
The Ability of Chlorophyll to Trap Carcinogen Aflatoxin B1: A Theoretical Approach

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Results
2.1. DFT Optimized Structures
Determination of Most Stable Conformer between chl a 1 and chl a 2
2.2. Structural and Energetic Parameters from Optimized Conformers
2.2.1. Atomic Charges of chl a 1, chl a 2, AFB1, and Their Complexes
2.2.2. Bond Distance (Å) of Optimized Geometries
2.2.3. Molecular Orbitals: HOMO–LUMO for chl a 1, chl a 2, and AFB1
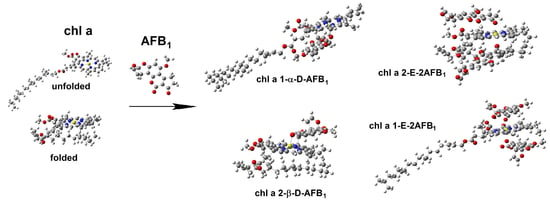
2.3. Geometry of Unfolded and Folded Chlorophyll with One AFB1 Molecule
2.3.1. Frontal View of Unfolded and Folded Chlorophyll
2.3.2. Geometry of Unfolded Chlorophyll with One AFB1 Molecule
2.3.3. Geometry of Unfolded Chlorophyll with Two AFB1 Molecules
2.3.4. Geometry of Folded Chlorophyll with One AFB1 Molecule
2.3.5. Geometry of Folded Chlorophyll with Two AFB1 Molecules
2.4. Interaction Energy of the Complexes in Gas Phase
2.4.1. Considering the Coupling of One AFB1 Molecule with chl a 1 and chl a 2
2.4.2. Considering the Coupling of Two AFB1 Molecules with chl a 1 and chl a 2
2.5. Interaction Energy of the Complexes in Water as a Solvent
2.6. Weak Hydrogen Bond Interactions between the Ester Functions of chl a 1 and chl a 2 with AFB1
2.6.1. Chl a 1–AFB1a (Three Hydrogen Bond Interactions)
2.6.2. Chl a 1–AFB1b (Three Hydrogen Bond Interactions)
2.6.3. Chl a 2–AFB1c (One Hydrogen Bond Interaction)
2.6.4. Chl a 2–AFB1d (Two Hydrogen Bond Interactions)
2.7. Docking Studies for chl a 1– and chl a 2–AFB1 Complexes
2.8. Molecular Dynamics (MD) Simulations for chl a 2
2.9. Correlation between Experimental and Theoretical Findings
3. Materials and Methods
3.1. Quantum Chemical Calculations
3.2. Docking Studies
3.3. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Asao, T.; Buchi, G.; Abdel-Kader, M.; Chang, S.B.; Wick, E.L.; Wogan, G. Aflatoxins b and g. J. Am. Chem. Soc. 1963, 85, 1706–1707. [Google Scholar] [CrossRef]
- Feibelman, T.P.; Cotty, P.J.; Doster, M.; Michailides, T. A morphologically distinct strain of Aspergillus nomius. Mycologia 1998, 90, 618–623. [Google Scholar] [CrossRef]
- Ostry, V.; Malir, F.; Toman, J.; Grosse, Y. Mycotoxins as human carcinogens-the IARC Monographs classification. Mycotoxin Res. 2017, 33, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Probst, C.; Njapau, H.; Cotty, P.J. Outbreak of an acute aflatoxicosis in Kenya in 2004: Identification of the causal agent. Appl. Environ. Microbiol. 2007, 73, 2762–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azziz-Baumgartner, E.; Lindblade, K.; Gieseker, K.; Rogers, H.S.; Kieszak, S.; Njapau, H.; Schleicher, R.; McCoy, L.F.; Misore, A.; DeCock, K.; et al. Case-control study of an acute aflatoxicosis outbreak, Kenya, 2004. Environ. Health Perspect. 2005, 113, 1779–1783. [Google Scholar] [CrossRef]
- Lewis, L.; Onsongo, M.; Njapau, H.; Schurz-Rogers, H.; Luber, G.; Kieszak, S.; Nyamongo, J.; Backer, L.; Dahiye, A.M.; Misore, A.; et al. Aflatoxin contamination of commercial maize products during an outbreak of acute aflatoxicosis in eastern and central Kenya. Environ. Health Perspect. 2005, 113, 1763–1767. [Google Scholar] [CrossRef]
- Kamala, A.; Shirima, C.; Jani, B.; Bakari, M.; Sillo, H.; Rusibamayila, N.; De Saeger, S.; Kimanya, M.; Gong, Y.; Simba, A. Outbreak of an acute aflatoxicosis in Tanzania during 2016. World Mycotoxin J. 2018, 11, 311–320. [Google Scholar] [CrossRef]
- Negishi, T.; Arimoto, S.; Nishizaki, C.; Hayatsu, H. Inhibitory effect of chlorophyll on the genotoxicity of 3-amino-1-methyl-5H-pyrido [4,3-b indole (Trp-P-2). Carcinogenesis 1989, 10, 145–149. [Google Scholar] [CrossRef]
- Kimm, S. Antimutagenic activity of chlorophyll to direct-and indirect-acting mutagens and its contents in vegetables. Korean J. Biochem. 1982, 14, 1–8. [Google Scholar]
- Barale, R.; Zucconi, D.; Bertani, R.; Loprieno, N. Vegetables inhibit, in vivo, the mutagenicity of nitrite combined with nitrosable compounds. Mutat. Res. 1983, 120, 145–150. [Google Scholar] [CrossRef]
- Lai, C.-N.; Butler, M.A.; Matney, T.S. Antimutagenic activities of common vegetables and their chlorophyll content. Mutat. Res./Gen. Toxicol. 1980, 77, 245–250. [Google Scholar] [CrossRef]
- Endo, Y.; Usuki, R.; Kaneda, T. Antioxidant effects of chlorophyll and pheophytin on the autoxidation of oils in the dark. II. The mechanism of antioxidative action of chlorophyll. J. Am. Oil Chem. Soc. 1985, 62, 1387–1390. [Google Scholar] [CrossRef]
- Endo, Y.; Usuki, R.; Kaneda, T. Antioxidant effects of chlorophyll and pheophytin on the autoxidation of oils in the dark. I. Comparison of the inhibitory effects. J. Am. Oil Chem. Soc. 1985, 62, 1375–1378. [Google Scholar] [CrossRef]
- Fahey, J.W.; Stephenson, K.K.; Dinkova-Kostova, A.T.; Egner, P.A.; Kensler, T.W.; Talalay, P. Chlorophyll, chlorophyllin and related tetrapyrroles are significant inducers of mammalian phase 2 cytoprotective genes. Carcinogenesis 2005, 26, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.Y.-W.; Tang, P.M.-K.; Hon, P.-M.; Au, S.W.-N.; Tsui, S.K.-W.; Waye, M.M.-Y.; Kong, S.-K.; Mak, T.C.-W.; Fung, K.-P. Pheophorbide a, a major antitumor component purified from Scutellaria barbata, induces apoptosis in human hepatocellular carcinoma cells. Planta Med. 2006, 72, 28–33. [Google Scholar] [CrossRef]
- Jubert, C.; Mata, J.; Bench, G.; Dashwood, R.; Pereira, C.; Tracewell, W.; Turteltaub, K.; Williams, D.; Bailey, G. Effects of chlorophyll and chlorophyllin on low-dose aflatoxin B1 pharmacokinetics in human volunteers. Cancer Prev. Res. 2009, 2, 1015–1022. [Google Scholar] [CrossRef] [Green Version]
- Simonich, M.T.; Egner, P.A.; Roebuck, B.D.; Orner, G.A.; Jubert, C.; Pereira, C.; Groopman, J.D.; Kensler, T.W.; Dashwood, R.H.; Williams, D.E.; et al. Natural chlorophyll inhibits aflatoxin B 1-induced multi-organ carcinogenesis in the rat. Carcinogenesis 2007, 8, 1294–1302. [Google Scholar] [CrossRef] [Green Version]
- Newmark, H.L. A hypothesis for dietary components as blocking agents of chemical carcinogenesis: Plant phenolics and pyrrole pigments. Nutr. Cancer 1984, 6, 58–70. [Google Scholar] [CrossRef]
- Simonich, M.T.; McQuistan, T.; Jubert, C.; Pereira, C.; Hendricks, J.D.; Schimerlik, M.; Zhu, B.; Dashwood, R.H.; Williams, D.E.; Bailey, G.S. Low-dose dietary chlorophyll inhibits multi-organ carcinogenesis in the rainbow trout. Food Chem. Toxicol. 2008, 46, 1014–1024. [Google Scholar] [CrossRef] [Green Version]
- Dashwood, R.; Negishi, T.; Hayatsu, H.; Breinholt, V.; Hendricks, J.; Bailey, G. Chemopreventive properties of chlorophylls towards aflatoxin B1: A review of the antimutagenicity and anticarcinogenicity data in rainbow trout. Mutat. Res. 1998, 399, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Huq, F.; Yunos, N. A molecular modelling analysis of the toxicity of Aflatoxin B1 and its modulation by chlorophyllin. Int. J. Pure Appl. Chem. 2007, 2, 371–376. [Google Scholar]
- Breinholt, V.; Schimerlik, M.; Dashwood, R.; Bailey, G. Mechanisms of chlorophyllin anticarcinogenesis against aflatoxin B1: Complex formation with the carcinogen. Chem. Res. Toxicol. 1995, 8, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Albores, A.; Nicolás-Vázquez, M.I.; Miranda-Ruvalcaba, R.; Moreno-Martínez, E. Mass spectrometry/mass spectrometry study on the degradation of B-aflatoxins in maize with aqueous citric acid. Am. J. Agric. Biol. Sci. 2008, 3, 482–489. [Google Scholar]
- Nicolas-Vazquez, I.; Mendez-Albores, A.; Moreno-Martınez, E.; Miranda, R.; Castro, M. Role of lactone ring in structural, electronic, and reactivity properties of aflatoxin B1: A theoretical study. Arch. Environ. Contam. Toxicol. 2010, 59, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Escobedo-González, R.; Méndez-Albores, A.; Villarreal-Barajas, T.; Aceves-Hernández, J.M.; Miranda-Ruvalcaba, R.; Nicolás-Vázquez, I. A theoretical study of 8-Chloro-9-hydroxy-Aflatoxin B1, the conversion product of aflatoxin B1 by neutral electrolyzed water. Toxins 2016, 8, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méndez-Albores, A.; Escobedo-González, R.; Aceves-Hernández, J.M.; García-Casillas, P.; Nicolás-Vázquez, M.I.; Miranda-Ruvalcaba, R. A theoretical study of the adsorption process of B-aflatoxins using Pyracantha koidzumii (Hayata) Rehder biomasses. Toxins 2020, 12, 283. [Google Scholar] [CrossRef]
- Nava-Ramírez, M.J.; Salazar, A.M.; Sordo, M.; López-Coello, C.; Téllez-Isaías, G.; Méndez-Albores, A.; Vázquez-Durán, A. Ability of low contents of biosorbents to bind the food carcinogen aflatoxin B1 in vitro. Food Chem. 2021, 345, 128863. [Google Scholar] [CrossRef]
- Vázquez-Durán, A.; Nava-Ramírez, M.D.J.; Hernández-Patlán, D.; Solís-Cruz, B.; Hernández-Gómez, V.; Téllez-Isaías, G.; Méndez-Albores, A. Potential of kale and lettuce residues as natural adsorbents of the carcinogen aflatoxin B1 in a dynamic gastrointestinal tract-simulated model. Toxins 2021, 13, 771. [Google Scholar] [CrossRef]
- Hayashi, T.; Schimerlik, M.; Bailey, G. Mechanisms of chlorophyllin anticarcinogenesis: Dose–responsive inhibition of aflatoxin uptake and biodistribution following oral co-administration in rainbow trout. Toxicol. Appl. Pharmacol. 1999, 158, 132–140. [Google Scholar] [CrossRef]
- Alvarado-González, M.; Flores-Holguín, N.; Glossman-Mitnik, D. Computational nanochemistry study of the molecular structure and properties of chlorophyll a. Int. J. Photoenergy 2013, 2013, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Schulte, T.; Hiller, R.G.; Hofmann, E. X-ray structures of the peridinin-chlorophyll-protein reconstituted with different chlorophylls. FEBS Lett. 2010, 584, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.; Steiner, T. The Weak Hydrogen Bond, in Structural Chemistry and Biology; Oxford University Press: New York, NY, USA, 2001; p. 506. [Google Scholar]
- Kobayashi, R.; Reimers, J.R. Free energies for the coordination of ligands to the magnesium of chlorophyll-a in solvents. Mol. Phys. 2015, 113, 1648–1654. [Google Scholar] [CrossRef]
- Chen, M.; Cai, Z.-L. Theoretical study on the thermodynamics properties of chlorophyll d-peptides coordinating ligand. Biochim. Biophys. Acta 2007, 1767, 603–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F.A. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Gross, K.C.; Seybold, P.G.; Peralta-Inga, Z.; Murray, J.S.; Politzer, P. Comparison of quantum chemical parameters and Hammett constants in correlating pKa values of substituted anilines. J. Org. Chem. 2001, 66, 6919–6925. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. The fundamental nature and role of the electrostatic potential in atoms and molecules. Theor Chim Acta 2002, 108, 134–142. [Google Scholar] [CrossRef]
- Nicolás, I.; Vilchis, M.; Aragón, N.; Miranda, R.; Hojer, G.; Castro, M. Theoretical study of the structure and antimicrobial activity of horminone. Int. J. Quantum Chem. 2003, 93, 411–421. [Google Scholar] [CrossRef]
- Bechaieb, R.; Lakhdar, Z.; Gérard, H. DFT and TD-DFT studies of Mg-substitution in chlorophyll by Cr(II), Fe(II) and Ni(II). Chem. Africa 2018, 1, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Chattaraj, P.K.; Sakar, U.; Roy, D.R. Electrophilicity index. Chem. Rev. 2006, 106, 2065–2091. [Google Scholar] [CrossRef]
- Pearson, R.G. The principle of maximum hardness. Acc. Chem. Res. 1993, 26, 250–255. [Google Scholar] [CrossRef]
- Eliel, E.L.; Wilen, S.H.; Monder, L.N. Stereochemistry of Organic Compounds; John Wiley & Sons, Inc.: New York, NY, USA, 1994; pp. 698–718. [Google Scholar]
- Rutkowska-Zbik, D.; Witko, M.; Fiedor, L. Ligation of water to magnesium chelates of biological importance. J. Mol. Model. 2013, 19, 4661–4667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Fredj, A.; Ben Lakhdar, Z.; Ruiz-López, M.F. Six-coordination in chlorophylls: The fundamental role of dispersion energy. Chem. Phys. Lett. 2009, 472, 243–247. [Google Scholar] [CrossRef]
- Heimdal, J.; Jensen, K.P.; Devarajan, A.; Ryde, U. The role of axial ligands for the structure and function of chlorophylls. J. Biol. Inorg. Chem. 2007, 2, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Timkovich, R.; Tulinsky, A. Structure of aquomagnesium tetraphenylporphyrin. J. Am. Chem. Soc. 1969, 71, 4430–4432. [Google Scholar] [CrossRef]
- Leonarski, F.; D’ascenzo, L.; Auffinger, P. Nucleobase carbonyl groups are poor Mg2+ inner-sphere binder but excellent monovalent ion binders—A critical PDB survey. RNA 2019, 25, 173–192. [Google Scholar] [CrossRef] [Green Version]
- Bechaieb, R.; Fredj, A.B.; Akacha, A.B.; Gérard, H. Interactions of copper (II) and zinc (II) with chlorophyll: Insights from Density Functional Theory studies. New J. Chem. 2016, 40, 4543–4549. [Google Scholar] [CrossRef]
- Ben Fredj, A. Theoretical study of the dimerization of chlorophyll(a) and its hydrates: Implication for chlorophyll (a) aggregation. Helv. Chim. Acta 2016, 99, 1–13. [Google Scholar] [CrossRef]
- Sharma Yamijala, S.R.K.C.; Periyasamy, G.; Pati, S.K. Computational studies on structural and excited-state properties of modified chlorophyll f with various axial ligands. J. Phys. Chem. A 2011, 115, 12298–12306. [Google Scholar] [CrossRef] [Green Version]
- Borah, K.D.; Bhuyan, J. Magnesium porphyrins with relevance to chlorophylls. Dalton Trans. 2017, 46, 6497–6509. [Google Scholar] [CrossRef]
- Zucchelli, G.; Brogioli, D.; Casazza, A.P.; Garlaschi, F.M.; Jennings, R.C. Chlorophyll ring deformation modulates Qy electronic energy in chlorophyll-protein complexes and generates spectral forms. Biophys. J. 2007, 93, 2240–2254. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Suzuki, T.; Ishikita, H. Absorption-energy calculations of chlorophyll a and b with an explicit solvent model. J. Photochem. Photobiol. A Chem. 2018, 358, 422–431. [Google Scholar] [CrossRef]
- Huheey, J.E. Inorganic Chemistry: Principles of Structure and Reactivity, 3rd ed.; Harper & Row, Publishers: New York, NY, USA, 1983; p. 506. [Google Scholar]
- Balaban, T.S.; Fromme, P.; Holzwarth, A.R.; Kraub, N.; Plokhorenko, V.I. Relevance of diastereotopic ligation of magnesium atoms of chlorophylls in photosystem I. Biochim. Biophys. Acta Bioenerg. 2002, 1556, 197–207. [Google Scholar] [CrossRef]
- Ghosh, A.; Mobin, S.M.; Fröhlich, R.; Butcher, R.J.; Maity, D.K.; Ravikanth, M. Effect of five membered versus six membered meso-substituents on structure and electronic properties of Mg(II) porphyrins: A combined experimental and theoretical study. Inorg. Chem. 2010, 49, 8287–8297. [Google Scholar] [CrossRef] [PubMed]
- Jing, Z.; Liu, C.; Qi, R.; Ren, P. Many-body effect determines the selectivity for Ca2+ and Mg2+ in proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E7495–E7501. [Google Scholar] [CrossRef] [Green Version]
- Stevens, M.J.; Rempe, S.L.B. Ion-specific effects in carboxylate binding sites. J. Phys. Chem. B 2016, 120, 12519–12530. [Google Scholar] [CrossRef]
- Chen, X.; Pu, H.; Fang, Y.; Wang, X.; Zhao, S.; Lin, Y.; Zhang, M.; Dai, H.E.; Gong, W.; Liu, L. Crystal structure of the catalytic subunit of magnesium chelatase. Nat. Plants 2015, 1, 15125. [Google Scholar] [CrossRef]
- Fredj, A.B.; Ruiz-López, M.F. Theoretical study of chlorophyll a hydrates formation in aqueous solvents. J. Phys. Chem. B 2010, 114, 681–687. [Google Scholar] [CrossRef]
- Agostiano, A.; Cosma, P.; Monica, M.D. Spectrocopic and electrochemical characterization of chlorophyll a in different water + organic solvent mixtures. J. Electroanal. Chem. 1990, 98, 311–324. [Google Scholar] [CrossRef]
- Hernández-Rodríguez, M.; Rosales-Hernández, M.C.; Mendieta-Wejebe, J.E.; Martínez-Archundia, M.; Basurto, J.C. Current tolos and methods in molecualr dynamics (MD) simulations for the drug design. Curr. Med. Chem. 2016, 23, 3909–3924. [Google Scholar] [CrossRef]
- Grossfield, A.; Zuckerman, D.M. Quantifying uncertainty and sampling quality in biomolecular simulations. Annu. Rep. Comput. Chem. 2009, 5, 23–48. [Google Scholar]
- Vincenzi, M.; Costantini, S.; Scala, S.; Tesauro, D.; Accardo, A.; Leone, M.; Colonna, G.; Guillon, J.; Portella, L.; Trotta, A.M.; et al. Conformational ensembles explored dynamically from disordered peptides targeting chemokine receptor CXCR4. Int. J. Mol. Sci. 2015, 16, 12159–12173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parr, R.G. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989; p. 333. [Google Scholar]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density functional theory of electronic structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef] [Green Version]
- Gaussian 09, Revision E.01. Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V. Gaussian, Inc.: Wallingford, UK, 2013.
- Deppmeier, B.J.; Driessen, A.J.; Hehre, T.S.; Hehre, W.J.; Johnson, J.A.; Klunzinger, P.E.; Leonard, J.M.; Pham, I.; Pietro, W.J.; Jianguo, Y.; et al. Spartan 06 for Windows; Wave Function Inc: Irvine, CA, USA, 2006. [Google Scholar]
- Frisch, A.; Nielsen, A.B.; Holder, A.J. Gaussview Users Manual; Gaussian Inc: Pittsburgh, PA, USA, 2000. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The Mo6 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functional sans systematic testing of four Mo6-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Torrent-Sucarrat, M.; Navarro, S.; Cossío, F.P.; Anglada, J.M.; Luis, J.M. Relevance of the DFT method to study expanded porphyrins with different topologies. J. Comput. Chem. 2017, 38, 2819–2828. [Google Scholar] [CrossRef] [Green Version]
- Jaramillo, P.; Coutinho, K.; Costa Cabral, B.J.; Canuto, S. Ionization of chlorophyll-c2 in liquid metanol. Chem. Phys. Lett. 2012, 546, 67–73. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. Chem. Phys. Lett. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Huczyński, A.; Janczak, J.; Łowicki, D.; Brzezinski, B. Monensin A acid complexes as a model of electrogenic transport of sodium cation. Biochim. Biophys. Acta 2012, 1818, 2108–2119. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.J.; Ahmad, S. Anharmonic vibrational studies of L-aspartic acid using HF and DFT calculations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2012, 96, 992–1004. [Google Scholar] [CrossRef]
- Ramalingam, S.; Babu, P.D.S.; Periandy, S.; Fereyduni, E. Vibrational investigation, molecular orbital studies and molecular electrostatic potential map analysis on 3-chlorobenzoic acid using hybrid computational calculations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2011, 4, 210–220. [Google Scholar] [CrossRef]
- Karelson, M.; Lobanov, V.S.; Katritzky, A.R. Quantum-chemical descriptors in QSAR/QSPR studies. Chem. Rev. 1996, 96, 1027–1044. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interactions of a solvent with a continuum. A direct utilization of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Cammi, R.; Tomasi, J. Remarks on the use of the apparent surface charges (ASC) methods in solvation problems: Iterative versus matrix-inversion procedures and the renormalization of the apparent charges. J. Comp. Chem. 1995, 16, 1449–1458. [Google Scholar] [CrossRef]
- Rubarani, P.; Gangadharana, S.; Sampath, K. First order hyperpolarizabilities, NPA and Fukui functions of cyclohexanone by density functional theory method. Acta Phys. Pol. A 2015, 127, 748–752. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4, AutoDock-Tools4, Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-Durán, L.A.; Rosales-Hernández, M.C.; Hernández-Rodríguez, M.; Mendieta-Wejebe, J.E.; Trujillo-Ferrara, J.; Correa-Basurto, J. Mapping myeloperoxidase to identify its promiscuity properties using docking and molecular dynamics simulations. Curr. Pharm. Des. 2013, 19, 2204–2215. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kal, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comp. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Zhu, X.; Lopez, P.E.M.; MacKerell, A.D. Recent developments and applications of the CHARMM force fields. WIREs 2012, 2, 167–185. [Google Scholar] [CrossRef]
- Batcho, P.F.; Case, D.A.; Schlick, T. Optimized particle-mesh Ewald/multiple-time step integration for molecular dynamics simulation. J. Chem. Phys. 2001, 115, 4003–4041. [Google Scholar] [CrossRef] [Green Version]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular-dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Glykos, N.M. Carma: A molecular dynamics analysis program. J. Comput. Chem. 2006, 27, 1765–1768. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Mg–O1 | Mg–N1 | Mg–N2 | Mg–N3 | Mg–N4 |
|---|---|---|---|---|---|
| chl a 2 | - | 2.131 | 2.013 | 2.061 | 2.001 |
| chl a 1 | - | 2.133 | 2.015 | 2.063 | 2.002 |
| chl a 1-α-D-AFB1 | 2.101 | 2.176 | 2.032 | 2.087 | 2.033 |
| chl a 1-α-E-AFB1 | 2.106 | 2.183 | 2.037 | 2.076 | 2.036 |
| chl a 1-β-D-AFB1 | 2.124 | 2.157 | 2.025 | 2.098 | 2.041 |
| chl a 1-β-E-AFB1 | 2.111 | 2.160 | 2.023 | 2.092 | 2.050 |
| chl a 2-α-D-AFB1 | 2.123 | 2.178 | 2.058 | 2.099 | 2.030 |
| chl a 2-α-E-AFB1 | 2.080 | 2.170 | 2.045 | 2.088 | 2.020 |
| chl a 2-β-E-AFB1 | 2.120 | 2.172 | 2.028 | 2.099 | 2.056 |
| chl a 2-β-E-AFB1 | 2.119 | 2.151 | 2.057 | 2.101 | 2.016 |
| chl a 1-D-2AFB1 | 2.230 | 2.135 | 2.043 | 2.084 | 2.024 |
| 2.428 * | |||||
| chl a 1-E-2AFB1 | 2.228 | 2.113 | 2.025 | 2.114 | 2.030 |
| 2.351 * | |||||
| chl a 2-D-2AFB1 | 2.285 | 2.128 | 2.054 | 2.084 | 2.019 |
| 2.167 * | |||||
| chl a 2-E-2AFB1 | 2.251 | 2.149 | 2.027 | 2.092 | 2.028 |
| 2.186 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez-Durán, A.; Téllez-Isaías, G.; Hernández-Rodríguez, M.; Ruvalcaba, R.M.; Martínez, J.; Nicolás-Vázquez, M.I.; Aceves-Hernández, J.M.; Méndez-Albores, A. The Ability of Chlorophyll to Trap Carcinogen Aflatoxin B1: A Theoretical Approach. Int. J. Mol. Sci. 2022, 23, 6068. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116068
Vázquez-Durán A, Téllez-Isaías G, Hernández-Rodríguez M, Ruvalcaba RM, Martínez J, Nicolás-Vázquez MI, Aceves-Hernández JM, Méndez-Albores A. The Ability of Chlorophyll to Trap Carcinogen Aflatoxin B1: A Theoretical Approach. International Journal of Molecular Sciences. 2022; 23(11):6068. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116068
Chicago/Turabian StyleVázquez-Durán, Alma, Guillermo Téllez-Isaías, Maricarmen Hernández-Rodríguez, René Miranda Ruvalcaba, Joel Martínez, María Inés Nicolás-Vázquez, Juan Manuel Aceves-Hernández, and Abraham Méndez-Albores. 2022. "The Ability of Chlorophyll to Trap Carcinogen Aflatoxin B1: A Theoretical Approach" International Journal of Molecular Sciences 23, no. 11: 6068. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116068