
APOE Molecular Spectrum in a French Cohort with Primary Dyslipidemia

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. New APOE Variants in Primary Dyslipidemia
2.2. Recurrent APOE Variants in ADH/FCHL Patients
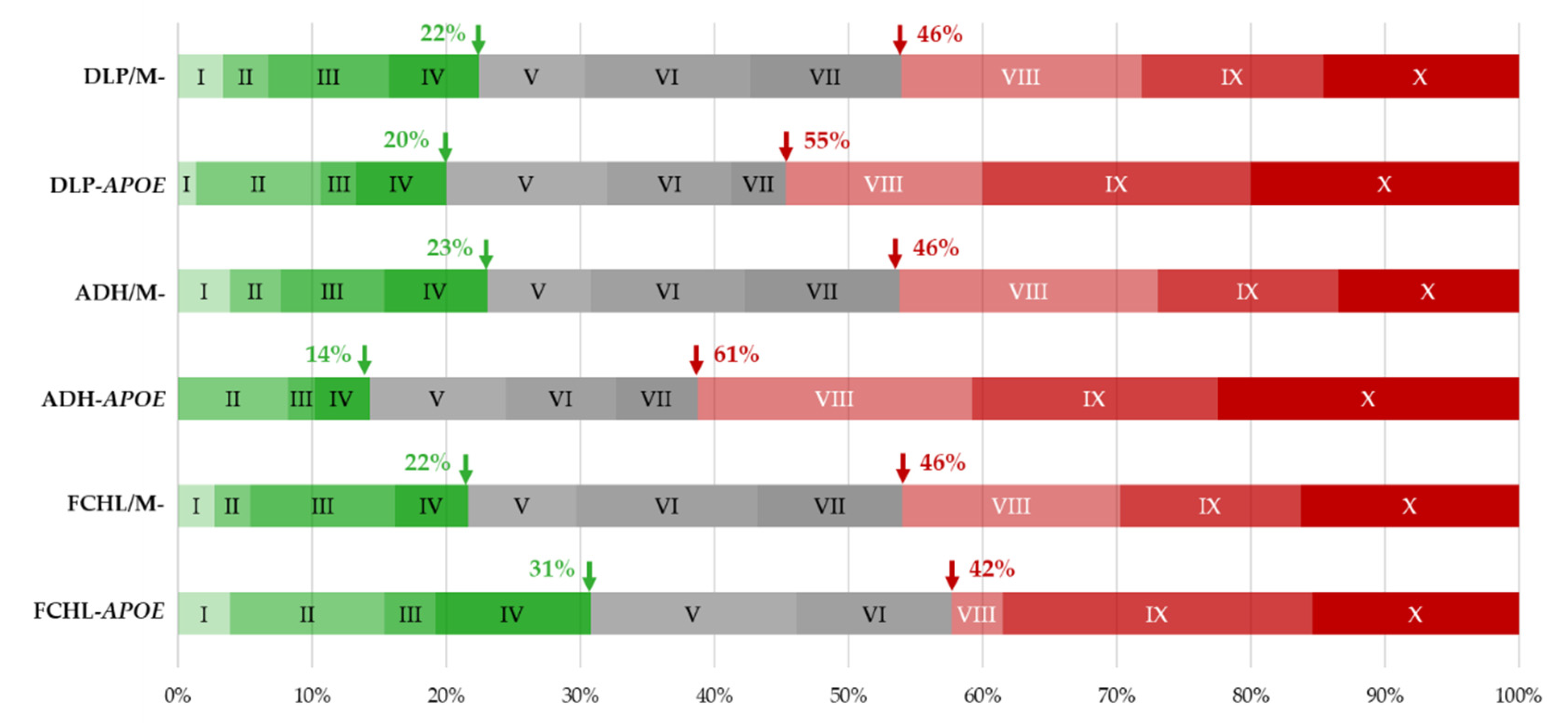
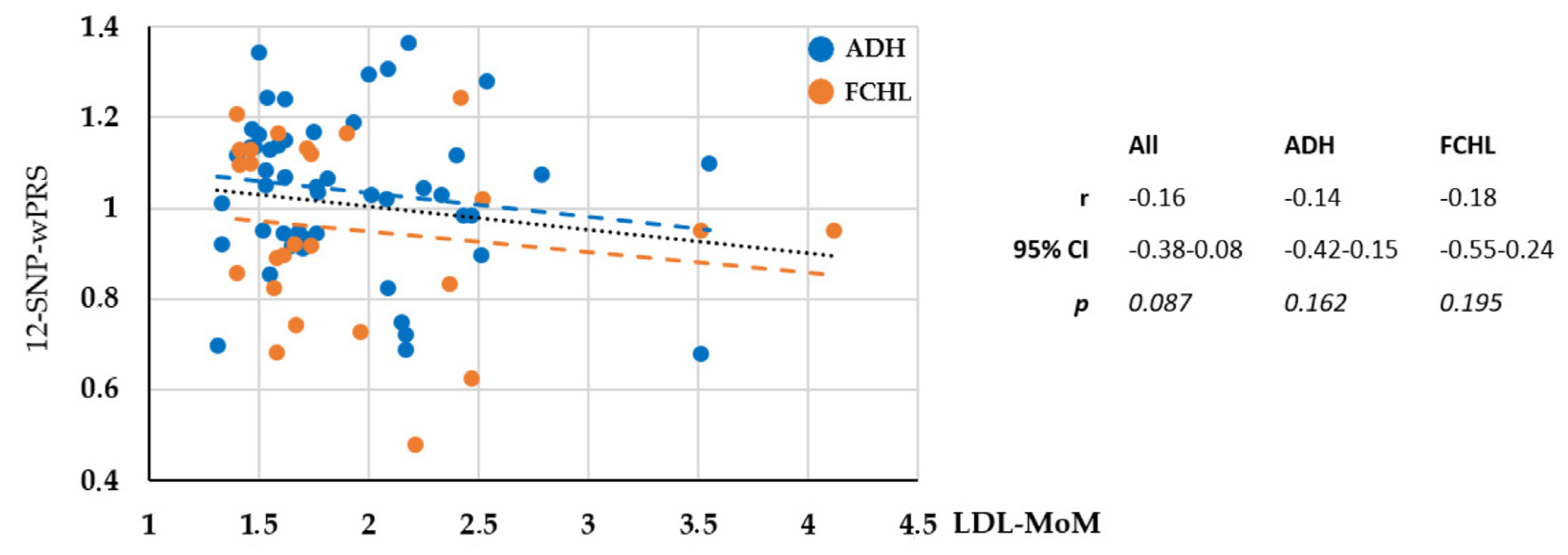
2.3. Monogenic or Polygenic Dyslipidemia?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| APOE Variant | LDL-MoM | TC-MoM | TG-MoM | Clinical Signs | Family History | Hyperlipidemia | ApoE Isoforms | 12-SNP wPRS | wPRS Decile b | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs1038445539 | c.-380A > G | p.? | 1.67 | 1.44 | 2.07 | Xanthelasma | Yes | FCHL | E3E4 | 0.743 | III |
| rs1038445539 | c.-380A > G | p.? | 2.52 | 1.91 | 6.1 | Corneal arcus | Yes | FCHL | E3E4 | 1.021 | VIII |
| c.-279G > A | p.? | 1.4 | 1.41 | 5.71 | Yes | FCHL | E3E4 | 1.207 | X | ||
| - | c.-233G > C | p.? | 2.00 | 1.71 | 1.30 | Yes | ADH | E3E4 | 1.296 | X | |
| c.-105A > G | p.? | 1.9 | 1.78 | 2.56 | Yes | FCHL | E3E3 | 1.164 | X | ||
| rs766215051 | c.-81G > A | p.? | 2.40 | 1.76 | 1.31 | ADH | E3E4 | 1.116 | IX | ||
| rs750782549 | c.-78C > G | p.? | 1.57 | 1.41 | 2.55 a | Yes | FCHL | E3E4 | 0.824 | IV | |
| rs750782549 | c.-78C > G | p.? | 1.59 | 1.60 | 2.04 | FCHL | E3E4 | 1.164 | X | ||
| rs750782549 | c.-78C > G | p.? | 1.52 | 1.33 | 1.65 | Xanthelasma | ADH | E3E4 | 0.950 | VI | |
| - | c.43+11G > A | p.? | 1.47 | 1.41 | 1.90 | ADH | E3E4 | 1.173 | X | ||
| rs770658351 | c.44-1G > C | p.? | 2.17 | 2.44 | na | ADH | E3E4 | 0.722 | II | ||
| rs144354013 | c.31A > G | p.Thr11Ala | 4.12 | na | 3.12 a | FCHL | E3E4 | 0.950 | VI | ||
| rs111833428 | c.69G > A | p.Ala23= | 1.41 | na | 5.89 a | FCHL | E3E3 | 1.097 | IX | ||
| rs776242156 | c.68C > T | p.Ala23Val | 1.48 | 1.29 | 0.57 | CVD | ADH | E3E4 | 1.136 | IX | |
| rs769452 | c.137T > C | p.Leu46Pro | 1.59 | 1.48 | 0.96 | ADH | E3E4 | 1.137 | IX | ||
| rs769452 | c.137T > C | p.Leu46Pro | 1.46 | 1.36 | 0.58 | Xanthoma | ADH | E4E4 | 1.136 | IX | |
| rs769452 | c.137T > C | p.Leu46Pro | 1.33 | 1.25 | 1.65 | ADH | E3E4 | 1.012 | VII | ||
| rs769452 | c.137T > C | p.Leu46Pro | 2.18 | 1.75 | 1.56 | ADH | E3E4 | 1.365 | X | ||
| rs769452 | c.137T > C | p.Leu46Pro | 1.62 | 1.41 | 0.75 | Yes | ADH | E3E4 | 1.149 | IX | |
| rs769452 | c.137T > C | p.Leu46Pro | 1.40 | 1.25 | 1.70 | Yes | ADH | E3E4 | 1.117 | IX | |
| rs769452 | c.137T > C | p.Leu46Pro | 1.62 | 1.38 | 1.56 | ADH | E3E4 | 1.240 | X | ||
| rs769452 | c.137T > C | p.Leu46Pro | 1.65 | 1.50 | 1.34 | ADH | E3E4 | 0.919 | V | ||
| rs769452 | c.137T > C | p.Leu46Pro | 1.53 | 1.86 | 0.77 | Yes | ADH | E3E4 | 1.049 | VIII | |
| rs769452 | c.137T > C | p.Leu46Pro | 2.25 | 1.90 | 0.75 | Yes | ADH | E3E4 | 1.045 | VIII | |
| rs769452 | c.137T > C | p.Leu46Pro | 1.62 | 1.43 | 1.09 | No | ADH | E3E4 | 1.068 | VIII | |
| rs769452 c | c.137T > C c | p.Leu46Pro c | 1.75 | 1.46 | 1.42 | Corneal arcus | ADH | E4E4 | 1.169 | X | |
| rs769452 | c.137T > C | p.Leu46Pro | 1.46 | 1.36 | 2.36 | Yes | FCHL | E2E4 | 1.128 | IX | |
| rs769452 | c.137T > C | p.Leu46Pro | 1.72 | 1.61 | 2.86 | CVD | FCHL | E3E4 | 1.133 | IX | |
| rs769452 | c.137T > C | p.Leu46Pro | 1.58 | 1.43 | 4.04 a | CVD | FCHL | E3E4 | 0.891 | V | |
| rs769452 | c.137T > C | p.Leu46Pro | 1.74 | 1.63 | 2.17 | Yes | FCHL | E3E4 | 1.120 | IX | |
| rs769452 | c.137T > C | p.Leu46Pro | 1.96 | 1.70 | 7.95 | Yes | FCHL | E3E4 | 0.727 | II | |
| rs767980905 | c.249C > T | p.Asp83= | 1.76 | 1.46 | 1.32 | ADH | E3E4 | 1.047 | VIII | ||
| rs11083750 | c.305C > T | p.Pro102Leu | 2.09 | 1.61 | 1.22 | ADH | E3E4 | 1.307 | X | ||
| rs573658040 | c.409C > T | p.Arg137Cys | 1.61 | 1.43 | 1.21 | ADH | E3E4 | 0.945 | VI | ||
| rs11542035 | c.410G > A | p.Arg137His | 1.70 | 1.51 | 1.40 | ADH | E3E3 | 0.912 | V | ||
| rs267606664 | c.434G > A | p.Gly145Asp | 1.4 | 1.43 | 3.03 | FCHL | E2E4 | 0.856 | IV | ||
| rs267606664 | c.434G > A | p.Gly145Asp | 2.21 | 2.72 | 5.5 a | Yes | FCHL | E3E4 | 0.480 | I | |
| rs1018669382 | c.463 C > T | p.Leu155Phe | 1.50 | 1.34 | 1.98 | ADH | E3E3 | 1.162 | X | ||
| rs769455 | c.487C > T | p.Arg163Cys | 1.68 | 1.54 | 1.60 | CVD | ADH | E3E3 | 0.948 | VI | |
| rs769455 | c.487C > T | p.Arg163Cys | 2.47 | na | 10 | Yes | FCHL | E3E3 | 0.625 | II | |
| rs769455 | c.487C > T | p.Arg163Cys | 1.41 | 1.23 | 2.52 | FCHL | E3E4 | 1.129 | IX | ||
| rs155726148 | c.500_502delTCC | p.Leu167del | 2.54 | 2.12 | 1.88 | CVD | ADH | E3E3 | 1.280 | X | |
| rs155726148 | c.500_502delTCC | p.Leu167del | 2.01 | 1.61 | 1.25 | ADH | E3E3 | 1.028 | VIII | ||
| rs155726148 | c.500_502delTCC | p.Leu167del | 2.33 | 2.23 | 1.47 | ADH | E3E3 | 1.030 | VIII | ||
| rs155726148 | c.500_502delTCC | p.Leu167del | 3.51 | 2.66 | 1.16 | Corneal arcus | ADH | E3E3 | 0.680 | II | |
| rs155726148 | c.500_502delTCC | p.Leu167del | 2.15 | 1.84 | 1.11 | ADH | E3E3 | 0.747 | III | ||
| rs155726148 | c.500_502delTCC | p.Leu167del | 3.55 | 2.52 | 1.03 | ADH | E3E3 | 1.098 | IX | ||
| rs155726148 | c.500_502delTCC | p.Leu167del | 2.43 | 2.03 | 1.11 | Yes | ADH | E3E3 | 0.985 | VII | |
| rs155726148 | c.500_502delTCC | p.Leu167del | 2.79 | 2.09 | 1.37 | ADH | E3E3 | 1.076 | VIII | ||
| rs155726148 | c.500_502delTCC | p.Leu167del | 1.33 | 1.21 | 0.66 | ADH | E3E3 | 0.920 | V | ||
| rs155726148 | c.500_502delTCC | p.Leu167del | 2.09 | 1.36 | 1.37 | Yes | ADH | E3E4 | 0.824 | IV | |
| rs155726148 | c.500_502delTCC | p.Leu167del | 2.47 | 2.12 | 0.88 | ADH | E3E4 | 0.983 | VII | ||
| rs155726148 | c.500_502delTCC | p.Leu167del | 1.93 | 1.61 | 1.05 | Corneal arcus | Yes | ADH | E3E4 | 1.190 | X |
| rs155726148 | c.500_502delTCC | p.Leu167del | 1.77 | 1.44 | 0.62 | Yes | ADH | E3E4 | 1.035 | VIII | |
| rs155726148 | c.500_502delTCC | p.Leu167del | 1.31 | 2.16 | 1.69 | Yes | ADH | E3E3 | 0.698 | II | |
| rs155726148 | c.500_502delTCC | p.Leu167del | 2.37 | 1.49 | 2.68 | FCHL | E3E4 | 0.832 | IV | ||
| rs155726148 | c.500_502delTCC | p.Leu167del | 1.58 | 1.47 | 2.02 | CVD | FCHL | E3E3 | 0.683 | II | |
| rs155726148 | c.500_502delTCC | p.Leu167del | 1.66 | 1.50 | 2.07 | Yes | FCHL | E3E4 | 0.921 | V | |
| rs155726148 | c.500_502delTCC | p.Leu167del | 3.51 | 2.61 | 3.23 | FCHL | E3E3 | 0.952 | VI | ||
| rs1239911444 | c.517C > T | p.Leu173= | 1.74 | 1.70 | 3.22 | Yes | FCHL | E3E3 | 0.918 | V | |
| rs1421977676 | c.536C > T | p.Val179Ala | 1.65 | 1.57 | 3.05 | CVD | FCHL | E3E3 | na | na | |
| rs781722239 | c.555C > T | p.Arg185= | 2.17 | 1.86 | 0.66 | Corneal arcus | ADH | E3E3 | 0.689 | II | |
| - | c.638T > A d | p.Val213Glu d | 2.51 | 1.99 | 1.07 | ADH | E3E3 | 0.896 | V | ||
| rs72654468 | c.651C > T | p.Ala217= | 1.54 | 1.42 | 1.13 | ADH | E3E3 | 1.243 | X | ||
| rs72654468 | c.651C > T | p.Ala217= | 2.08 | 1.69 | 1.85 | ADH | E3E3 | 1.020 | VIII | ||
| rs72654468 | c.651C > T | p.Ala217= | 1.55 | 1.40 | 1.30 | ADH | E3E3 | 1.130 | IX | ||
| - | c.652G > T | p.Gly218Cys | 1.76 | 1.48 | 1.24 | ADH | E3E4 | 0.945 | VI | ||
| rs762906934 | c.745G > A | p.Glu249Lys | Na | 1.62 | 2.19 | CVD | FCHL | E3E3 | 0.962 | VI | |
| - | c.754G > A | p.Glu252Lys | 1.61 | 1.45 | 2.53 | Yes | FCHL | E3E4 | 0.897 | V | |
| rs267606661 | c.805C > G | p.Arg269Gly | 2.42 | 1.99 | 2.13 | CVD | FCHL | E3E4 | 1.243 | X | |
| rs267606661 | c.805C > G | p.Arg269Gly | 1.55 | 1.30 | 1.95 | CVD | ADH | E4E4 | 0.855 | IV | |
| rs267606661 | c.805C > G | p.Arg269Gly | 1.81 | 1.50 | 1.51 | ADH | E3E4 | 1.067 | VIII | ||
| rs267606661 | c.805C > G | p.Arg269Gly | 1.71 | 1.52 | 1.43 | ADH | E3E4 | 0.933 | V | ||
| rs374329439 | c.*25C > T | 3’UTR variant | 1.53 | 1.42 | 1.54 | ADH | E3E3 | 1.083 | IX | ||
| rs374329439 | c.*25C > T | 3’UTR variant | 1.46 | 1.41 | 2.1 | FCHL | E3E3 | 1.099 | IX | ||
| - | c.*36C > G | 3’UTR variant | 1.50 | 1.32 | 1.52 | ADH | E3E4 | 1.344 | X | ||
| Median [First quartile–third quartile] | 1.71 [1.54–2.17] | 1.50 [1.41–1.81] | 1.56 [1.21–2.36] | ||||||||
| rs Number | cDNA Position (NM_000041.4) | Protein Position (NP_000032.1) | Hyperlipidemia | AF a in the ADH/FCHL Cohort | FREX Total AF a | GnomAD Total AF a | PolyPhen 2 b | SIFT c | Mutation Taster d | CADD e | Provean f | Splice Site Affected g | ACMG (Varsome) h | References |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs1038445539 | c.-380A > G | 5’UTR variant | FCHL | 0.017 (2/11,486) | 0 | 0.005 (7/152,092) | na | na | na | 7.106 | na | no | na | |
| - | c.-279G > A | 5’UTR variant | FCHL | 0.009 (1/11,486) | 0 | 0 | na | na | na | 5.676 | na | no | na | |
| - | c.-233G > C | 5’UTR variant | ADH | 0.009 (1/11,486) | 0 | 0 | na | na | na | 10.31 (top 10%) | na | no | na | |
| - | c.-105A > G | 5’UTR variant | FCHL | 0.009 (1/11,486) | 0 | 0 | na | na | DC | 22.7 (top 1%) | na | no | VUS | |
| rs766215051 | c.-81G > A | 5’UTR variant | ADH | 0.009 (1/11,486) | 0 | 0.003 (5/152,130) | na | na | DC | 14.13 (top 10%) | na | no | VUS | |
| rs750782549 | c.-78C > G | 5’UTR variant | ADH, FCHL | 0.026 (3/11,486) i | 0 | 0.001 (2/152,116) | na | na | DC | 14.91 (top 10%) | na | no | VUS | |
| rs770658351 | c.43+11G > A | p.? | ADH | 0.009 (1/11,486) | 0 | 0 | na | na | SNP | 13.12 (top 10%) | na | no | VUS | |
| - | c.44-1G > C | p.? | ADH | 0.009 (1/11,486) | 0 | 0 | na | na | DC | 33 (top 0.1%) | na | Yes | P | |
| rs144354013 | c.31A > G | p.Thr11Ala | FCHL | 0.009 (1/11,486) | 0 | 0.009 (13/151,914) | B | T | SNP | 0.294 | N (0.8) | no | VUS/P | |
| rs776242156 | c.68C > T | p.Ala23Val | ADH | 0.009 (1/11,486) | 0 | 0.001 (1/152,206) | B | T | SNP | 0.047 | N (−0.2) | no | VUS/LP | |
| rs111833428 | c.69G > A | p.Ala23= | FCHL | 0.009 (1/11,486) | 0 | 0.023 (35/152,212) | na | na | SNP | 5.195 | N (0) | no | LB | |
| rs769452 | c.137T > C | p.Leu46Pro | ADH, FCHL | 0.157 (18/11,486) | 0.174 (2/1148) | 0.193 (293/152,188) | P | T | DC | 0.72 | N (−1.1) | no | LB | [10] |
| rs767980905 | c.249C > T | p.Asp83= | ADH | 0.009 (1/11,486) | 0 | 0.003 (4/152,218) | na | na | DC | 0.615 | N (0) | no | LB | |
| rs11083750 | c.305C > T | p.Pro102Leu | ADH | 0.009 (1/11,486) | 0 | 0 | PD | D | DC | 23.4 (top 1%) | D (−8.7) | no | LP | |
| rs573658040 | c.409C > T | p.Arg137Cys | ADH | 0.009 (1/11,486) | 0 | 0.002 (3/152,132) | PD | T | DC | 25.8 (top 1%) | N (−2.4) | no | VUS/P | |
| rs11542035 | c.410G > A | p.Arg137His | ADH | 0.009 (1/11,486) | 0 | 0.003(5/152,112) | P | T | SNP | 22.1 (top 1%) | N (−1.0) | no | VUS/P | |
| rs267606664 | c.434G > A | p.Gly145Asp | FCHL | 0.017 (2/11,486) | 0.087 (1/1148) | 0.015 (22/152,152) | PD | T | DC | 24.5 (top 1%) | N (0.656) | no | VUS/P | [27] |
| rs1018669382 | c.463 C > T | p.Leu155Phe | ADH | 0.009 (1/11,486) i | 0 | 0.001 (2/152,148) | B | T | SNP | 5.538 | N (−1.6) | no | VUS/P | |
| rs769455 | c.487C > T | p.Arg163Cys | ADH, FCHL | 0.026 (3/11,486) j | 0 | 0.643 (978/152,126) | PD | D | DC | 28.4 (top 1%) | D (−4.9) | no | VUS/P | [10] |
| rs515726148 | c.500_502delTCC | p.Leu167del | ADH, FCHL | 0.157 (18/11,486) i | 0 | 0.003 (4/152,132) | na | na | SNP | na | D (−7.4) | no | LP | [9,10,16,28,29,30] |
| rs1239911444 | c.517C > T | p.Leu173= | FCHL | 0.009 (1/11,486) | 0 | 0 | na | na | DC | 7.641 | N (0) | no | LB | |
| rs1421977676 | c.536T > C | p.Val179Ala | FCHL | 0.009 (1/11,486) | 0 | 0 | PD | T | SNP | 23.5 (top 1%) | N (−1.0) | no | VUS/P | |
| rs781722239 | c.555C > T | p.Arg185= | ADH | 0.009 (1/11,486) | 0 | 0.009 (13/151,932) | na | na | SNP | 7.192 | N (0) | no | LB | |
| - | c.638T > A | p.Val213Glu | ADH | 0.009 (1/11,486) | 0 | 0 | P | D | SNP | 11.3 (top 10%) | N (−0.6) | no | VUS/P | |
| rs72654468 | c.651C > T | p.Ala217= | ADH | 0.026 (3/11,486) j | 0.182 (2/1,094) | 0.089 (135/151,926) | na | na | SNP | 6.242 | N (0) | no | LB | |
| - | c.652G > T | p.Gly218Cys | ADH | 0.009 (1/11,486) | 0 | 0 | PD | T | SNP | 6.506 | N (−1.4) | no | VUS/P | |
| rs762906934 | c.745G > A | p.Glu249Lys | FCHL | 0.009 (1/11,486) | 0 | 0.001 (1/152,172) | B | T | SNP | 19.7 (top 10%) | N (−1.4) | no | VUS/P | |
| - | c.754G > A | p.Glu252Lys | FCHL | 0.009 (1/11,486) | 0 | 0 | P | D | SNP | 22.2 (top 1%) | D (−2.9) | no | VUS/P | |
| rs267606661 | c.805C > G | p.Arg269Gly | ADH, FCHL | 0.035 (4/11,486) | 0.087 (1/1148) | 0.030 (46/152,200) | P | D | DC | 23.3 (top 1%) | D (−2.9) | no | VUS/P | [10] |
| rs374329439 | c.*25C > T | 3’UTR variant | ADH, FCHL | 0.017 (2/11,486) | 0 | 0.071 (108/152,194) | na | na | SNP | 5.508 | na | no | VUS | |
| - | c.*36C > G | 3’UTR variant | ADH | 0.009 (1/11,486) | 0 | 0 | na | na | SNP | 6.597 | na | no | VUS |
2.4. Genotype–Phenotype Correlation
2.5. Lipid-Lowering Treatment Response
3. Discussion
4. Materials and Methods
4.1. Proband Inclusion
4.2. Molecular Analysis
4.3. Variant Nomenclature
4.4. In Silico Variant Analyses
4.5. Variant Classifications
4.6. Multiple of Median for Total Cholesterol, LDL-C, and Triglyceride Level Calculation
4.7. Weighted Polygenic Risk Score (wPRS)
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Do, R.; Stitziel, N.O.; Won, H.-H.; Jørgensen, A.B.; Duga, S.; Angelica Merlini, P.; Kiezun, A.; Farrall, M.; Goel, A.; Zuk, O.; et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature 2015, 518, 102–106. [Google Scholar] [CrossRef]
- Chemello, K.; García-Nafría, J.; Gallo, A.; Martín, C.; Lambert, G.; Blom, D. Lipoprotein metabolism in familial hypercholesterolemia. J. Lipid Res. 2021, 62, 100062. [Google Scholar] [CrossRef]
- Berberich, A.J.; Hegele, R.A. The complex molecular genetics of familial hypercholesterolaemia. Nat. Rev. Cardiol. 2019, 16, 9–20. [Google Scholar] [CrossRef]
- Beheshti, S.O.; Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J. Am. Coll. Cardiol. 2020, 75, 2553–2566. [Google Scholar] [CrossRef] [PubMed]
- Defesche, J.C.; Gidding, S.S.; Harada-Shiba, M.; Hegele, R.A.; Santos, R.D.; Wierzbicki, A.S. Familial hypercholesterolaemia. Nat. Rev. Dis. Primer 2017, 3, 17093. [Google Scholar] [CrossRef] [PubMed]
- Rabès, J.-P.; Béliard, S.; Carrié, A. Familial hypercholesterolemia: Experience from France. Curr. Opin. Lipidol. 2018, 29, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Talmud, P.J.; Shah, S.; Whittall, R.; Futema, M.; Howard, P.; Cooper, J.A.; Harrison, S.C.; Li, K.; Drenos, F.; Karpe, F.; et al. Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: A case-control study. Lancet 2013, 381, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Varret, M.; Abifadel, M.; Rabès, J.-P.; Boileau, C. Genetic heterogeneity of autosomal dominant hypercholesterolemia. Clin. Genet. 2008, 73, 1–13. [Google Scholar] [CrossRef]
- Marduel, M.; Ouguerram, K.; Serre, V.; Bonnefont-Rousselot, D.; Marques-Pinheiro, A.; Erik Berge, K.; Devillers, M.; Luc, G.; Lecerf, J.-M.; Tosolini, L.; et al. Description of a large family with autosomal dominant hypercholesterolemia associated with the APOE p.Leu167del mutation. Hum. Mutat. 2013, 34, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Wintjens, R.; Bozon, D.; Belabbas, K.; MBou, F.; Girardet, J.-P.; Tounian, P.; Jolly, M.; Boccara, F.; Cohen, A.; Karsenty, A.; et al. Global molecular analysis and APOE mutations in a cohort of autosomal dominant hypercholesterolemia patients in France. J. Lipid Res. 2016, 57, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Versmissen, J.; Oosterveer, D.M.; Yazdanpanah, M.; Defesche, J.C.; Basart, D.C.G.; Liem, A.H.; Heeringa, J.; Witteman, J.C.; Lansberg, P.J.; Kastelein, J.J.P.; et al. Efficacy of statins in familial hypercholesterolaemia: A long term cohort study. BMJ 2008, 337, a2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bea, A.M.; Lamiquiz-Moneo, I.; Marco-Benedí, V.; Mateo-Gallego, R.; Pérez-Calahorra, S.; Jarauta, E.; Martín, C.; Cenarro, A.; Civeira, F. Lipid-lowering response in subjects with the p.(Leu167del) mutation in the APOE gene. Atherosclerosis 2019, 282, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Bello-Chavolla, O.Y.; Kuri-García, A.; Ríos-Ríos, M.; Vargas-Vázquez, A.; Cortés-Arroyo, J.E.; Tapia-González, G.; Cruz-Bautista, I.; Aguilar-Salinas, C.A. Familial combined hyperlipidemia: Current knowledge, perspectives, and controversies. Rev. Investig. Clin. Organo Hosp. Enferm. Nutr. 2018, 70, 224–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwers, M.C.G.J.; van Greevenbroek, M.M.J.; Stehouwer, C.D.A.; de Graaf, J.; Stalenhoef, A.F.H. The genetics of familial combined hyperlipidaemia. Nat. Rev. Endocrinol. 2012, 8, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Civeira, F.; Jarauta, E.; Cenarro, A.; García-Otín, A.L.; Tejedor, D.; Zambón, D.; Mallen, M.; Ros, E.; Pocoví, M. Frequency of low-density lipoprotein receptor gene mutations in patients with a clinical diagnosis of familial combined hyperlipidemia in a clinical setting. J. Am. Coll. Cardiol. 2008, 52, 1546–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solanas-Barca, M.; de Castro-Orós, I.; Mateo-Gallego, R.; Cofán, M.; Plana, N.; Puzo, J.; Burillo, E.; Martín-Fuentes, P.; Ros, E.; Masana, L.; et al. Apolipoprotein E gene mutations in subjects with mixed hyperlipidemia and a clinical diagnosis of familial combined hyperlipidemia. Atherosclerosis 2012, 222, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Marais, A.D. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology 2019, 51, 165–176. [Google Scholar] [CrossRef]
- Huang, Y.; Mahley, R.W. Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis. 2014, 72, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breslow, J.L.; Zannis, V.I.; SanGiacomo, T.R.; Third, J.L.; Tracy, T.; Glueck, C.J. Studies of familial type III hyperlipoproteinemia using as a genetic marker the apoE phenotype E2/2. J. Lipid Res. 1982, 23, 1224–1235. [Google Scholar] [CrossRef]
- Bennet, A.M.; Di Angelantonio, E.; Ye, Z.; Wensley, F.; Dahlin, A.; Ahlbom, A.; Keavney, B.; Collins, R.; Wiman, B.; de Faire, U.; et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA 2007, 298, 1300–1311. [Google Scholar] [CrossRef]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Mahlab-Aviv, S.; Zohar, K.; Cohen, Y.; Peretz, A.R.; Eliyahu, T.; Linial, M.; Sperling, R. Spliceosome-Associated microRNAs Signify Breast Cancer Cells and Portray Potential Novel Nuclear Targets. Int. J. Mol. Sci. 2020, 21, 8132. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.L.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. APOE and dementia—Resequencing and genotyping in 105,597 individuals. Alzheimers Dement. J. Alzheimers Assoc. 2020, 16, 1624–1637. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Weng, W.; Funke, H.; Steinmetz, A.; Assmann, G.; Nauck, M.; Dierkes, J.; Ambrosch, A.; Weisgraber, K.H.; Mahley, R.W.; et al. Effects of a frequent apolipoprotein E isoform, ApoE4Freiburg (Leu28-- > Pro), on lipoproteins and the prevalence of coronary artery disease in whites. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Biesecker, L.G. Correspondence on: Homozygous familial hypercholesterolemia in Italy: Clinical and molecular features. Atherosclerosis 2021, 326, 63–64. [Google Scholar] [CrossRef]
- Yue, P.; Averna, M.; Lin, X.; Schonfeld, G. The c.43_44insCTG variation in PCSK9 is associated with low plasma LDL-cholesterol in a Caucasian population. Hum. Mutat. 2006, 27, 460–466. [Google Scholar] [CrossRef]
- Richard, P.; Beucler, I.; Pascual De Zulueta, M.; Biteau, N.; De Gennes, J.L.; Iron, A. Compound heterozygote for both rare apolipoprotein E1 (Gly127-- > Asp, Arg158-- > Cys) and E3(Cys112-- > Arg, Arg251-- > Gly) alleles in a multigeneration pedigree with hyperlipoproteinaemia. Clin. Sci. Lond. Engl. 1979 1997, 93, 89–95. [Google Scholar] [CrossRef]
- Cenarro, A.; Etxebarria, A.; de Castro-Orós, I.; Stef, M.; Bea, A.M.; Palacios, L.; Mateo-Gallego, R.; Benito-Vicente, A.; Ostolaza, H.; Tejedor, T.; et al. The p.Leu167del Mutation in APOE Gene Causes Autosomal Dominant Hypercholesterolemia by Down-regulation of LDL Receptor Expression in Hepatocytes. J. Clin. Endocrinol. Metab. 2016, 101, 2113–2121. [Google Scholar] [CrossRef] [Green Version]
- Faivre, L.; Saugier-Veber, P.; Pais de Barros, J.-P.; Verges, B.; Couret, B.; Lorcerie, B.; Thauvin, C.; Charbonnier, F.; Huet, F.; Gambert, P.; et al. Variable expressivity of the clinical and biochemical phenotype associated with the apolipoprotein E p.Leu149del mutation. Eur. J. Hum. Genet. 2005, 13, 1186–1191. [Google Scholar] [CrossRef]
- Awan, Z.; Choi, H.Y.; Stitziel, N.; Ruel, I.; Bamimore, M.A.; Husa, R.; Gagnon, M.-H.; Wang, R.-H.L.; Peloso, G.M.; Hegele, R.A.; et al. APOE p.Leu167del mutation in familial hypercholesterolemia. Atherosclerosis 2013, 231, 218–222. [Google Scholar] [CrossRef]
- Abou Khalil, Y.; Rabès, J.-P.; Boileau, C.; Varret, M. APOE gene variants in primary dyslipidemia. Atherosclerosis 2021, 328, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Argyri, L.; Dafnis, I.; Theodossiou, T.A.; Gantz, D.; Stratikos, E.; Chroni, A. Molecular basis for increased risk for late-onset Alzheimer disease due to the naturally occurring L28P mutation in apolipoprotein E4. J. Biol. Chem. 2014, 289, 12931–12945. [Google Scholar] [CrossRef] [Green Version]
- Andersen, L.H.; Miserez, A.R.; Ahmad, Z.; Andersen, R.L. Familial defective apolipoprotein B-100: A review. J. Clin. Lipidol. 2016, 10, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Haralambos, K.; Whatley, S.D.; Edwards, R.; Gingell, R.; Townsend, D.; Ashfield-Watt, P.; Lansberg, P.; Datta, D.B.N.; McDowell, I.F.W. Clinical experience of scoring criteria for Familial Hypercholesterolaemia (FH) genetic testing in Wales. Atherosclerosis 2015, 240, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Wardell, M.R.; Rall, S.C.; Schaefer, E.J.; Kane, J.P.; Weisgraber, K.H. Two apolipoprotein E5 variants illustrate the importance of the position of additional positive charge on receptor-binding activity. J. Lipid Res. 1991, 32, 521–528. [Google Scholar] [CrossRef]
- Weisgraber, K.H.; Rall, S.C.; Innerarity, T.L.; Mahley, R.W.; Kuusi, T.; Ehnholm, C. A novel electrophoretic variant of human apolipoprotein E. Identification and characterization of apolipoprotein E1. J. Clin. Investig. 1984, 73, 1024–1033. [Google Scholar] [CrossRef] [Green Version]
- Whiffin, N.; Minikel, E.; Walsh, R.; O’Donnell-Luria, A.H.; Karczewski, K.; Ing, A.Y.; Barton, P.J.R.; Funke, B.; Cook, S.A.; MacArthur, D.; et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. Off. J. Am. Coll. Med. Genet. 2017, 19, 1151–1158. [Google Scholar] [CrossRef] [Green Version]
- Chora, J.R.; Medeiros, A.M.; Alves, A.C.; Bourbon, M. Analysis of publicly available LDLR, APOB, and PCSK9 variants associated with familial hypercholesterolemia: Application of ACMG guidelines and implications for familial hypercholesterolemia diagnosis. Genet. Med. Off. J. Am. Coll. Med. Genet. 2018, 20, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Masana, L.; Ibarretxe, D.; Rodríguez-Borjabad, C.; Plana, N.; Valdivielso, P.; Pedro-Botet, J.; Civeira, F.; López-Miranda, J.; Guijarro, C.; Mostaza, J.; et al. Toward a new clinical classification of patients with familial hypercholesterolemia: One perspective from Spain. Atherosclerosis 2019, 287, 89–92. [Google Scholar] [CrossRef] [Green Version]
- Decourt, C.; Janin, A.; Moindrot, M.; Chatron, N.; Nony, S.; Muntaner, M.; Dumont, S.; Divry, E.; Dauchet, L.; Meirhaeghe, A.; et al. PCSK9 post-transcriptional regulation: Role of a 3’UTR microRNA-binding site variant in linkage disequilibrium with c.1420G. Atherosclerosis 2020, 314, 63–70. [Google Scholar] [CrossRef]
- Mellerio, H.; Alberti, C.; Druet, C.; Capelier, F.; Mercat, I.; Josserand, E.; Vol, S.; Tichet, J.; Lévy-Marchal, C. Novel modeling of reference values of cardiovascular risk factors in children aged 7 to 20 years. Pediatrics 2012, 129, e1020–e1029. [Google Scholar] [CrossRef] [PubMed]
- Balder, J.W.; de Vries, J.K.; Nolte, I.M.; Lansberg, P.J.; Kuivenhoven, J.A.; Kamphuisen, P.W. Lipid and lipoprotein reference values from 133,450 Dutch Lifelines participants: Age- and gender-specific baseline lipid values and percentiles. J. Clin. Lipidol. 2017, 11, 1055–1064. [Google Scholar] [CrossRef] [Green Version]
- Marmontel, O.; Charrière, S.; Simonet, T.; Bonnet, V.; Dumont, S.; Mahl, M.; Jacobs, C.; Nony, S.; Chabane, K.; Bozon, D.; et al. Single, short in-del, and copy number variations detection in monogenic dyslipidemia using a next-generation sequencing strategy. Clin. Genet. 2018, 94, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Marmontel, O.; Rollat-Farnier, P.A.; Wozny, A.-S.; Charrière, S.; Vanhoye, X.; Simonet, T.; Chatron, N.; Collin-Chavagnac, D.; Nony, S.; Dumont, S.; et al. Development of a new expanded next-generation sequencing panel for genetic diseases involved in dyslipidemia. Clin. Genet. 2020, 98, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinforma. Oxf. Engl. 2005, 21, 263–265. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]





| APOE Variant | Pathogenic Prediction a | Gender | Age b | LDL-C without Preatment c | Treatment | Age d | LDL-C under Preatment c | Estimated Reduction e | Observed Reduction | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs776242156 | c.68C > T | p.Ala23Val | VUS/LP | M | 43 | 5.17 | Atorvastatin 20 | 46 | 1.42 | 1.8 | 3.6 |
| rs11542035 | c.410G > A | p.Arg137His | VUS/P | F | 61 | 6.45 | Simvastatin 20 | 62 | 3.06 | 1.6 | 2.1 |
| rs769455 | c.487C > T | p.Arg163Cys | VUS/P | M | 40 | 5.88 | Atorvastatin 80 Ezetimibe 10 | 41 | 1.69 | 2.5 | 3.5 |
| rs155726148 | c.500_502delTCC | p.Leu167del | LP | M | 69 | 7.24 | Atorvastatin 20 | 70 | 2.74 | 1.8 | 2.6 |
| rs155726148 | c.500_502delTCC | p.Leu167del | LP | F | 38 | 9.44 | Atorvastatin 80 | 56 | 3.59 | 2.2 | 2.6 |
| rs155726148 | c.500_502delTCC | p.Leu167del | LP | F | 31 | 6.18 | Atorvastatin 80 | 32 | 5.20 | 2.2 | 1.2 |
| rs155726148 | c.500_502delTCC | p.Leu167del | LP | M | 31 | 7.55 | Simvastatin 20 Ezetimibe 10 | 38 | 2.87 | 1.8 | 2.6 |
| rs155726148 | c.500_502delTTC | p.Leu167del | LP | M | 20 | 6.99 | Rosuvastatin 5 | 27 | 3.74 | 1.8 | 1.9 |
| rs781722239 | c.555C > T | p.Arg185= | LB | M | 65 | 7.81 | Atorvastatin 20 | 65 | 4.29 | 1.8 | 1.8 |
| rs267606661 | c.805C > G | p.Arg269Gly | VUS/P | M | 51 | 5.73 | Atorvastatin 20 | 58 | 3.18 | 1.8 | 1.8 |
| rs267606661 | c.805C > G | p.Arg269Gly | VUS/P | M | 59 | 6.33 | Atorvastatin 10 | 59 | 2.49 | 1.6 | 2.5 |
| Mean | 1.90 | 2.39 | |||||||||
| SD | 0.28 | 0.74 | |||||||||
| Wilcoxon matched-pairs test | p = 0.0426 | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abou Khalil, Y.; Marmontel, O.; Ferrières, J.; Paillard, F.; Yelnik, C.; Carreau, V.; Charrière, S.; Bruckert, E.; Gallo, A.; Giral, P.; et al. APOE Molecular Spectrum in a French Cohort with Primary Dyslipidemia. Int. J. Mol. Sci. 2022, 23, 5792. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105792
Abou Khalil Y, Marmontel O, Ferrières J, Paillard F, Yelnik C, Carreau V, Charrière S, Bruckert E, Gallo A, Giral P, et al. APOE Molecular Spectrum in a French Cohort with Primary Dyslipidemia. International Journal of Molecular Sciences. 2022; 23(10):5792. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105792
Chicago/Turabian StyleAbou Khalil, Yara, Oriane Marmontel, Jean Ferrières, François Paillard, Cécile Yelnik, Valérie Carreau, Sybil Charrière, Eric Bruckert, Antonio Gallo, Philippe Giral, and et al. 2022. "APOE Molecular Spectrum in a French Cohort with Primary Dyslipidemia" International Journal of Molecular Sciences 23, no. 10: 5792. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105792