
Genome-Wide Identification of Long Noncoding RNA and Their Potential Interactors in ISWI Mutants


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
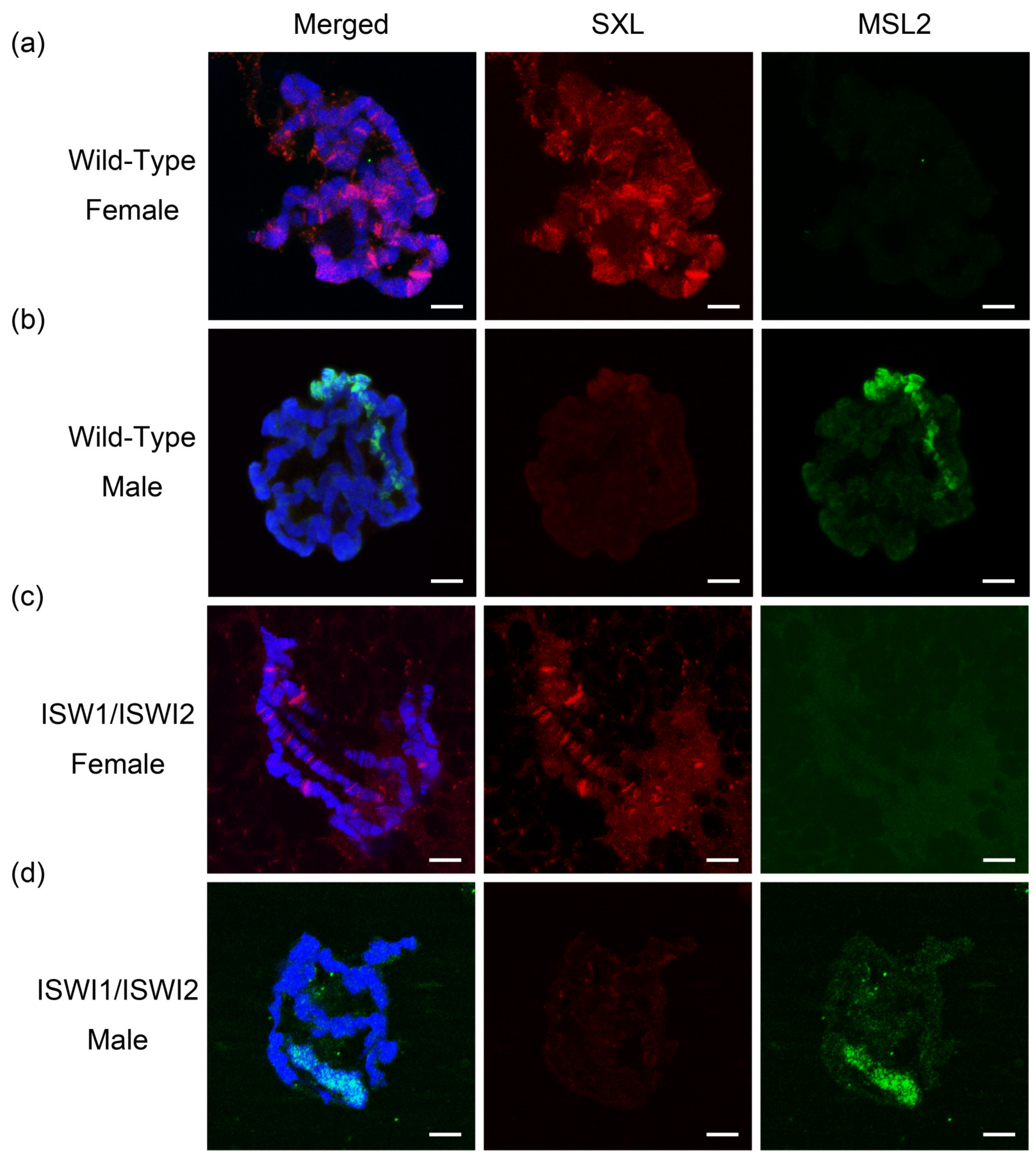
2.1. Hybridization and Identification of ISWI Mutation Lines
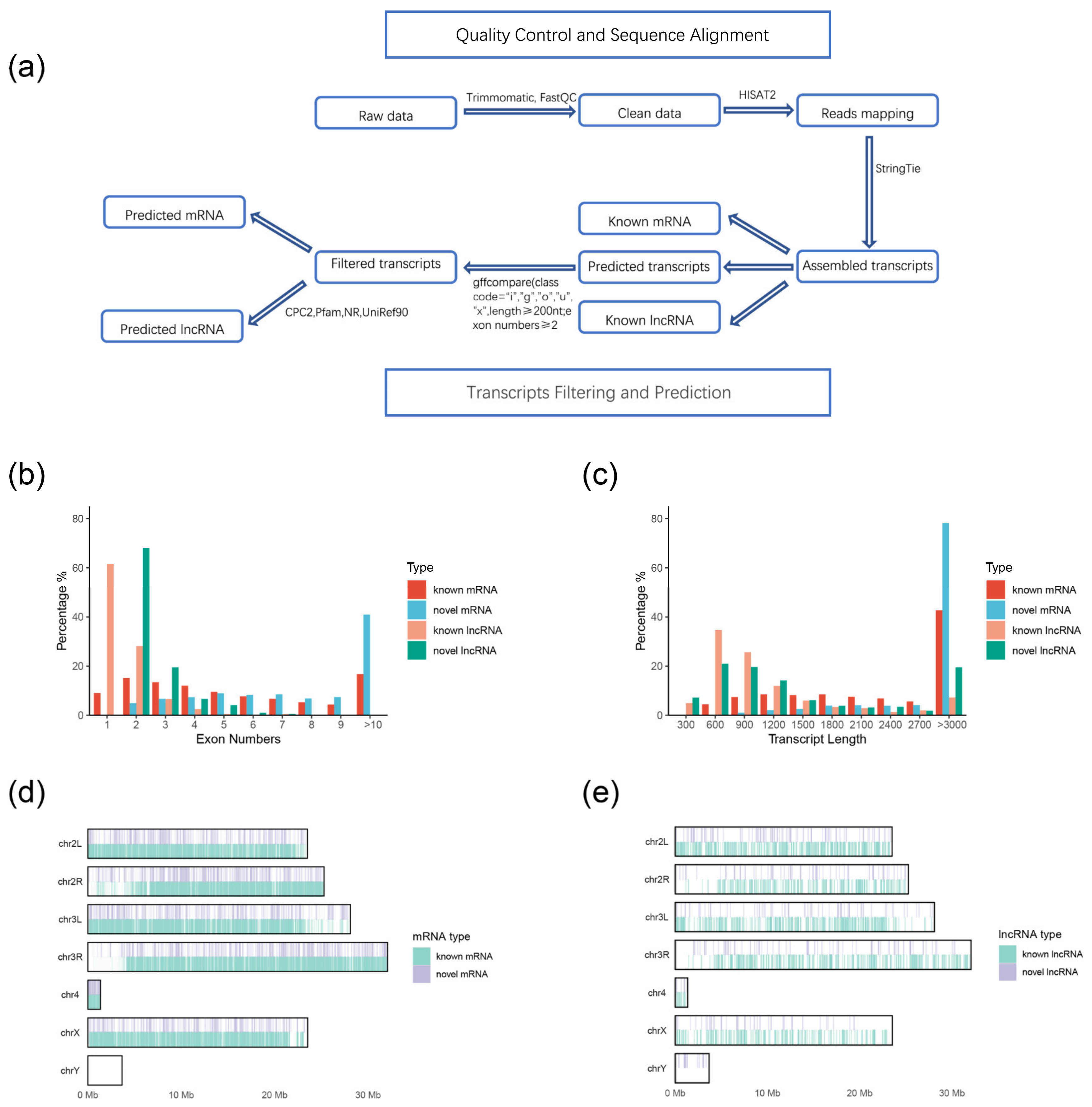
2.2. Identification and Genomic Characteristics of lncRNAs and mRNAs
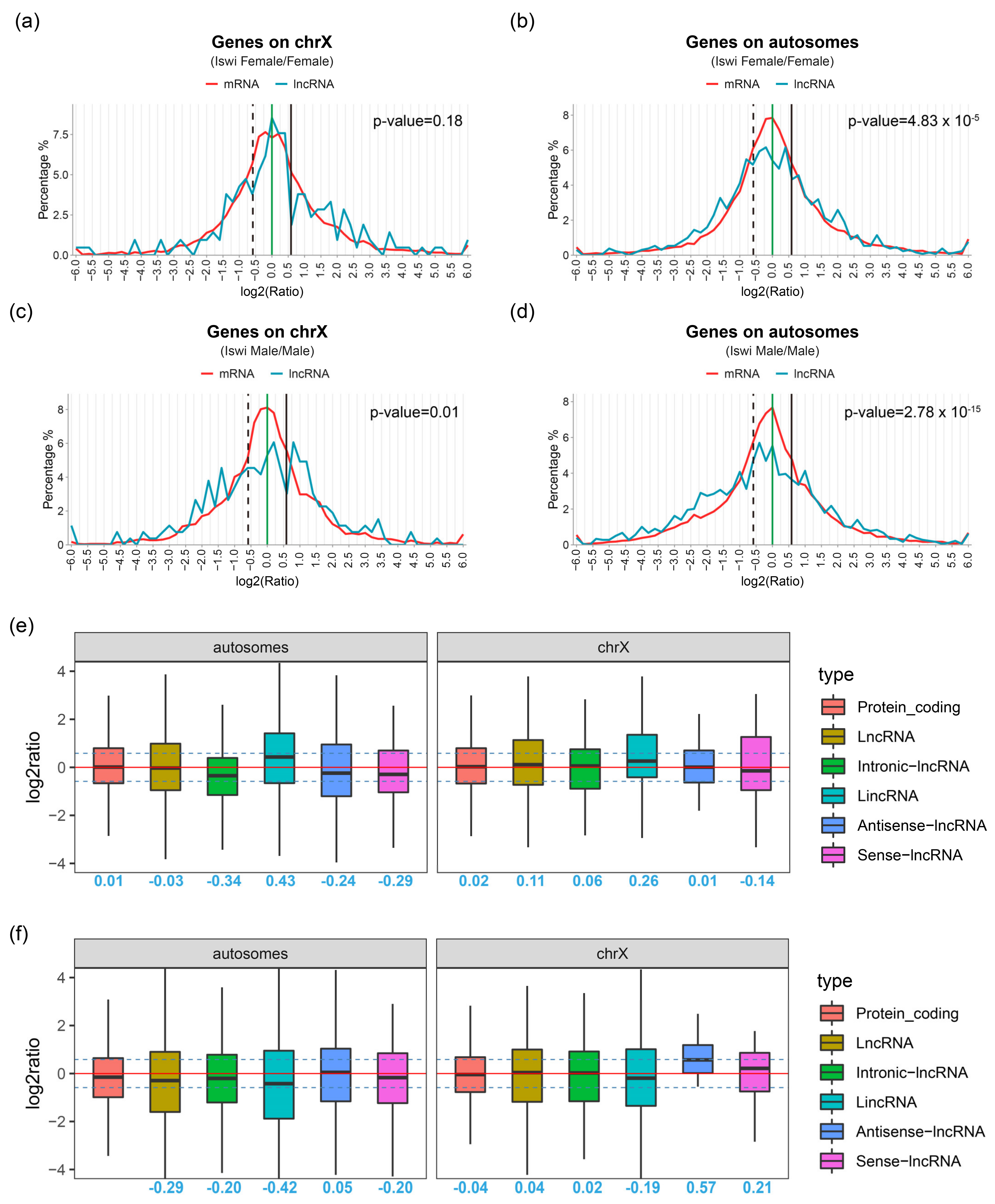
2.3. LncRNAs Expression Changes Significantly in ISWI-Mutated Background
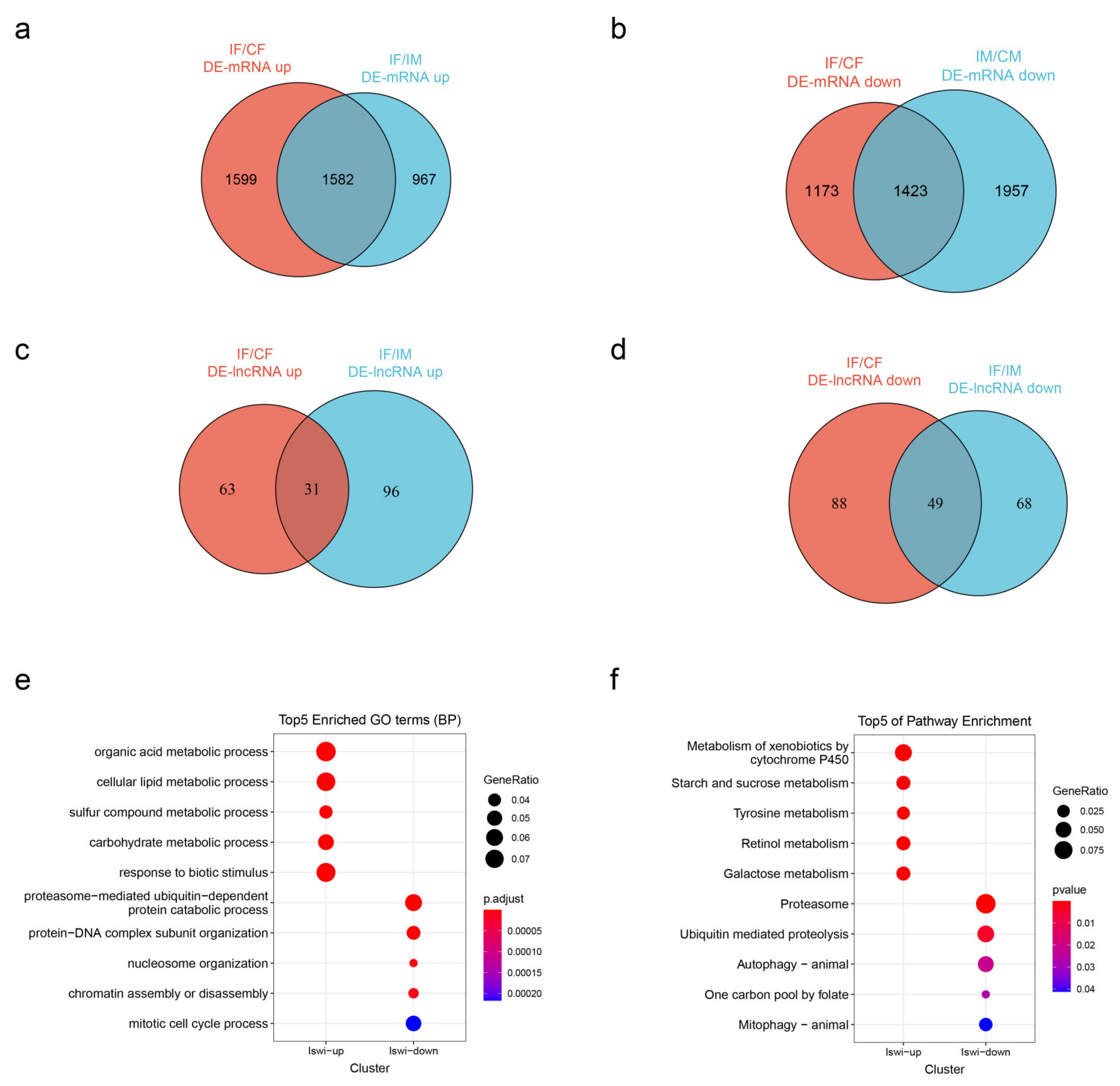
2.4. Differentially Expressed mRNAs and lncRNAs
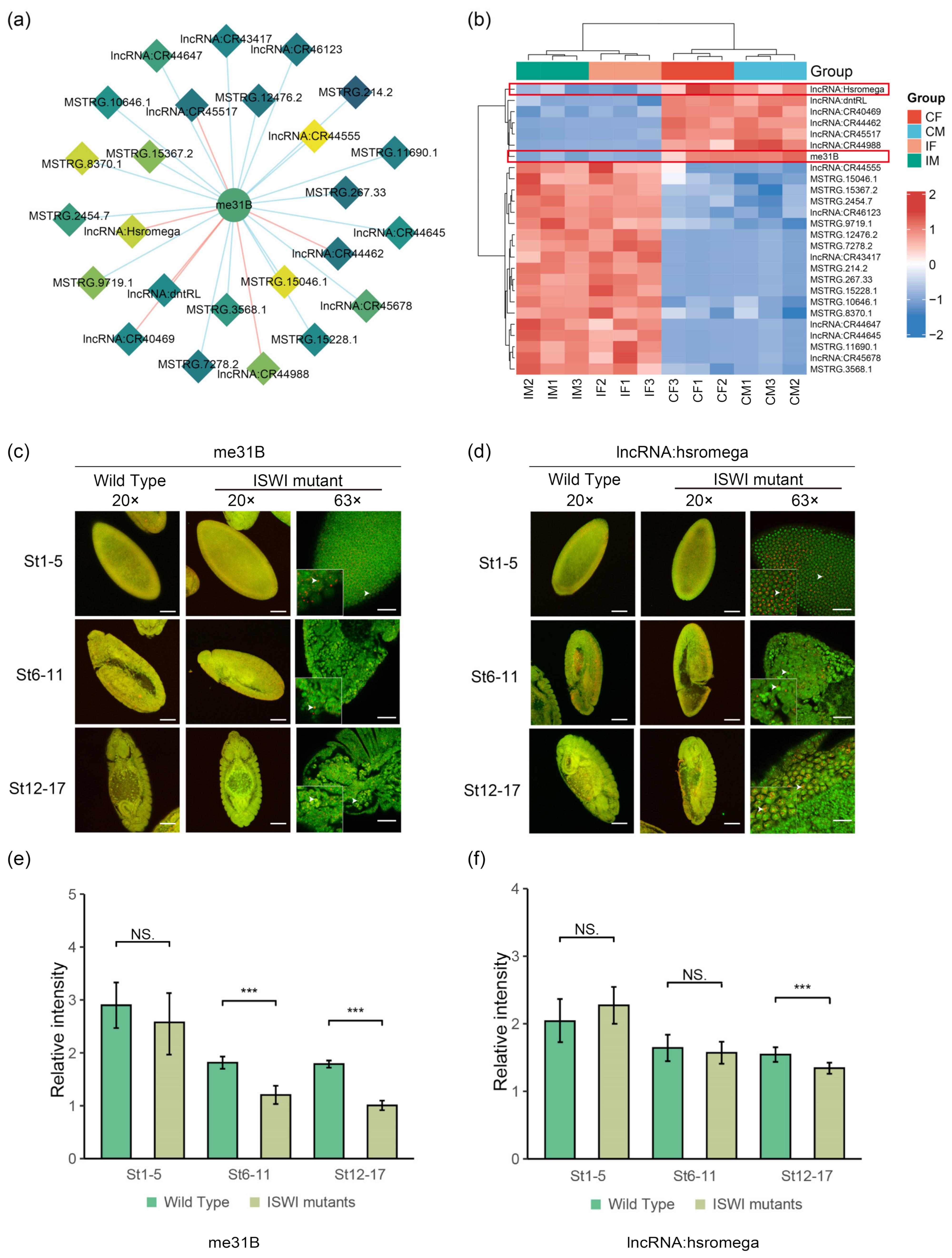
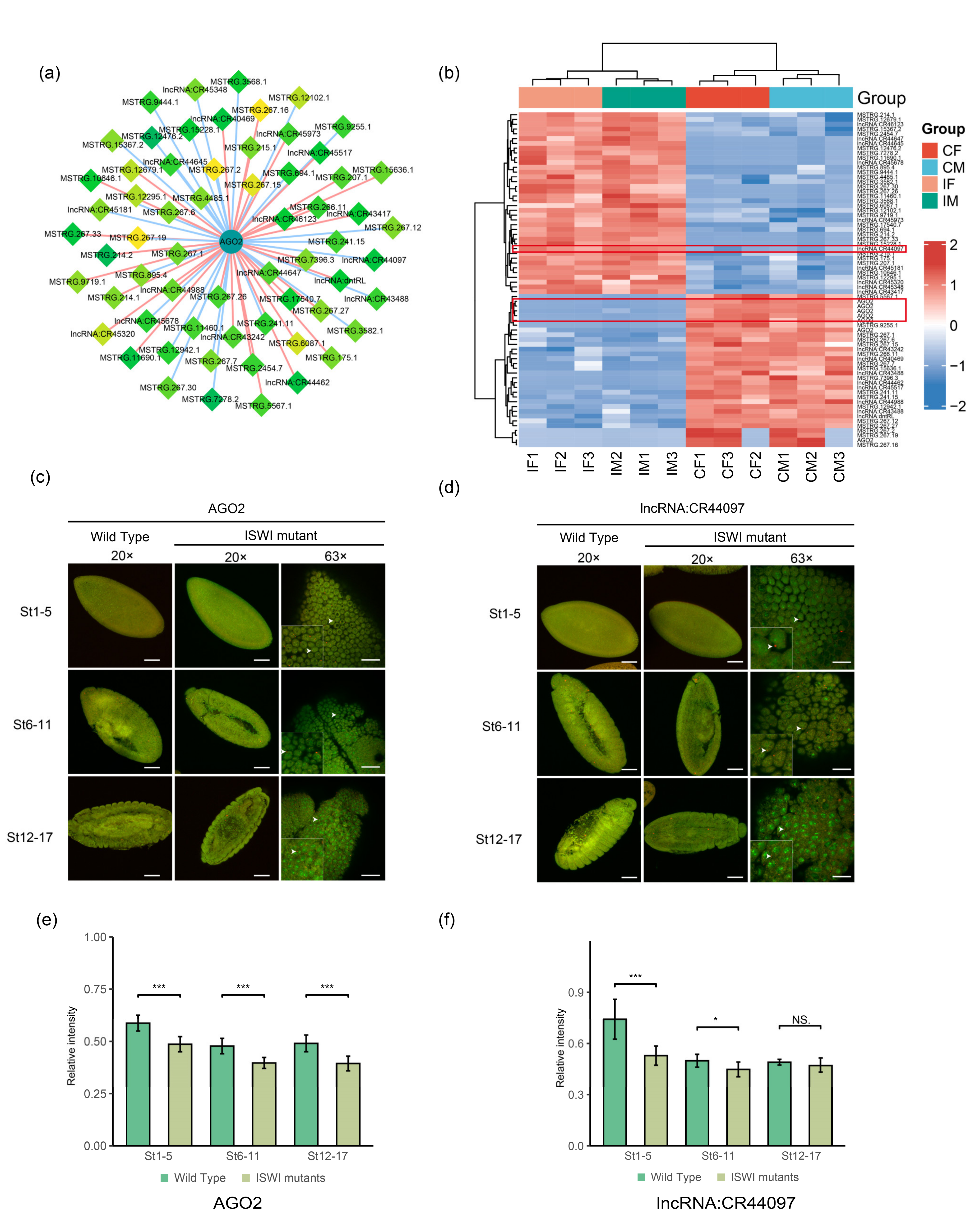
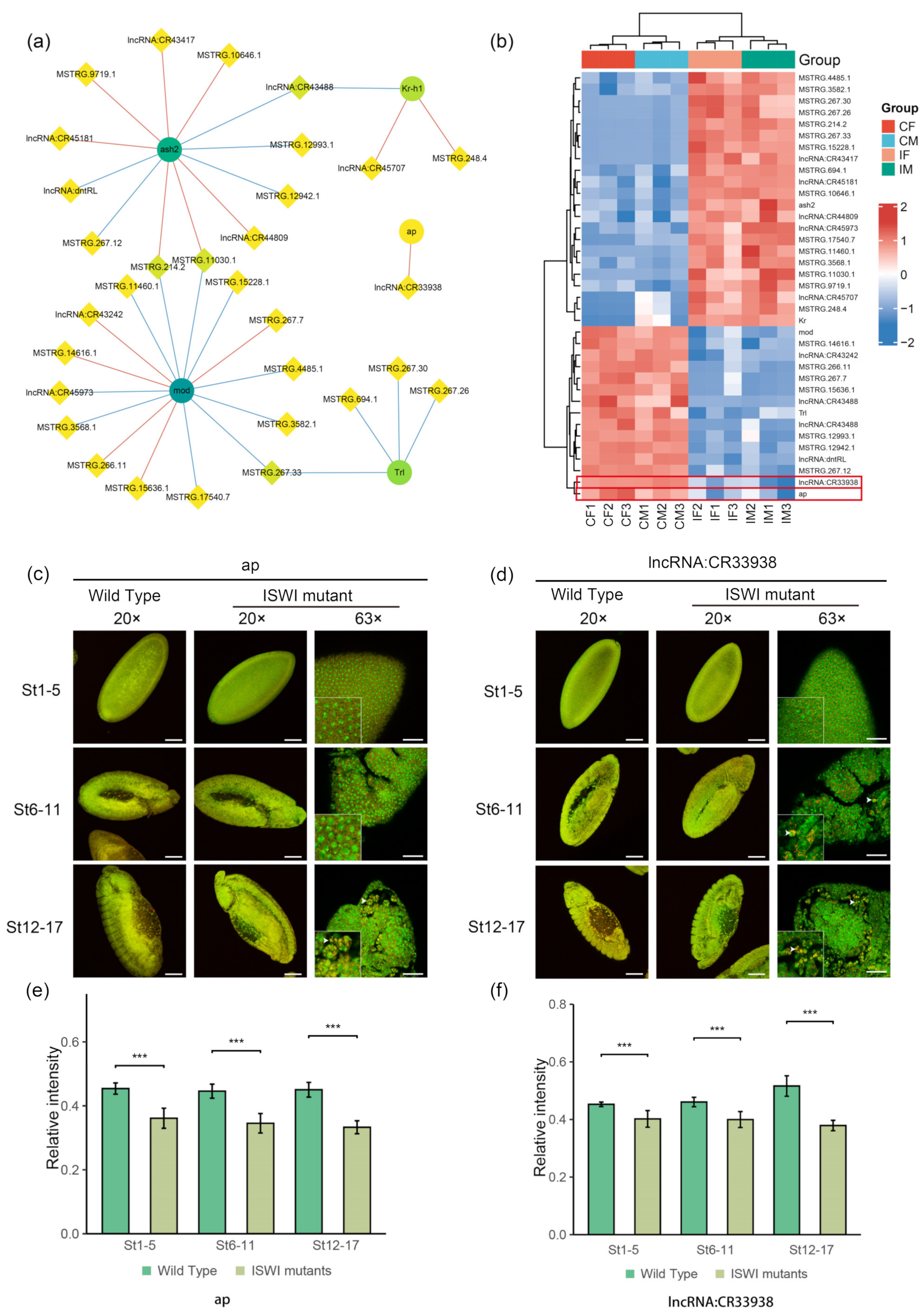
2.5. Potential Interactors of DE-lncRNAs
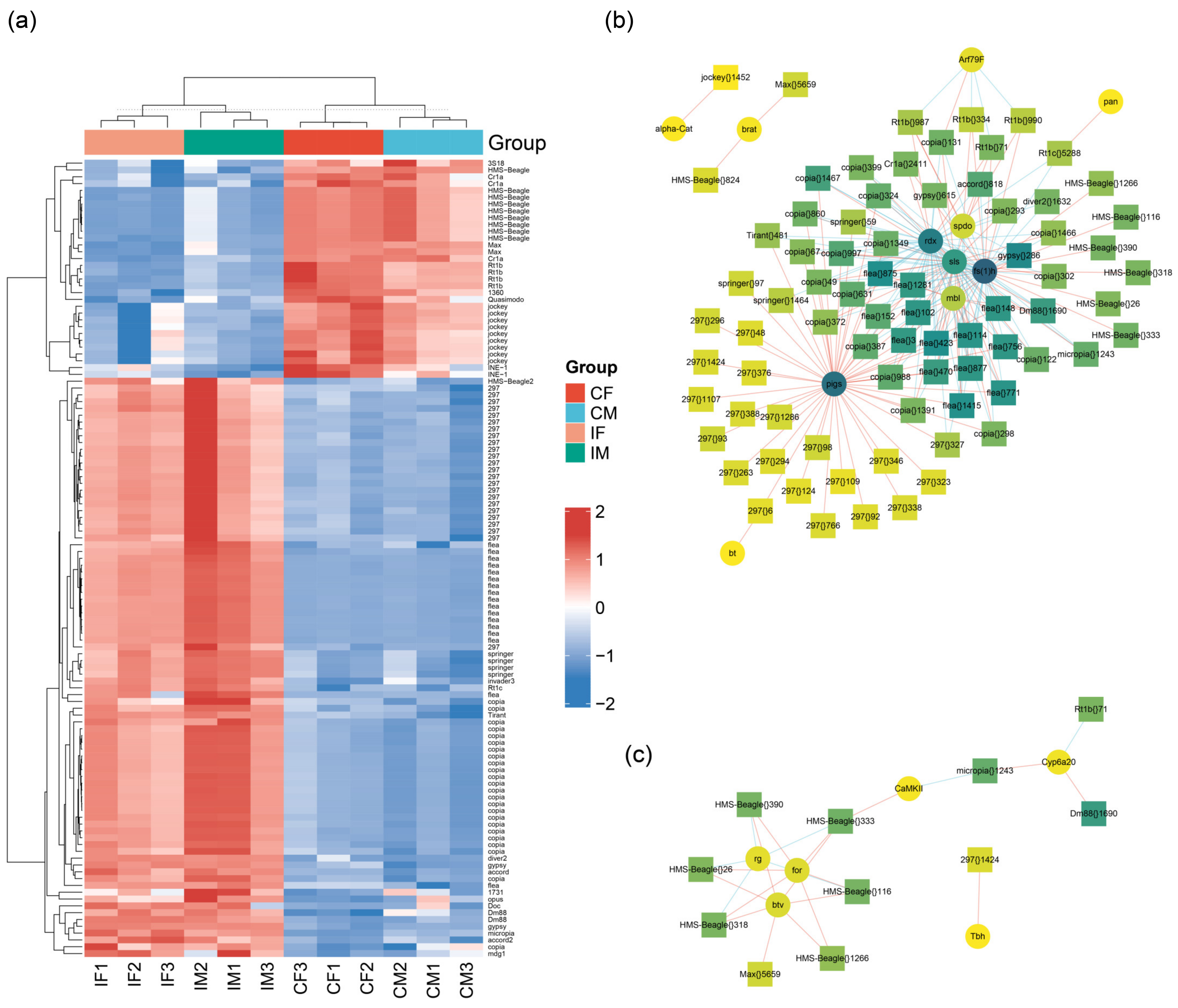
2.6. Differentially Expressed Transposons
3. Discussion
4. Materials and Methods
4.1. Drosophila Stocks and Crosses
4.2. Immunostaining of Chromosomes
4.3. Embryo TSA-FISH
4.4. RNA Extraction and Sequencing
4.5. RNA-Seq Reads Mapping and Transcriptome Assembly
4.6. Transcripts Filtering and Prediction
4.7. Comparisons between lncRNAs and mRNA Transcripts
4.8. Ratio Distribution
4.9. Differential Expression Analysis
4.10. Potential Reactors of DE-lncRNA
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsukiyama, T.; Danial, C.; Tamkun, J.; Wu, C. ISWI, a member of the SWI2/SNF2 ATPase family, encodes the 140 kDa subunit of the nucleosome remodeling factor. Cell 1995, 83, 1021–1026. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Gong, H.; Wang, P.; Zhu, Y.; Peng, H.; Cui, Y.; Li, H.; Liu, J.; Wang, Z. The emerging role of ISWI chromatin remodeling complexes in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 346. [Google Scholar] [CrossRef] [PubMed]
- Borner, K.; Jain, D.; Vazquez-Pianzola, P.; Vengadasalam, S.; Steffen, N.; Fyodorov, D.V.; Tomancak, P.; Konev, A.; Suter, B.; Becker, P.B. A role for tuned levels of nucleosome remodeler subunit ACF1 during Drosophila oogenesis. Dev. Biol. 2016, 411, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Klinker, H.; Mueller-Planitz, F.; Yang, R.L.; Forne, I.; Liu, C.F.; Nordenskiold, L.; Becker, P.B. ISWI Remodelling of Physiological Chromatin Fibres Acetylated at Lysine 16 of Histone H4. PLoS ONE 2014, 9, e88411. [Google Scholar] [CrossRef] [Green Version]
- Mizuguchi, G.; Tsukiyama, T.; Wisniewski, J.; Wu, C. Role of nucleosome remodeling factor NURF in transcriptional activation of chromatin. Mol. Cell 1997, 1, 141–150. [Google Scholar] [CrossRef]
- Mizutani, R.; Wakamatsu, A.; Tanaka, N.; Yoshida, H.; Tochigi, N.; Suzuki, Y.; Oonishi, T.; Tani, H.; Tano, K.; Ijiri, K.; et al. Identification and characterization of novel genotoxic stress-inducible nuclear long noncoding RNAs in mammalian cells. PLoS ONE 2012, 7, e34949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Mo, X.; Fu, L.; Xiao, B.; Guo, J. Molecular mechanisms of long noncoding RNAs on gastric cancer. Oncotarget 2016, 7, 8601–8612. [Google Scholar] [CrossRef] [Green Version]
- Scheuermann, J.C.; Boyer, L.A. Getting to the heart of the matter: Long non-coding RNAs in cardiac development and disease. EMBO J. 2013, 32, 1805–1816. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Camilleri-Robles, C.; Amador, R.; Klein, C.C.; Guigo, R.; Corominas, M.; Ruiz-Romero, M. Genomic and functional conservation of lncRNAs: Lessons from flies. Mamm. Genome 2022, 33, 328–342. [Google Scholar] [CrossRef]
- Yang, F.; Deng, X.; Ma, W.; Berletch, J.B.; Rabaia, N.; Wei, G.; Moore, J.M.; Filippova, G.N.; Xu, J.; Liu, Y.; et al. The lncRNA Firre anchors the inactive X chromosome to the nucleolus by binding CTCF and maintains H3K27me3 methylation. Genome Biol. 2015, 16, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seila, A.C.; Calabrese, J.M.; Levine, S.S.; Yeo, G.W.; Rahl, P.B.; Flynn, R.A.; Young, R.A.; Sharp, P.A. Divergent transcription from active promoters. Science 2008, 322, 1849–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartford CCR, L.A. When Long Noncoding Becomes Protein Coding. Mol. Cell Biol. 2020, 40, e00528-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meller, V.H.; Rattner, B.P. The roX genes encode redundant male-specific lethal transcripts required for targeting of the MSL complex. EMBO J. 2002, 21, 1084–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Y.; Hu, F.; Zhou, Y.; Wu, F.; Gaut, B.S. Maize transposable elements contribute to long non-coding RNAs that are regulatory hubs for abiotic stress response. BMC Genom. 2019, 20, 864. [Google Scholar] [CrossRef]
- Marsano, R.M.; Dimitri, P. Constitutive Heterochromatin in Eukaryotic Genomes: A Mine of Transposable Elements. Cells 2022, 11, 761. [Google Scholar] [CrossRef]
- Percharde, M.; Sultana, T.; Ramalho-Santos, M. What Doesn’t Kill You Makes You Stronger: Transposons as Dual Players in Chromatin Regulation and Genomic Variation. Bioessays 2020, 42, e1900232. [Google Scholar] [CrossRef] [Green Version]
- McCullers, T.J.; Steiniger, M. Transposable elements in Drosophila. Mob. Genet. Elem. 2017, 7, 1–18. [Google Scholar] [CrossRef] [Green Version]
- SanMiguel, P.; Tikhonov, A.; Jin, Y.K.; Motchoulskaia, N.; Zakharov, D.; Melake-Berhan, A.; Springer, P.S.; Edwards, K.J.; Lee, M.; Avramova, Z.; et al. Nested retrotransposons in the intergenic regions of the maize genome. Science 1996, 274, 765–768. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [Green Version]
- Tenaillon, M.I.; Hufford, M.B.; Gaut, B.S.; Ross-Ibarra, J. Genome size and transposable element content as determined by high-throughput sequencing in maize and Zea luxurians. Genome Biol. Evol. 2011, 3, 219–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barron, M.G.; Fiston-Lavier, A.S.; Petrov, D.A.; Gonzalez, J. Population genomics of transposable elements in Drosophila. Annu. Rev. Genet. 2014, 48, 561–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lannoy, N.; Hermans, C. Principles of genetic variations and molecular diseases: Applications in hemophilia A. Crit. Rev. Oncol. Hematol. 2016, 104, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pedro, D.L.F.; Lorenzetti, A.P.R.; Domingues, D.S.; Paschoal, A.R. PlaNC-TE: A comprehensive knowledgebase of non-coding RNAs and transposable elements in plants. Database 2018, 2018, bay078. [Google Scholar] [CrossRef]
- Moschall, R.; Gaik, M.; Medenbach, J. Promiscuity in post-transcriptional control of gene expression: Drosophila sex-lethal and its regulatory partnerships. FEBS Lett. 2017, 591, 1471–1488. [Google Scholar] [CrossRef] [Green Version]
- Haussmann, I.U.; Bodi, Z.; Sanchez-Moran, E.; Mongan, N.P.; Archer, N.; Fray, R.G.; Soller, M. m(6)A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature 2016, 540, 301–304. [Google Scholar] [CrossRef] [Green Version]
- Corona, D.F.; Clapier, C.R.; Becker, P.B.; Tamkun, J.W. Modulation of ISWI function by site-specific histone acetylation. EMBO Rep. 2002, 3, 242–247. [Google Scholar] [CrossRef]
- Judd, J.; Duarte, F.M.; Lis, J.T. Pioneer-like factor GAF cooperates with PBAP (SWI/SNF) and NURF (ISWI) to regulate transcription. Genes Dev. 2021, 35, 147–156. [Google Scholar] [CrossRef]
- Prabhakaran, M.; Kelley, R.L. Mutations in the transcription elongation factor SPT5 disrupt a reporter for dosage compensation in Drosophila. PLoS Genet. 2012, 8, e1003073. [Google Scholar] [CrossRef] [Green Version]
- Espinas, M.L.; Canudas, S.; Fanti, L.; Pimpinelli, S.; Casanova, J.; Azorin, F. The GAGA factor of Drosophila interacts with SAP18, a Sin3-associated polypeptide. EMBO Rep. 2000, 1, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, C.D.; Stephens, G.E.; Thompson, B.A.; Funches, L.; Bernat, J.A.; Craig, C.A.; Elgin, S.C. Heterochromatin protein 2 (HP2), a partner of HP1 in Drosophila heterochromatin. Proc. Natl. Acad. Sci. USA 2002, 99, 14332–14337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, K.A.; Khoury, M.J.; Wirtz-Peitz, F.; Bilder, D. Evidence for a nuclear role for Drosophila Dlg as a regulator of the NURF complex. Mol. Biol. Cell 2021, 32, ar23. [Google Scholar] [CrossRef] [PubMed]
- Erickson, J.W. Primary Sex Determination in Drosophila melanogaster Does Not Rely on the Male-Specific Lethal Complex. Genetics 2016, 202, 541–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slack, C.; Alic, N.; Foley, A.; Cabecinha, M.; Hoddinott, M.P.; Partridge, L. The Ras-Erk-ETS-Signaling Pathway Is a Drug Target for Longevity. Cell 2015, 162, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Yoo, E.; Lee, H.; Park, K.; Hur, J.H.; Lim, C. LSM12 and ME31B/DDX6 Define Distinct Modes of Posttranscriptional Regulation by ATAXIN-2 Protein Complex in Drosophila Circadian Pacemaker Neurons. Mol. Cell 2017, 66, 129–140.e7. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, A.; Amikura, R.; Hanyu, K.; Kobayashi, S. Me31B silences translation of oocyte-localizing RNAs through the formation of cytoplasmic RNP complex during Drosophila oogenesis. Development 2001, 128, 3233–3242. [Google Scholar] [CrossRef]
- Ray, M.; Acharya, S.; Shambhavi, S.; Lakhotia, S.C. Over-expression of Hsp83 in grossly depleted hsromge lncRNA background causes synthetic lethality and l(2)gl phenocopy in Drosophila. J. Biosci. 2019, 44, 36. [Google Scholar] [CrossRef]
- Lécuyer, E.; Yoshida, H.; Parthasarathy, N.; Alm, C.; Babak, T.; Cerovina, T.; Hughes, T.R.; Tomancak, P.; Krause, H.M. Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell 2007, 131, 174–187. [Google Scholar] [CrossRef] [Green Version]
- Wilk, R.; Hu, J.; Blotsky, D.; Krause, H.M. Diverse and pervasive subcellular distributions for both coding and long noncoding RNAs. Genes Dev. 2016, 30, 594–609. [Google Scholar] [CrossRef] [Green Version]
- Meyer, W.J.; Schreiber, S.; Guo, Y.; Volkmann, T.; Welte, M.A.; Muller, H.A. Overlapping functions of argonaute proteins in patterning and morphogenesis of Drosophila embryos. PLoS Genet. 2006, 2, e134. [Google Scholar] [CrossRef]
- Hammond, S.M.; Boettcher, S.; Caudy, A.A.; Kobayashi, R.; Hannon, G.J. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science 2001, 293, 1146–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhadra, M.P.; Bhadra, U.; Kundu, J.; Birchler, J.A. Gene expression analysis of the function of the male-specific lethal complex in Drosophila. Genetics 2005, 169, 2061–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, W.; Birchler, J.A. Identification of Inverse Regulator-a (Inr-a) as Synonymous with Pre-mRNA Cleavage Complex II Protein (Pcf11) in Drosophila. G3 2012, 2, 701–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, B.; Kim, H.Y.; Chen, X.; Shen, W.; Jang, J.S.; Stein, S.N.; Cormier, O.; Pereira, L.; Shih, C.R.Y.; Krieger, C.; et al. 20-hydroxyecdysone (20E) signaling regulates amnioserosa morphogenesis during Drosophila dorsal closure: EcR modulates gene expression in a complex with the AP-1 subunit, Jun. Biol. Open 2021, 10, bio058605. [Google Scholar] [CrossRef] [PubMed]
- Keegan, S.E.; Hughes, S.C. Role of nuclear-cytoplasmic protein localization during Drosophila neuroblast development. Genome 2021, 64, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Arama, E.; Dickman, D.; Kimchie, Z.; Shearn, A.; Lev, Z. Mutations in the beta-propeller domain of the Drosophila brain tumor (brat) protein induce neoplasm in the larval brain. Oncogene 2000, 19, 3706–3716. [Google Scholar] [CrossRef] [Green Version]
- Perkins, A.D.; Tanentzapf, G. An ongoing role for structural sarcomeric components in maintaining Drosophila melanogaster muscle function and structure. PLoS ONE 2014, 9, e99362. [Google Scholar] [CrossRef]
- Li, D.; Liu, J.; Liu, W.; Li, G.; Yang, Z.; Qin, P.; Xu, L. The ISWI remodeler in plants: Protein complexes, biochemical functions, and developmental roles. Chromosoma 2017, 126, 365–373. [Google Scholar] [CrossRef]
- Yaniv, M. Chromatin remodeling: From transcription to cancer. Cancer Genet. 2014, 207, 352–357. [Google Scholar] [CrossRef]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Misteli, T. Beyond the sequence: Cellular organization of genome function. Cell 2007, 128, 787–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapier, C.R. Sophisticated Conversations between Chromatin and Chromatin Remodelers, and Dissonances in Cancer. Int. J. Mol. Sci. 2021, 22, 5578. [Google Scholar] [CrossRef] [PubMed]
- Jandura, A.; Krause, H.M. The New RNA World: Growing Evidence for Long Noncoding RNA Functionality. Trends Genet. 2017, 33, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Währisch, S.; Beisaw, A.; Macura, K.; Bläss, G.; Kellis, M.; Werber, M.; et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell 2013, 24, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Constanty, F.; Shkumatava, A. lncRNAs in development and differentiation: From sequence motifs to functional characterization. Development 2021, 148, dev182741. [Google Scholar] [CrossRef]
- Li, K.; Tian, Y.; Yuan, Y.; Fan, X.; Yang, M.; He, Z.; Yang, D. Insights into the Functions of LncRNAs in Drosophila. Int. J. Mol. Sci. 2019, 20, 4646. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Geng, D.; Li, S.; Chen, Z.; Sun, M. LncRNA HOTAIR influences cell growth, migration, invasion, and apoptosis via the miR-20a-5p/HMGA2 axis in breast cancer. Cancer Med. 2018, 7, 842–855. [Google Scholar] [CrossRef]
- Drummond-Barbosa, D. Local and Physiological Control of Germline Stem Cell Lineages in Drosophila melanogaster. Genetics 2019, 213, 9–26. [Google Scholar] [CrossRef]
- Birchler, J.A. Parallel Universes for Models of X Chromosome Dosage Compensation in Drosophila: A Review. Cytogenet. Genome Res. 2016, 148, 52–67. [Google Scholar] [CrossRef]
- Finnegan, D.J. Eukaryotic transposable elements and genome evolution. Trends Genet. 1989, 5, 103–107. [Google Scholar] [CrossRef]
- Pradhan, R.K.; Ramakrishna, W. Transposons: Unexpected players in cancer. Gene 2022, 808, 145975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qi, H.; Huang, C.; Yuan, L.; Zhang, L.; Wang, R.; Tian, Y.; Sun, L. Interaction of Male Specific Lethal complex and genomic imbalance on global gene expression in Drosophila. Sci. Rep. 2021, 11, 19679. [Google Scholar] [CrossRef] [PubMed]
- Lecuyer, E.; Parthasarathy, N.; Krause, H.M. Fluorescent in situ hybridization protocols in Drosophila embryos and tissues. Methods Mol. Biol. 2008, 420, 289–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Yuan, Z.; Guo, D.; Hou, B.; Yin, C.; Zhang, W.; Li, F. Genome-wide identification of long noncoding RNA genes and their potential association with fecundity and virulence in rice brown planthopper, Nilaparvata lugens. BMC Genom. 2015, 16, 749. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Shi, G.; Chen, G.; Li, J.; Li, M.; Zou, C.; Fang, C.; Li, C. Transcriptome Analysis Suggests the Roles of Long Intergenic Non-coding RNAs in the Growth Performance of Weaned Piglets. Front. Genet. 2019, 10, 196. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Arndt, W.; Miller, B.L.; Wheeler, T.J.; Schreiber, F.; Bateman, A.; Eddy, S.R. HMMER web server: 2015 update. Nucleic Acids Res. 2015, 43, W30–W38. [Google Scholar] [CrossRef]
- Pirooznia, M.; Perkins, E.J.; Deng, Y. Batch Blast Extractor: An automated blastx parser application. BMC Genom. 2008, 9 (Suppl. S2), S10. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Zhang, S.; Wang, R.; Sun, L. Genome-Wide Identification of Long Noncoding RNA and Their Potential Interactors in ISWI Mutants. Int. J. Mol. Sci. 2022, 23, 6247. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116247
Zhang L, Zhang S, Wang R, Sun L. Genome-Wide Identification of Long Noncoding RNA and Their Potential Interactors in ISWI Mutants. International Journal of Molecular Sciences. 2022; 23(11):6247. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116247
Chicago/Turabian StyleZhang, Ludan, Shuai Zhang, Ruixue Wang, and Lin Sun. 2022. "Genome-Wide Identification of Long Noncoding RNA and Their Potential Interactors in ISWI Mutants" International Journal of Molecular Sciences 23, no. 11: 6247. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116247