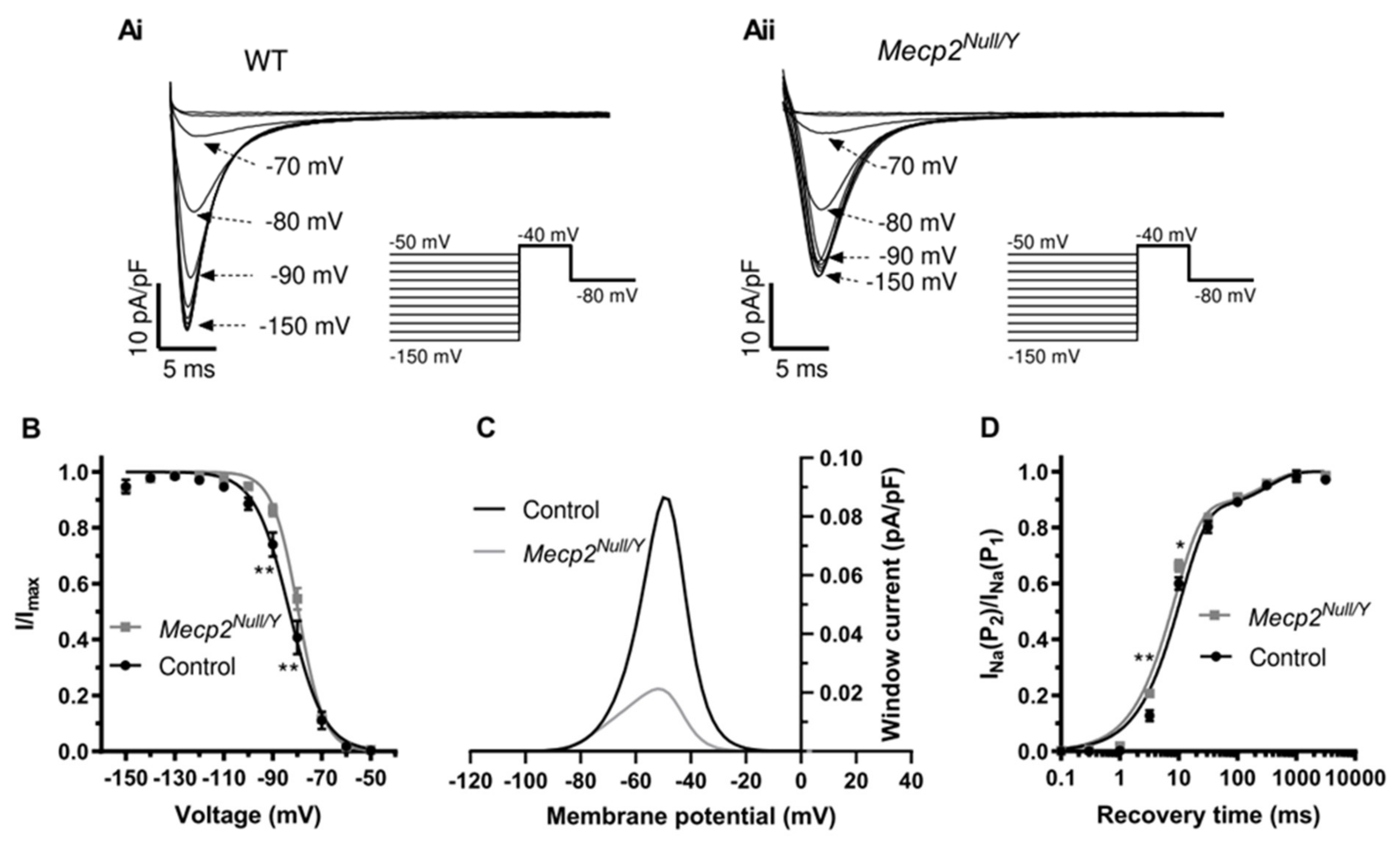
2.3. Alterations to Fast INa in Mecp2Null/Y Mice
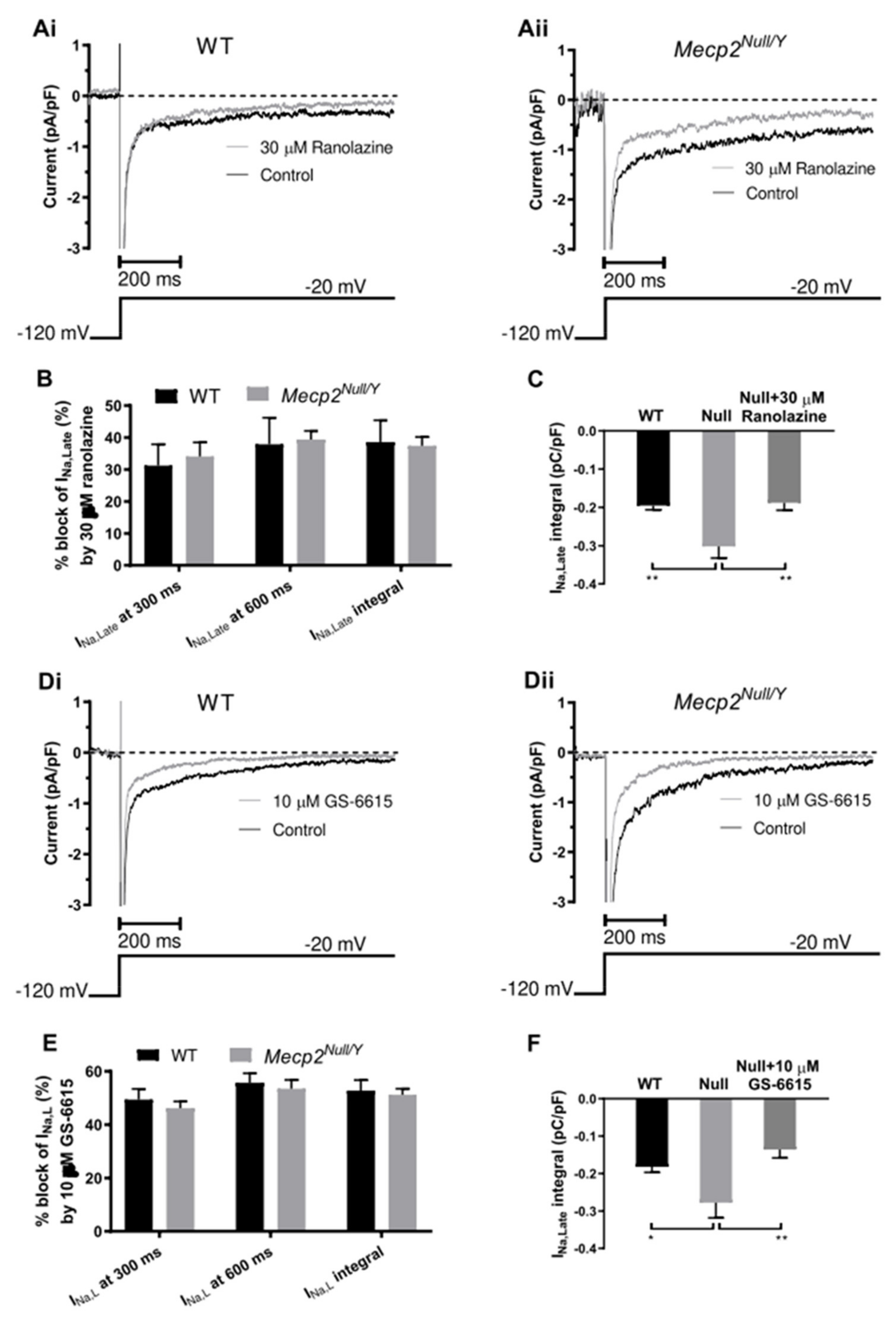
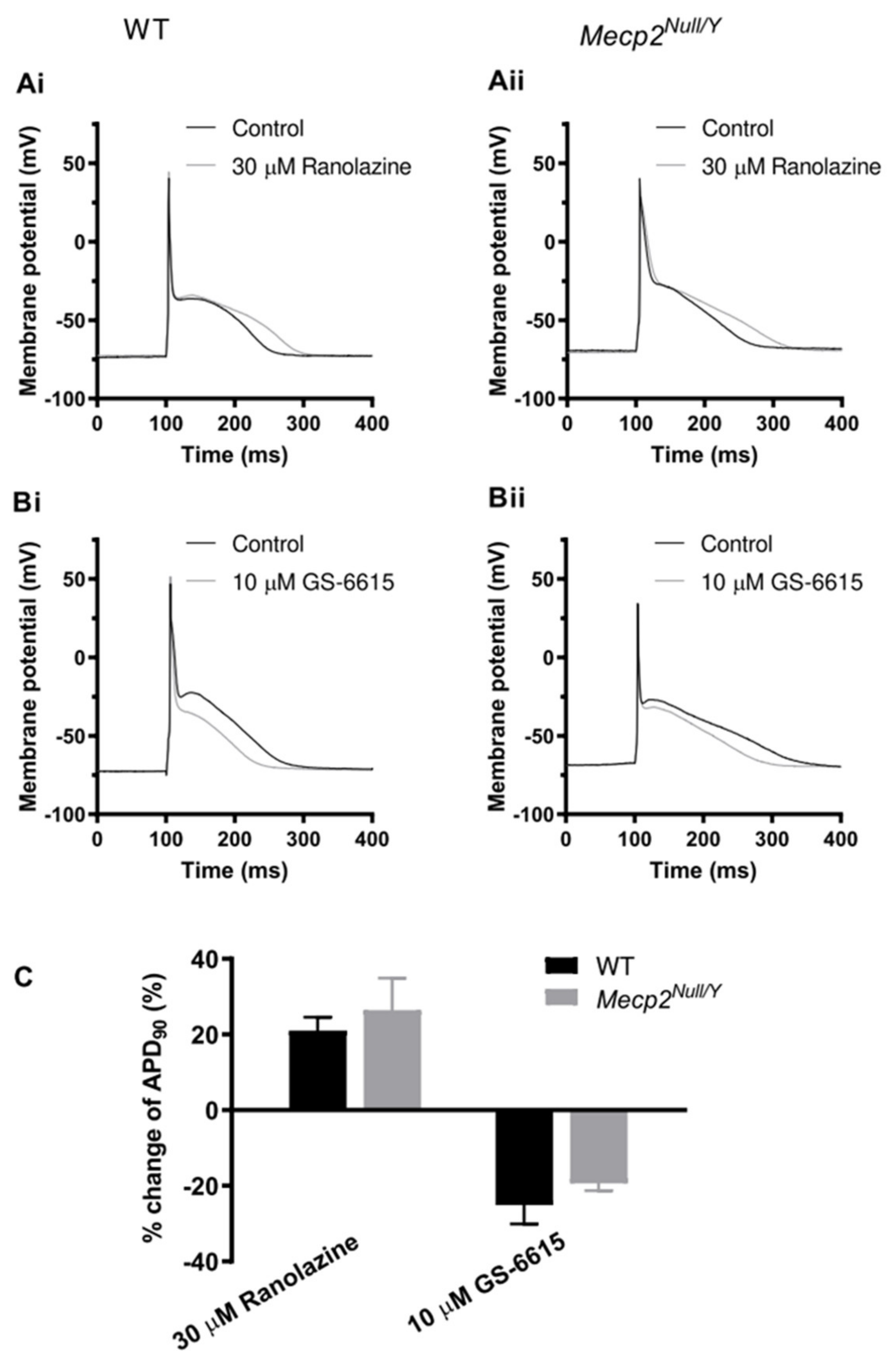
2.4. Effects of the INa,Late Inhibitors Ranolazine and Eleclazine (GS-6615)
3. Discussion
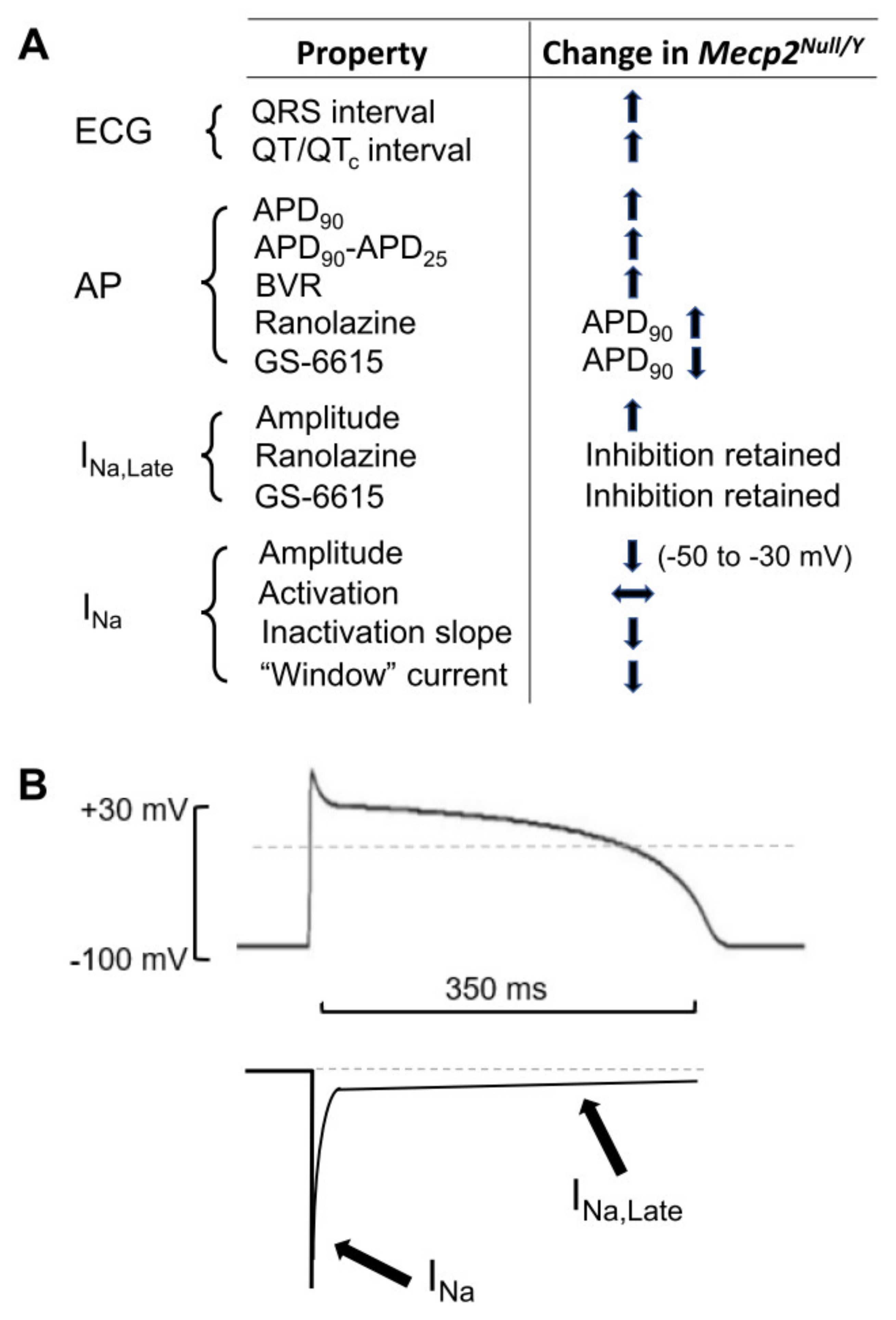
3.1. Summary of Main Findings
3.2. QT Interval and AP Prolongation
3.3. Changes to Fast INa and INa,Late
3.4. Actions of Ranolazine and GS-6615
3.5. Limitations and Conclusions
4. Materials and Methods
4.1. Mouse Model of RTT Used
4.2. Electrocardiogram (ECG) Measurement
4.3. Unrestrained Whole Body Plethysmography
4.4. Ventricular Myocyte Isolation
4.5. Cellular Electrophysiology
4.6. Action Potential (AP) Measurement
4.7. INa,Late Measurement
4.8. INa Measurement
4.9. Data Analysis and Statistics
4.10. Drugs
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rett, A. On a remarkable syndrome of cerebral atrophy associated with hyperammonaemia in childhood. Wien. Med. Wochenschr. 2016, 166, 322–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rett, A. [Hyperammonaemia and cerebral atrophy in childhood]. Folia Hered. Pathol. 1969, 18, 115–123. [Google Scholar]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.S.; Schanen, N.C.; Zappella, M.; et al. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liyanage, V.R.; Rastegar, M. Rett syndrome and MeCP2. Neuromolecular. Med. 2014, 16, 231–264. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, J.-M.; Ward, C.S.; Neul, J.L. Breathing challenges in Rett Syndrome: Lessons learned from humans and animal models. Respir. Physiol. Neurobiol. 2013, 189, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Amir, R.E.; Van, V.I.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Kim, S.J.; Cook, E.H., Jr. Novel de novo nonsense mutation of MECP2 in a patient with Rett syndrome. Hum. Mutat. 2000, 15, 382–383. [Google Scholar] [CrossRef]
- Huppke, P.; Laccone, F.; Kramer, N.; Engel, W.; Hanefeld, F. Rett syndrome: Analysis of MECP2 and clinical characterization of 31 patients. Hum. Mol. Genet. 2000, 22, 1369–1375. [Google Scholar] [CrossRef]
- Bienvenu, T.; Carrié, A.; de Roux, N.; Vinet, M.-C.; Jonveaux, P.; Couvert, P.; Villard, L.; Arzimanoglou, A.; Beldjord, C.; Fontes, M.; et al. MECP2 mutations account for most cases of typical forms of Rett syndrome. Hum. Mol. Genet. 2000, 9, 1377–1384. [Google Scholar] [CrossRef]
- Neul, J.L.; Fang, P.; Barrish, J.; Lane, J.; Caeg, E.B.; Smith, E.O.; Zoghbi, H.; Percy, A.; Glaze, D.G. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008, 70, 1313–1321. [Google Scholar] [CrossRef] [Green Version]
- Moog, U.; Smeets, E.E.; van Roozendaal, K.E.; Schoenmakers, S.; Herbergs, J.; Schoonbrood-Lenssen, A.M.; Schrander-Stumpel, C.T. Neurodevelopmental disorders in males related to the gene causing Rett syndrome in females (MECP2). Eur. J. Paediatr. Neurol. 2003, 7, 5–12. [Google Scholar] [CrossRef]
- Kerr, A.M.; Armstrong, D.D.; Prescott, R.J.; Doyle, D.; Kearney, D.L. Rett syndrome: Analysis of deaths in the British survey. Eur. Child Adolesc. Psychiatry 1997, 6, 71–74. [Google Scholar] [PubMed]
- Sekul, E.A.; Moak, J.P.; Schultz, R.J.; Glaze, D.G.; Dunn, J.K.; Percy, A.K. Electrocardiographic findings in Rett syndrome: An explanation for sudden death? J. Pediatr. 1994, 125, 80–82. [Google Scholar] [CrossRef]
- Ellaway, C.J.; Sholler, G.; Leonard, H.; Christodoulou, J. Prolonged QT interval in Rett syndrome. Arch. Dis. Child 1999, 80, 470–472. [Google Scholar] [CrossRef] [Green Version]
- Guideri, F.; Acampa, M.; DiPerri, T.; Zappella, M.; Hayek, Y. Progressive cardiac dysautonomia observed in patients affected by classic Rett syndrome and not in the preserved speech variant. J. Child Neurol. 2001, 16, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Crosson, J.; Srivastava, S.; Bibat, G.M.; Gupta, S.; Kantipuly, A.; Smith-Hicks, C.; Myers, S.M.; Sanyal, A.; Yenokyan, G.; Brenner, J.; et al. Evaluation of QTc in Rett syndrome: Correlation with age, severity, and genotype. Am. J. Med. Genet. Part A 2017, 173, 1495–1501. [Google Scholar] [CrossRef]
- Clark, B.C.; Kopp, A.; Morey, W.; Djukic, A. Serial follow-up of corrected QT interval in Rett syndrome. Dev. Med. Child Neurol. 2020, 62, 833–836. [Google Scholar] [CrossRef]
- McCauley, M.D.; Wang, T.; Mike, E.; Herrera, J.; Beavers, D.L.; Huang, T.-W.; Ward, C.S.; Skinner, S.; Percy, A.K.; Glaze, D.G.; et al. Pathogenesis of Lethal Cardiac Arrhythmias in Mecp2 Mutant Mice: Implication for Therapy in Rett Syndrome. Sci. Transl. Med. 2011, 3, 113ra125. [Google Scholar] [CrossRef] [Green Version]
- Herrera, J.A.; Ward, C.S.; Pitcher, M.R.; Percy, A.K.; Skinner, S.; Kaufmann, W.E.; Glaze, D.G.; Wehrens, X.H.T.; Neul, J.L. Treatment of cardiac arrhythmias in a mouse model of Rett syndrome with Na+-channel-blocking antiepileptic drugs. Dis. Model. Mech. 2015, 8, 363–371. [Google Scholar]
- Mucerino, S.; Di Salle, A.; Alessio, N.; Margarucci, S.; Nicolai, R.; Melone, M.A.B.; Galderisi, U.; Peluso, G. Alterations in the carnitine cycle in a mouse model of Rett syndrome. Sci. Rep. 2017, 7, 41824. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yu, J.; Niu, Y.; Qin, D.; Liu, H.; Li, G.; Hu, Y.; Wang, J.; Lu, Y.; Kang, Y.; et al. Modeling Rett Syndrome Using TALEN-Edited MECP2 Mutant Cynomolgus Monkeys. Cell 2017, 169, 945–955.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapleau, C.A.; Lane, J.; Larimore, J.; Li, W.; Pozzo-Miller, L.; Percy, A.K. Recent progress in Rett syndrome and MECP2 dysfunction: Assessment of potential treatment options. Futur. Neurol. 2013, 8, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of Neurological Defects in a Mouse Model of Rett Syndrome. Science 2007, 315, 1143–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, M.; Takahashi, T.; Mitsumasu, C.; Igata, S.; Takano, M.; Minami, T.; Yasukawa, H.; Okayama, H.; Nakamura, K.; Okabe, Y.; et al. Disturbance of cardiac gene expression and cardiomyocyte structure predisposes Mecp2-null mice to arrhythmias. Sci. Rep. 2015, 5, 11204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, G.F.; Jeron, A.; Koren, G. Measurement of heart rate and Q-T interval in the conscious mouse. Am. J. Physiol. 1998, 274, H747–H751. [Google Scholar] [CrossRef]
- Speerschneider, T.; Thomsen, M.B. Physiology and analysis of the electrocardiographic T wave in mice. Acta Physiol. 2013, 209, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Abdala, A.P.; Lioy, D.T.; Garg, S.K.; Knopp, S.J.; Paton, J.F.R.; Bissonnette, J.M. Effect of Sarizotan, a 5-HT1aand D2-Like Receptor Agonist, on Respiration in Three Mouse Models of Rett Syndrome. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Varró, A.; Tomek, J.; Nagy, N.; Virág, L.; Passini, E.; Rodriguez, B.; Baczkó, I. Cardiac transmembrane ion channels and action potentials: Cellular physiology and arrhythmogenic behavior. Physiol. Rev. 2021, 101, 1083–1176. [Google Scholar] [CrossRef]
- Derangeon, M.; Montnach, J.; Baró, I.; Charpentier, F. Mouse Models of SCN5A-Related Cardiac Arrhythmias. Front. Physiol. 2012, 3, 210. [Google Scholar] [CrossRef] [Green Version]
- Zaza, A. [The late sodium current: Pathophysiology and pharmacology of a new therapeu-tic target]. G. Ital. Cardiol. 2011, 10, 3S–11S. [Google Scholar]
- Yang, T.; Atack, T.C.; Stroud, D.M.; Zhang, W.; Hall, L.; Roden, D.M. Blocking Scn10a Channels in Heart Reduces Late Sodium Current and Is Antiarrhythmic. Circ. Res. 2012, 111, 322–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuill, K.H.; Convery, M.K.; Dooley, P.C.; Doggrell, S.A.; Hancox, J.C. Effects of BDF 9198 on action potentials and ionic currents from guinea-pig isolated ventricular myocytes. J. Cereb. Blood Flow Metab. 2000, 130, 1753–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, C.I.; Yuill, K.H.; Borg, J.J.; Hancox, J.C.; Kozlowski, R.Z. Actions of pyrethroid insecticides on sodium currents, action potentials and contractile rhythm in isolated mammalian ventricular myocytes and perfused hearts. J. Pharmacol. Exp. Ther. 2001, 298, 1067–1082. [Google Scholar] [PubMed]
- Caves, R.E.; Carpenter, A.; Choisy, S.C.; Clennell, B.; Cheng, H.; McNiff, C.; Mann, B.; Milnes, J.T.; Hancox, J.C.; James, A.F. Inhibition of voltage-gated Na+ currents by eleclazine in rat atrial and ventricular myocytes. Heart Rhythm O2 2020, 1, 206–214. [Google Scholar] [CrossRef]
- Hancox, J.C.; Doggrell, S.A. Perspective: Does ranolazine have potential for the treatment of atrial fibrillation? Expert Opin. Investig. Drugs 2010, 19, 1465–1474. [Google Scholar] [CrossRef]
- Dhalla, A.K.; Wang, W.-Q.; Dow, J.; Shryock, J.C.; Belardinelli, L.; Bhandari, A.; Kloner, R.A. Ranolazine, an antianginal agent, markedly reduces ventricular arrhythmias induced by ischemia and ischemia-reperfusion. Am. J. Physiol. Circ. Physiol. 2009, 297, H1923–H1929. [Google Scholar] [CrossRef] [Green Version]
- Murdock, D.K.; Kersten, M.; Kaliebe, J.; Larrain, G. The Use Of Oral Ranolazine To Convert New Or Paroxysmal Atrial Fibrillation: A Review Of Experience With Implications For Possible “Pill In The Pocket” Approach To Atrial Fibrillation. Indian Pacing Electrophysiol. J. 2009, 9, 260–267. [Google Scholar]
- Sicouri, S.; Glass, A.; Belardinelli, L.; Antzelevitch, C. Antiarrhythmic effects of ranolazine in canine pulmonary vein sleeve preparations. Heart Rhythm 2008, 5, 1019–1026. [Google Scholar] [CrossRef] [Green Version]
- Caves, R.E.; Cheng, H.; Choisy, S.C.; Gadeberg, H.C.; Bryant, S.M.; Hancox, J.C.; James, A.F. Atrial-ventricular differences in rabbit cardiac voltage-gated Na + currents: Basis for atrial-selective block by ranolazine. Heart Rhythm 2017, 14, 1657–1664. [Google Scholar] [CrossRef]
- Rajamani, S.; El-Bizri, N.; Shryock, J.C.; Makielski, J.C.; Belardinelli, L. Use-dependent block of cardiac late Na+ current by ranolazine. Heart Rhythm 2009, 6, 1625–1631. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Rajamani, S.; Li, H.; January, C.T.; Shryock, J.C.; Belardinelli, L. Reduction of repolarization reserve unmasks the proarrhythmic role of endogenous late Na+ current in the heart. Am. J. Physiol. Circ. Physiol. 2009, 297, H1048–H1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zablocki, J.A.; Elzein, E.; Li, X.; Koltun, D.O.; Parkhill, E.Q.; Kobayashi, T.; Martinez, R.; Corkey, B.; Jiang, H.; Perry, T.; et al. Discovery of Dihydrobenzoxazepinone (GS-6615) Late Sodium Current Inhibitor (Late INai), a Phase II Agent with Demonstrated Preclinical Anti-Ischemic and Antiarrhythmic Properties. J. Med. Chem. 2016, 59, 9005–9017. [Google Scholar] [CrossRef] [PubMed]
- El-Bizri, N.; Xie, C.; Liu, L.; Limberis, J.; Krause, M.; Hirakawa, R.; Nguyen, S.; Tabuena, D.R.; Belardinelli, L.; Kahlig, K.M. Eleclazine exhibits enhanced selectivity for long QT syndrome type 3–associated late Na + current. Heart Rhythm 2018, 15, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Hondeghem, L.M.; Carlsson, L.; Duker, G. Instability and Triangulation of the Action Potential Predict Serious Proarrhythmia, but Action Potential Duration Prolongation Is Antiarrhythmic. Circulation 2001, 103, 2004–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hondeghem, L.M. Relative Contributions of TRIaD and QT to Proarrhythmia. J. Cardiovasc. Electrophysiol. 2007, 18, 655–657. [Google Scholar] [CrossRef]
- Zaza, A.; Rocchetti, M. The Late Na+ Current-Origin and Pathophysiological Relevance. Cardiovasc. Drugs Ther. 2013, 27, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, D.; Noble, P. Late sodium current in the pathophysiology of cardiovascular disease: Consequences of sodium-calcium overload. Heart 2006, 92, iv1–iv5. [Google Scholar] [CrossRef]
- Kiyosue, T.; Arita, M. Late sodium current and its contribution to action potential configuration in guinea pig ventricular myocytes. Circ. Res. 1989, 6, 389–397. [Google Scholar] [CrossRef] [Green Version]
- Patlak, J.B.; Ortiz, M. Slow currents through single sodium channels of the adult rat heart. J. Gen. Physiol. 1985, 86, 89–104. [Google Scholar] [CrossRef] [Green Version]
- Maltsev, V.; Undrovinas, A.I. A multi-modal composition of the late Na+ current in human ventricular cardiomyocytes. Cardiovasc. Res. 2006, 69, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Jaryal, A.; Gulati, S.; Chakrabarty, B.; Singh, A.; Deepak, K.; Pandey, R.; Gupta, N.; Sapra, S.; Kabra, M.; et al. Cardiovascular autonomic dysfunction in children and adolescents with Rett Syndrome. Pediatr. Neurol. 2017, 70, 61–66. [Google Scholar] [CrossRef] [PubMed]
- De Felice, C.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Durand, T.; Valacchi, G.; Ciccoli, L.; Hayek, J. The role of oxidative stress in Rett syndrome: An overview. Ann. N. Y. Acad. Sci. 2012, 1259, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Zaza, A.; Belardinelli, L.; Shryock, J.C. Pathophysiology and pharmacology of the cardiac “late sodium current”. Pharmacol. Ther. 2008, 119, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Fredj, S.; Sampson, K.J.; Liu, H.; Kass, R.S. Molecular basis of ranolazine block of LQT-3 mutant sodium channels: Evidence for site of action. J. Cereb. Blood Flow Metab. 2006, 148, 16–24. [Google Scholar] [CrossRef]
- Lowe, J.S.; Stroud, D.M.; Yang, T.; Hall, L.; Atack, T.C.; Roden, D.M. Increased late sodium current contributes to long QT-related arrhythmia susceptibility in female mice. Cardiovasc. Res. 2012, 95, 300–307. [Google Scholar] [CrossRef] [Green Version]
- Antzelevitch, C.; Belardinelli, L.; Wu, L.; Fraser, H.; Zygmunt, A.C.; Burashnikov, A.; Di Diego, J.M.; Fish, J.M.; Cordeiro, J.M.; Goodrow, R.J., Jr.; et al. Electrophysiologic properties and antiarrhythmic actions of a novel antianginal agent. J. Cardiovasc. Pharmacol. Ther. 2004, 9 (Suppl. 1), S65–S83. [Google Scholar] [CrossRef]
- Rajamani, S.; Shryock, J.C.; Belardinelli, L. Rapid kinetic interactions of ranolazine with HERG K+ current. J. Cardiovasc. Pharmacol. 2008, 51, 581–589. [Google Scholar] [CrossRef]
- Du, C.; Zhang, Y.; EL Harchi, A.; Dempsey, C.; Hancox, J.C. Ranolazine inhibition of hERG potassium channels: Drug–pore interactions and reduced potency against inactivation mutants. J. Mol. Cell. Cardiol. 2014, 74, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Nerbonne, J.M.; Nichols, C.G.; Schwarz, T.L.; Escande, D. Genetic manipulation of cardiac K(+) channel function in mice: What have we learned, and where do we go from here? Circ. Res. 2001, 89, 944–956. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Lv, T.; Li, S.; Zhang, P. Sodium channel blockers in the management of long QT syndrome types 3 and 2: A system review and meta-analysis. J. Cardiovasc. Electrophysiol. 2021, 32, 3057–3067. [Google Scholar] [CrossRef]
- Kaufmann, W.E. Long QT interval in Rett syndrome: Expanding the knowledge of a poorly understood phenomenon. Dev. Med. Child Neurol. 2020, 62, 775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Abdala Sheikh, A.P. Algorhythms for the Analysis of Unrestrained Whole-Body Plethysmography in Mice. Figshare. Software. 2022. Available online: https://0-doi-org.brum.beds.ac.uk/10.6084/m9.figshare.19518877.v2 (accessed on 17 April 2022).
- Gadeberg, H.C.; Kong, C.H.T.; Bryant, S.M.; James, A.F.; Orchard, C.H. Sarcolemmal distribution of ICa and INCX and Ca2+ auto-regulation in mouse ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H190–H199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, A.J.; Hancox, J.C.; Howarth, F.C.; Croker, J.; Vinnicombe, J. A method for making rapid changes of superfusate whilst main-taining temperature at 37oC. Pflugers Arch. 1996, 432, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-H.; Xu, X.-H.; Sun, H.-Y.; Du, X.-L.; Liu, H.; Yang, L.; Xiao, G.-S.; Wang, Y.; Jin, M.-W.; Li, G.-R. Distinctive property and pharmacology of voltage-gated sodium current in rat atrial vs ventricular myocytes. Heart Rhythm 2015, 13, 762–770. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | WT | Mecp2Null/Y | t-Test p Value |
|---|---|---|---|
| RR (ms) | 130.7 ± 2.4 | 125.8 ± 5.3 | 0.4132 |
| HR (bpm) | 460.8 ± 8.6 | 485.8 ± 19.3 | 0.2569 |
| PR (ms) | 38.4 ± 0.8 | 36.4 ± 1.0 | 0.1271 |
| QRS (ms) | 10.8 ± 0.2 | 12.2 ± 0.5 | 0.0094 |
| QT (ms) | 51.9 ± 0.9 | 56.9 ± 1.8 | 0.0233 |
| QTc (ms; Equation (1)) | 45.5 ± 0.9 | 50.8 ± 1.0 | 0.0006 |
| QTc (ms; Equation (2)) | 64.4 ± 1.1 | 71.0 ± 1.1 | 0.0005 |
| Respiratory rate (bpm) | 136.7 ± 4.7 | 104.3 ± 8.2 | 0.0029 |
| Parameter | WT | Mecp2Null/Y |
|---|---|---|
| Resting potential (mV) | 72.2 ± 0.8 | −69.5 ± 0.9 * |
| Overshoot (mV) | 48.0 ± 2.0 | 45.4 ± 2.2 |
| Amplitude (mV) | 120.2 ± 2.4 | 115.0 ± 2.8 |
| Vmax (V s−1) | 139.8 ± 6.1 | 128.1 ± 7.7 |
| APD10 (ms) | 0.6 ± 0.1 | 0.8 ± 0.2 |
| APD25 (ms) | 2.1 ± 0.4 | 2.5 ± 0.3 |
| APD50 (ms) | 6.7 ± 1.1 | 8.1 ± 0.9 |
| APD75 (ms) | 71.1 ± 8.2 | 99.6 ± 7.9 * |
| APD90 (ms) | 115.6 ± 9.5 | 166.0 ± 10.8 ** |
| APD90-APD25 (ms) | 113.5 ± 9.4 | 163.5 ± 10.8 ** |
| Threshold stimulus (pA) | 907.4 ± 40.9 | 597.4 ± 29.3 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, H.; Charles, I.; James, A.F.; Abdala, A.P.; Hancox, J.C. Delayed Ventricular Repolarization and Sodium Channel Current Modification in a Mouse Model of Rett Syndrome. Int. J. Mol. Sci. 2022, 23, 5735. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105735
Cheng H, Charles I, James AF, Abdala AP, Hancox JC. Delayed Ventricular Repolarization and Sodium Channel Current Modification in a Mouse Model of Rett Syndrome. International Journal of Molecular Sciences. 2022; 23(10):5735. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105735
Chicago/Turabian StyleCheng, Hongwei, Ian Charles, Andrew F. James, Ana P. Abdala, and Jules C. Hancox. 2022. "Delayed Ventricular Repolarization and Sodium Channel Current Modification in a Mouse Model of Rett Syndrome" International Journal of Molecular Sciences 23, no. 10: 5735. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105735