
Biochemical, Structural Analysis, and Docking Studies of Spiropyrazoline Derivatives

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
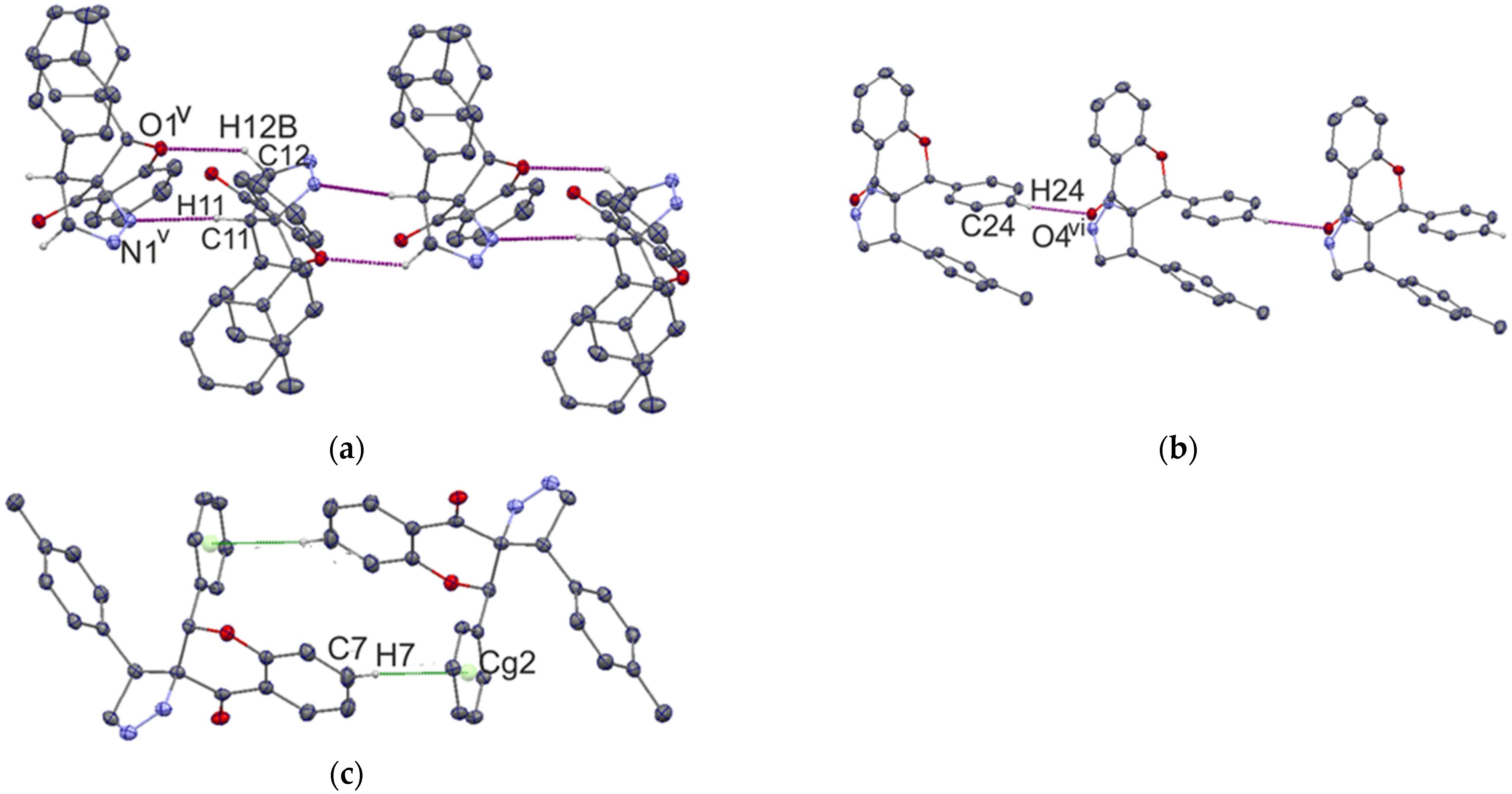
2.1. Molecular and Crystal Structures
Structure of Compound I and II
2.2. Molecular Docking
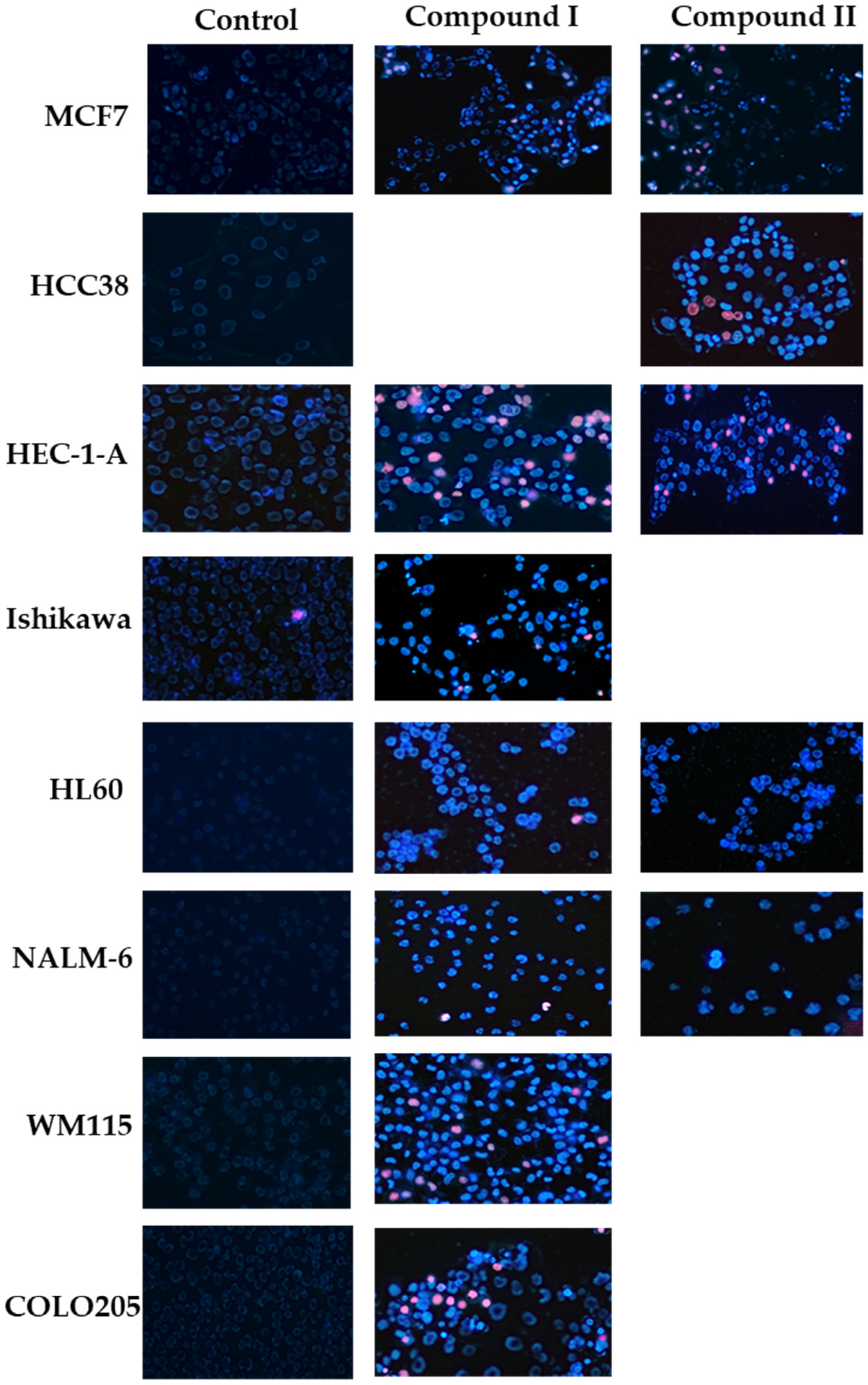
2.3. Biological Assay
2.3.1. Cytotoxic Activity
2.3.2. Analysis of DNA Damage Using the Alkaline Version of the Comet Assay (Single Cell Electrophoresis)—DNA Comet Assay
2.3.3. Determination of Apoptotic and Necrotic Cell Fractions by Fluorescence Microscopy (Double Staining of Cells with Fluorescent Dyes Hoechst 33258 and Propidium Iodide)
2.3.4. Measurement of Cleaved PARP1 Levels
2.3.5. Changes in the Transmembrane Mitochondrial Potential (ΔΨm)
3. Materials and Methods
3.1. Refinement of X-ray Data
3.2. Molecular Docking Calculation
3.2.1. Ligand Preparation
3.2.2. Protein Preparation
- Removal of B, C, and D chains of initial receptor structure;
- Removal of co-crystalized ligands (including co-crystalized inhibitor);
- Removal of water molecules;
- Addition of hydrogen atoms and Gasteiger charges to each atom.
3.2.3. Molecular Docking
3.3. Cell Lines and Cell Culture
3.4. Cytotoxicity Assay
3.5. Measurement of the Changes in Mitochondrial Potential (ΔΨm) Using the Microplate Spectrofluorimetric Method with the JC-1 Fluorescent Probe
3.6. Determination of Apoptotic and Necrotic Cell Fractions by Fluorescence Microscopy (Double Staining of Cells with Fluorescent Dyes Hoechst 33258 and Propidium Iodide)
- Live cells (weak, dull, light blue fluorescence);
- Cells in the early phase of PCD (bright, light blue fluorescence);
- Cells in the late phase of PCD (pink-purple fluorescence);
- Necrotic cells (intense red fluorescence).
3.7. Analysis of DNA Damage Using the Alkaline Version of the Comet Assay (Single Cell Electrophoresis)
3.8. Measurement of Cleaved PARP Levels
3.9. Statistical Analysis
3.10. Chemistry
3.10.1. Synthesis of 5′-(3-Methylphenyl)-2-phenyl-4′,5′-dihydro-4H-spiro[chromano-3,3′-pirazol]-4-one (Compound I)
3.10.2. Synthesis of 5′-(4-Methylphenyl)-2-phenyl-4′,5′-dihydro-4H-spiro[chromano-3,3′-pirazol]-4-one (Compound II)
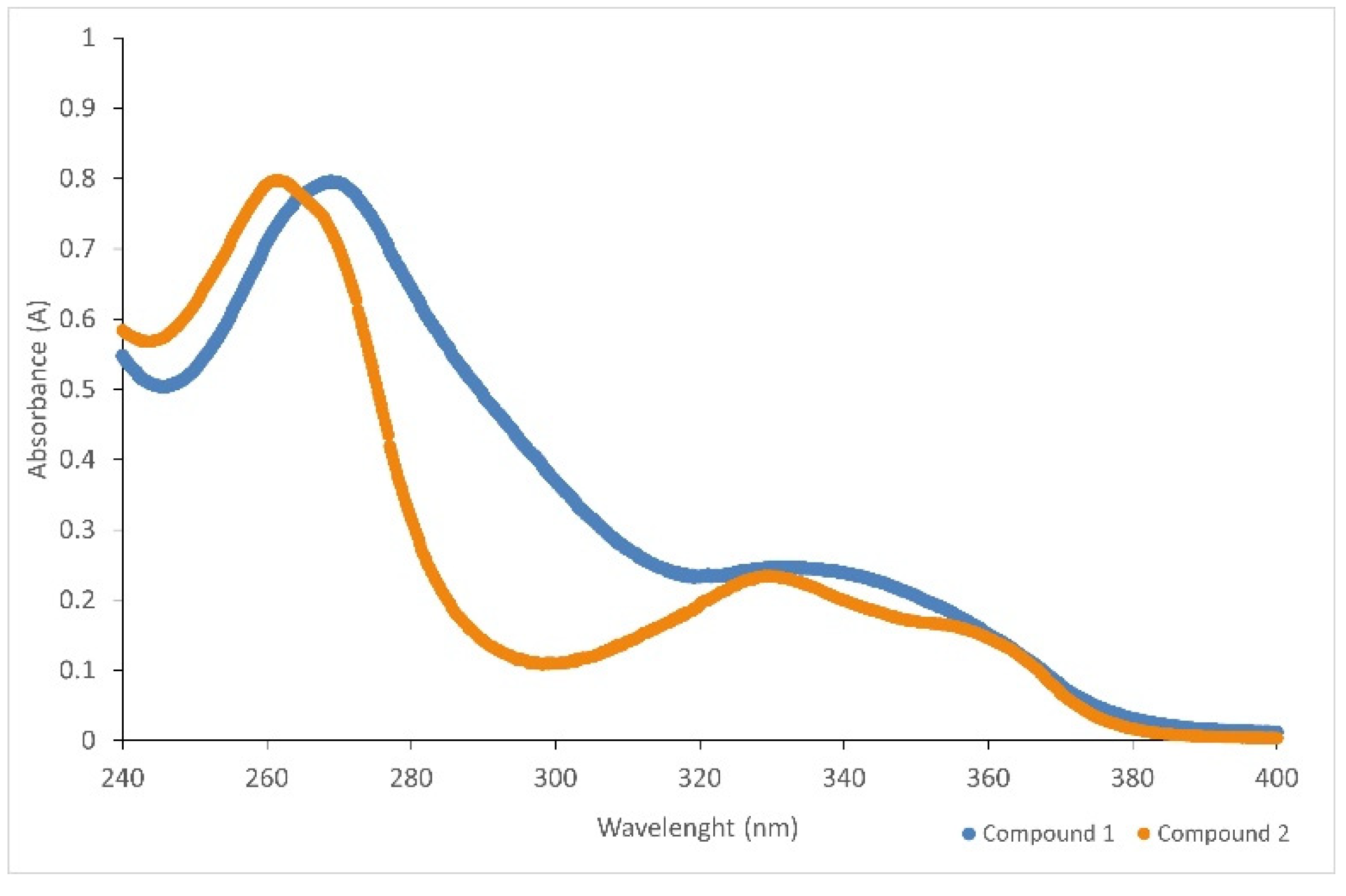
3.10.3. Spectroscopy UV–VIS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Malyuchenko, N.V.; Kotova, E.Y.; Kulaeva, O.I.; Kirpichnikov, M.P.; Studitskiy, V.M. PARP1 Inhibitors: Antitumor Drug Design. Acta Nat. 2015, 7, 27–37. [Google Scholar] [CrossRef]
- Mao, K.; Zhang, G. The Role of PARP1 in Neurodegenerative Diseases and Aging. FEBS J. 2022, 289, 2013–2024. [Google Scholar] [CrossRef]
- Kamaletdinova, T.; Fanaei-Kahrani, Z.; Wang, Z.-Q. The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers. Cells 2019, 8, 1625. [Google Scholar] [CrossRef] [Green Version]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the Repair of DNA Damage by Homologous Recombination and Sensitivity to Poly(ADP-Ribose) Polymerase Inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [Green Version]
- Morales, J.C.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of Poly (ADP-Ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Vinayak, S.; Ford, J.M. PARP Inhibitors for the Treatment and Prevention of Breast Cancer. Curr. Breast Cancer Rep. 2010, 2, 190–197. [Google Scholar] [CrossRef]
- de la Lastra, C.A.; Villegas, I.; Sánchez-Fidalgo, S. Poly(ADP-Ribose) Polymerase Inhibitors: New Pharmacological Functions and Potential Clinical Implications. Curr. Pharm. Des. 2007, 13, 933–962. [Google Scholar] [CrossRef]
- Chen, Y.; Du, H. The Promising PARP Inhibitors in Ovarian Cancer Therapy: From Olaparib to Others. Biomed. Pharmacother. Biomed. Pharmacother. 2018, 99, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. PARP Inhibitors for Cancer Therapy. Expert Rev. Mol. Med. 2005, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kopeć, B.; Sikora, M.; Słuczanowska-Głąbowska, S.; Pawlik, A. Znaczenie inhibitorów PARP w terapii wybranych nowotworów złośliwych związanych z mutacjami w genach BRCA1 i BRCA2. Farm Pol. 2019, 75, 129–133. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirley, M. Rucaparib: A Review in Ovarian Cancer. Target. Oncol. 2019, 14, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Niraparib: First Global Approval. Drugs 2017, 77, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Carmichael, J. Phase II Trial of the Oral PARP Inhibitor Olaparib in BRCA-Deficient Advanced Breast Cancer. J. Clin. Oncol. 2009, 27, CRA501. [Google Scholar] [CrossRef]
- Dębska, S.; Kubicka, J.; Czyżykowski, R.; Habib, M.; Potemski, P. PARP Inhibitors—Theoretical Basis and Clinical Application. Postępy Hig. Med. Dośw. 2012, 66, 311–321. [Google Scholar] [CrossRef]
- Yarchoan, M.; Myzak, M.C.; Johnson, B.A.; De Jesus-Acosta, A.; Le, D.T.; Jaffee, E.M.; Azad, N.S.; Donehower, R.C.; Zheng, L.; Oberstein, P.E.; et al. Olaparib in Combination with Irinotecan, Cisplatin, and Mitomycin C in Patients with Advanced Pancreatic Cancer. Oncotarget 2017, 8, 44073–44081. [Google Scholar] [CrossRef]
- Adamus-Grabicka, A.A.; Markowicz-Piasecka, M.; Cieślak, M.; Królewska-Golińska, K.; Hikisz, P.; Kusz, J.; Małecka, M.; Budzisz, E. Biological Evaluation of 3-Benzylidenechromanones and Their Spiropyrazolines-Based Analogues. Molecules 2020, 25, 1613. [Google Scholar] [CrossRef] [Green Version]
- Grazul, M.; Budzisz, E. Biological Activity of Metal Ions Complexes of Chromones, Coumarins and Flavones. Coord. Chem. Rev. 2009, 21–22, 2588–2598. [Google Scholar] [CrossRef]
- Kumar, S.; Pandey, A.K. Chemistry and Biological Activities of Flavonoids: An Overview. Available online: https://www.hindawi.com/journals/tswj/2013/162750/ (accessed on 22 May 2020).
- Pietta, P.-G. Flavonoids as Antioxidants. J. Nat. Prod. 2000, 63, 1035–1042. [Google Scholar] [CrossRef]
- Matiadis, D.; Sagnou, M. Pyrazoline Hybrids as Promising Anticancer Agents: An Up-to-Date Overview. Int. J. Mol. Sci. 2020, 21, 5507. [Google Scholar] [CrossRef]
- Berhow, C.A.; Vaughn, S.F. Higher Plants Flavonoids: Biosynthesis and Chemical Ecology. In Principles and Practices in Plant Ecology, Allochemical Interactions; CRC Press: Boca Raton, FL, USA, 1999; pp. 423–437. [Google Scholar]
- Jucá, M.M.; Cysne Filho, F.M.S.; de Almeida, J.C.; da Silva Mesquita Mesquita, D.; de Moraes Barriga Barriga, J.R.; Dias, K.C.F.; Barbosa, T.M.; Vasconcelos, L.C.; Leal, L.K.A.M.; Ribeiro, J.E.; et al. Flavonoids: Biological Activities and Therapeutic Potential. Nat. Prod. Res. 2020, 34, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Kopustinskiene, D.M.; Jakstas, V.; Savickas, A.; Bernatoniene, J. Flavonoids as Anticancer Agents. Nutrients 2020, 12, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamus-Grabicka, A.A.; Markowicz-Piasecka, M.; Ponczek, M.B.; Kusz, J.; Małecka, M.; Krajewska, U.; Budzisz, E. Interaction of Arylidenechromanone/Flavanone Derivatives with Biological Macromolecules Studied as Human Serum Albumin Binding, Cytotoxic Effect, Biocompatibility Towards Red Blood Cells. Molecules 2018, 23, 3172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremer, D.; Pople, J.A. General Definition of Ring Puckering Coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Duax, W.L.; Norton, D.A. Atlas of Steroid Structure, 1st ed.; IFI/Plenum: New York, NY, USA, 1975; Volume 1, ISBN 978-0-306-66101-3. [Google Scholar]
- Etter, M.C. Encoding and Decoding Hydrogen-Bond Patterns of Organic Compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Cossarizza, A.; Baccarani-Contri, M.; Kalashnikova, G.; Franceschi, C. A New Method for the Cytofluorimetric Analysis of Mitochondrial Membrane Potential Using the J-Aggregate Forming Lipophilic Cation 5,5’,6,6’-Tetrachloro-1,1’,3,3’-Tetraethylbenzimidazolcarbocyanine Iodide (JC-1). Biochem. Biophys. Res. Commun. 1993, 197, 40–45. [Google Scholar] [CrossRef]
- Parkin, S.; Moezzi, B.; Hope, H. XABS2: An Empirical Absorption Correction Program. J. Appl. Crystallogr. 1995, 28, 53–56. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction, CrysAlisPro Software System, Version 1.171; Rigaku Corporation: Oxford, UK, 2015.
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. Diamond—Crystal and Molecular Structure Visualization; Crystal Impact: Bonn, Germany, 1999. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Wang, X.; Chu, Z.; He, G.; Dong, G.; Xu, Y. Design, Synthesis and Biological Evaluation of Novel Imidazo[4,5-c]Pyridinecarboxamide Derivatives as PARP-1 Inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 1993–1996. [Google Scholar] [CrossRef] [PubMed]
- Salmas, R.E.; Unlu, A.; Yurtsever, M.; Noskov, S.Y.; Durdagi, S. In Silico Investigation of PARP-1 Catalytic Domains in Holo and Apo States for the Design of High-Affinity PARP-1 Inhibitors. J. Enzyme Inhib. Med. Chem. 2016, 31, 112–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, M.B.; Nielsen, S.E.; Berg, K. Re-Examination and Further Development of a Precise and Rapid Dye Method for Measuring Cell Growth/Cell Kill. J. Immunol. Methods 1989, 119, 203–210. [Google Scholar] [CrossRef]
- Sakamuru, S.; Li, X.; Attene-Ramos, M.S.; Huang, R.; Lu, J.; Shou, L.; Shen, M.; Tice, R.R.; Austin, C.P.; Xia, M. Application of a Homogenous Membrane Potential Assay to Assess Mitochondrial Function. Physiol. Genom. 2012, 44, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Sung, H.J.; Ma, W.; Wang, P.; Hynes, J.; O’Riordan, T.C.; Combs, C.A.; McCoy, J.P.; Bunz, F.; Kang, J.; Hwang, P.M. Mitochondrial Respiration Protects against Oxygen-Associated DNA Damage. Nat. Commun. 2010, 1, 5. [Google Scholar] [CrossRef]
- Olive, P.L.; Banáth, J.P. The Comet Assay: A Method to Measure DNA Damage in Individual Cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef]
- Pijewska, L.; Kamecki, J.; Perka-Karolczak, W. 3-Arylideneflavanones. II: Reaction with Diazomethane. 3-Arylideneflavanones II React. Diazomethane 1993, 48, 254–257. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D–H···A | D–H | H···A | D···A | D–H···A |
|---|---|---|---|---|
| Compound I | ||||
| C5–H5···O4i | 0.95 | 2.51 | 3.235(3) | 133 |
| C25–H25···O1ii | 0.95 | 2.69 | 3.587(3) | 159 |
| C6–H6···N2iii | 0.95 | 2.66 | 3.308(4) | 126 |
| C27–H27A···N2iv | 0.98 | 2.71 | 3.483(4) | 136 |
| Compound II | ||||
| C12–H12B··O1v | 0.99 | 2.46 | 3.224(2) | 134 |
| C24–H24···O4vi | 0.95 | 2.43 | 3.170(2) | 135 |
| C11–H11···N1v | 1.00 | 2.58 | 3.575(2) | 172 |
| No | Structure | Free Energy of Binding (kcal mol−1) |
|---|---|---|
| Compound I |  | −9.7 |
| Compound II |  | −8.7 |
| Quercetin |  | −8.9 |
| Number of Compound | IC50 Concentration (µM) | |||
|---|---|---|---|---|
| MCF7 | HCC38 | Ishikawa | HEC-1-A | |
| Compound I | 43.1 ± 1.5 | 57.3 ± 1.1 | 34.2 ± 1.7 | 39.3 ± 1.3 |
| Compound II | 31.4 ± 0.9 | 37.2 ± 1.9 | 56.7 ± 2.2 | 27.4 ± 0.8 |
| 4-Chromanone * | >500 | >500 | >500 | >500 |
| Cisplatin * | 11.4 ± 1.3 | 29.6 ± 2.2 | 16.1 ± 2.5 | 89.5 ± 4.7 |
| (Compound I) | (Compound II) | |
|---|---|---|
| Crystal data | ||
| Chemical formula | C24H20N2O2 | C24H20N2O2 |
| Mr | 368.44 | 368.42 |
| Crystal system, space group | Monoclinic, P21/c | Monoclinic, P21/c |
| Temperature (K) | 100 | 100 |
| a, b, c (Å) | 9.2373 (5), 9.7336 (5), 20.4183 (17) | 9.2071 (2), 10.5662 (2), 19.6790 (4) |
| Β (°) | 90.641 (6) | 93.778 (2) |
| V (Å3) | 1835.7 (2) | 1910.29 (7) |
| Z | 4 | 4 |
| Radiation type | Mo Kα | Mo Kα |
| Μ (mm−1) | 0.09 | 0.08 |
| Crystal size (mm) | 0.4 × 0.05 × 0.03 | 0.32 × 0.03 × 0.02 |
| Data collection | ||
| Diffractometer | SuperNova, Dual, Cu at zero, Atlas | SuperNova, Dual, Cu at zero, Atlas |
| Absorption correction | XABS2 [30] | Multi-scan CrysAlis PRO 1.171.38.41q [24] Empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. |
| Tmin, Tmax | 0.484, 0.979 | 0.808, 1.000 |
| No. of measured independent and observed [I > 2σ(I)] reflections | 3803, 3803, 2670 | 15831, 3957, 3351 |
| Rint | 0.070 | 0.029 |
| (sin θ/λ)max (Å−1) | 0.628 | 0.628 |
| Refinement | ||
| R[F2 > 2σ(F2)], wR(F2), S | 0.068, 0.200, 1.03 | 0.037, 0.100, 1.04 |
| No. of reflections | 3803 | 3957 |
| No. of parameters | 254 | 254 |
| No. of restraints | 1 | 0 |
| H atom treatment | H atom parameters constrained | H atom parameters constrained |
| Δρmax, Δρmin (e Å−3) | 0.60, −0.39 | 0.25, −0.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamus-Grabicka, A.A.; Daśko, M.; Hikisz, P.; Kusz, J.; Malecka, M.; Budzisz, E. Biochemical, Structural Analysis, and Docking Studies of Spiropyrazoline Derivatives. Int. J. Mol. Sci. 2022, 23, 6061. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116061
Adamus-Grabicka AA, Daśko M, Hikisz P, Kusz J, Malecka M, Budzisz E. Biochemical, Structural Analysis, and Docking Studies of Spiropyrazoline Derivatives. International Journal of Molecular Sciences. 2022; 23(11):6061. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116061
Chicago/Turabian StyleAdamus-Grabicka, Angelika A., Mateusz Daśko, Pawel Hikisz, Joachim Kusz, Magdalena Malecka, and Elzbieta Budzisz. 2022. "Biochemical, Structural Analysis, and Docking Studies of Spiropyrazoline Derivatives" International Journal of Molecular Sciences 23, no. 11: 6061. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116061