
Genome-Wide Identification and Analysis of Ariadne Gene Family Reveal Its Genetic Effects on Agronomic Traits of Brassica napus

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Identification of ARI Genes in B. napus
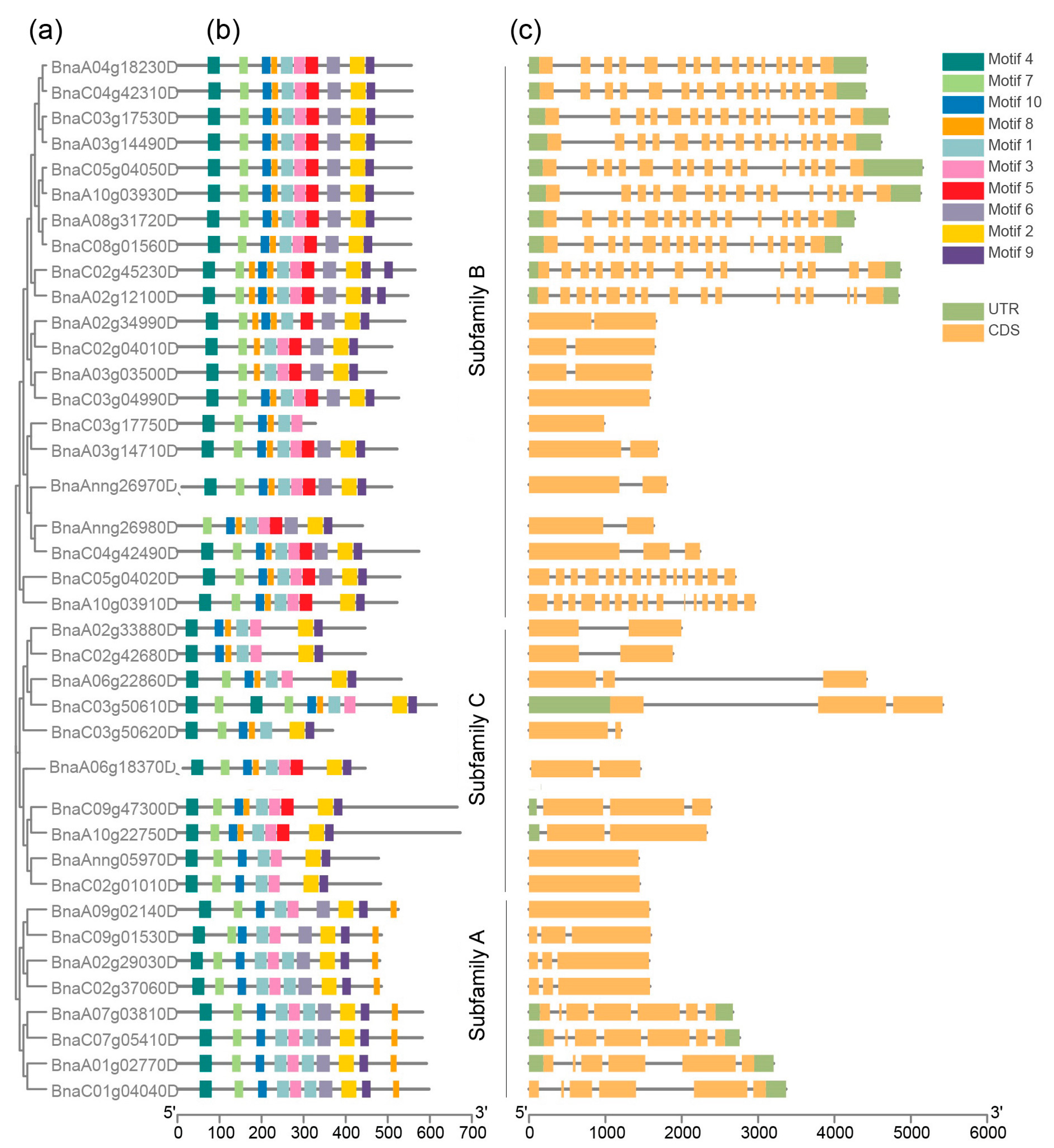
2.2. Phylogenetic Analysis of BnARI Proteins
2.3. Chromosomal Location and ARI Genes Duplication in B. napus
2.4. Gene Structure and Conserved Motif Analysis of BnARIs
2.5. Cis-Elements and Protein Interaction Analysis of BnARI Genes
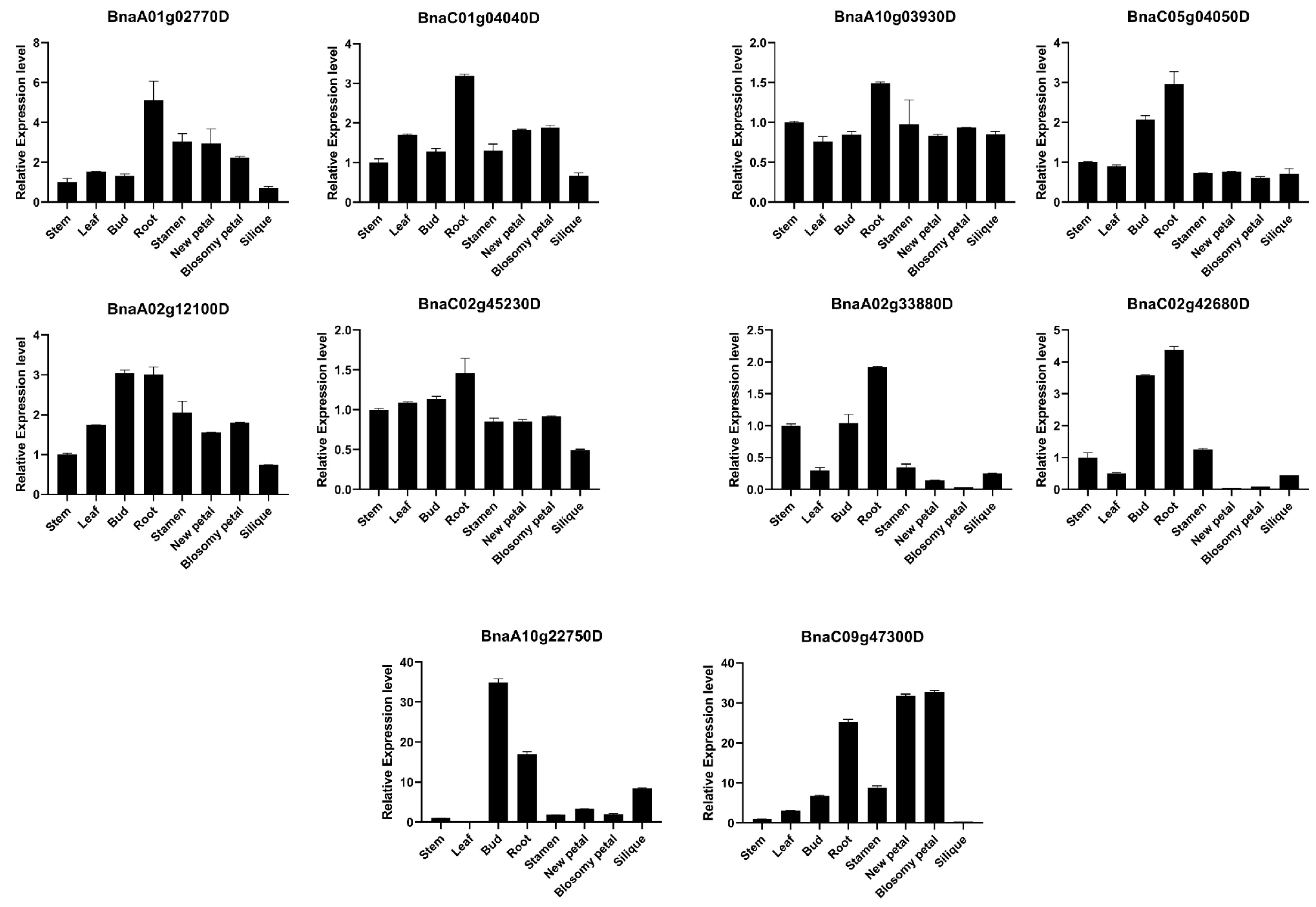
2.6. Expression Patterns of BnARI Genes in Different Tissues and under Abiotic Stresses
2.7. Functional Importance of BnARIs Using Association Mapping Analysis in Natural Population
3. Discussion
4. Materials and Methods
4.1. Identification of ARI Gene Family in B. napus
4.2. Phylogenetic Analysis of BnARI Family
4.3. Chromosomal Distribution, Duplication Status, Ka/Ks Ratio
4.4. Identification of Conserved Motifs and Gene Structure in BnARI Gene Family
4.5. Identification of Cis-Acting Regulatory Elements and Protein–Protein Interaction in BnARI Gene Family
4.6. Expression Analysis of BnARI Genes in Different Tissues and under Abiotic Stress
4.7. RNA Extraction and Reverse Transcription-Quantitative PCR (qRT PCR)
4.8. Functional Significance of BnARIs by Using Association Mapping in Natural Population
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, L.; Hellmann, H. Plant E3 ligases: Flexible enzymes in a sessile world. Mol. Plant 2013, 6, 1388–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smalle, J.; Vierstra, R.D. The ubiquitin 26S proteasome proteolytic pathway. Annu. Rev. Plant Biol. 2004, 55, 555–590. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, H.; Estelle, M. Plant development: Regulation by protein degradation. Science 2002, 297, 793–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vierstra, R.D. The ubiquitin–26S proteasome system at the nexus of plant biology. Nat. Rev. Mol. Cell Biol. 2009, 10, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Mazzucotelli, E.; Belloni, S.; Marone, D.; De Leonardis, A.M.; Guerra, D.; Di Fonzo, N.; Cattivelli, L.; Mastrangelo, A.M. The E3 ubiquitin ligase gene family in plants: Regulation by degradation. Curr. Genom. 2006, 7, 509–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, S.L.; Hauksdóttir, H.; Troy, A.; Herschleb, J.; Kraft, E.; Callis, J. Functional analysis of the RING-type ubiquitin ligase family of Arabidopsis. Plant Physiol. 2005, 137, 13–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, B.; Joshi, D.; Yadav, P.K.; Gupta, A.K.; Bhatt, T.K. Role of ubiquitin-mediated degradation system in plant biology. Front. Plant Sci. 2016, 7, 806. [Google Scholar] [CrossRef]
- Callis, J. The ubiquitination machinery of the ubiquitin system. Arab. Book Am. Soc. Plant Biol. 2014, 12, e0174. [Google Scholar] [CrossRef] [Green Version]
- Mladek, C.; Guger, K.; Hauser, M.T. Identification and characterization of the ARIADNE gene family in Arabidopsis. A group of putative E3 ligases. Plant Physiol. 2003, 131, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Borden, K.L.B. RING domains: Master builders of molecular scaffolds? J. Mol. Biol. 2000, 295, 1103–1112. [Google Scholar] [CrossRef]
- Lim, S.D.; Yim, W.C.; Moon, J.-C.; Kim, D.S.; Lee, B.-M.; Jang, C.S. A gene family encoding RING finger proteins in rice: Their expansion, expression diversity, and co-expressed genes. Plant Mol. Biol. 2010, 72, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.D.; Hwang, J.G.; Jung, C.G.; Hwang, S.G.; Moon, J.C.; Jang, C.S. Comprehensive analysis of the rice RING E3 ligase family reveals their functional diversity in response to abiotic stress. DNA Res. 2013, 20, 299–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.K.; Ryu, M.Y.; Seo, D.H.; Kang, B.G.; Kim, W.T. The Arabidopsis RING E3 ubiquitin ligase AtAIRP2 plays combinatory roles with AtAIRP1 in abscisic acid-mediated drought stress responses. Plant Physiol. 2011, 157, 2240–2257. [Google Scholar] [CrossRef] [Green Version]
- Piñeiro, M.; Jarillo, J.A. Ubiquitination in the control of photoperiodic flowering. Plant Sci. 2013, 198, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Eisenhaber, B.; Chumak, N.; Eisenhaber, F.; Hauser, M.T. The ring between ring fingers (RBR) protein family. Genome Biol. 2007, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Marín, I. Diversification and specialization of plant RBR ubiquitin ligases. PLoS ONE 2010, 5, e11579. [Google Scholar] [CrossRef] [Green Version]
- Marin, I.; Ferrús, A. Comparative genomics of the RBR family, including the Parkinson’s disease–related gene Parkin and the genes of the Ariadne subfamily. Mol. Biol. Evol. 2002, 19, 2039–2050. [Google Scholar] [CrossRef] [Green Version]
- Beasley, S.A.; Hristova, V.A.; Shaw, G.S. Structure of the Parkin in-between-ring domain provides insights for E3-ligase dysfunction in autosomal recessive Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 3095–3100. [Google Scholar] [CrossRef] [Green Version]
- Morett, E.; Bork, P. A novel transactivation domain in parkin. Trends Biochem. Sci. 1999, 24, 229–231. [Google Scholar] [CrossRef]
- Marín, I.; Lucas, J.I.; Gradilla, A.-C.; Ferrús, A. Parkin and relatives: The RBR family of ubiquitin ligases. Physiol. Genom. 2004, 17, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Marteijn, J.A.F.; van Emst, L.; Erpelinck-Verschueren, C.A.J.; Nikoloski, G.; Menke, A.; de Witte, T.; Löwenberg, B.; Jansen, J.H.; van der Reijden, B.A. The E3 ubiquitin-protein ligase Triad1 inhibits clonogenic growth of primary myeloid progenitor cells. Blood 2005, 106, 4114–4123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardley, H.C.; Tan, N.G.S.; Rose, S.A.; Markham, A.F.; Robinson, P.A. Features of the parkin/ariadne-like ubiquitin ligase, HHARI, that regulate its interaction with the ubiquitin-conjugating enzyme, Ubch7. J. Biol. Chem. 2001, 276, 19640–19647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duda, D.M.; Olszewski, J.L.; Schuermann, J.P.; Kurinov, I.; Miller, D.J.; Nourse, A.; Alpi, A.F.; Schulman, B.A. Structure of HHARI, a RING-IBR-RING ubiquitin ligase: Autoinhibition of an Ariadne-family E3 and insights into ligation mechanism. Structure 2013, 21, 1030–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilera, M.; Oliveros, M.; Martínez-Padrón, M.; Barbas, J.A.; Ferrús, A. Ariadne-1: A vital Drosophila gene is required in development and defines a new conserved family of ring-finger proteins. Genetics 2000, 155, 1231–1244. [Google Scholar] [CrossRef]
- Itier, J.-M.; Ibáñez, P.; Mena, M.A.; Abbas, N.; Cohen-Salmon, C.; Bohme, G.A.; Laville, M.; Pratt, J.; Corti, O.; Pradier, L. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum. Mol. Genet. 2003, 12, 2277–2291. [Google Scholar] [CrossRef]
- Lang-Mladek, C.; Xie, L.; Nigam, N.; Chumak, N.; Binkert, M.; Neubert, S.; Hauser, M.T. UV-B signaling pathways and fluence rate dependent transcriptional regulation of ARIADNE12. Physiol. Plant. 2012, 145, 527–539. [Google Scholar] [CrossRef]
- Xie, L.; Lang-Mladek, C.; Richter, J.; Nigam, N.; Hauser, M.T. UV-B induction of the E3 ligase ARIADNE12 depends on CONSTITUTIVELY PHOTOMORPHOGENIC 1. Plant Physiol. Biochem. 2015, 93, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, N.; Chen, P.; Gao, M.; Liu, J.; Wang, Y.; Zhao, T.; Li, Y.; Gai, J. Overexpression of a soybean ariadne-like ubiquitin ligase gene GmARI1 enhances aluminum tolerance in Arabidopsis. PLoS ONE 2014, 9, e111120. [Google Scholar] [CrossRef] [Green Version]
- Schallau, A.; Arzenton, F.; Johnston, A.J.; Hähnel, U.; Koszegi, D.; Blattner, F.R.; Altschmied, L.; Haberer, G.; Barcaccia, G.; Bäumlein, H. Identification and genetic analysis of the APOSPORY locus in Hypericum perforatum L. Plant J. 2010, 62, 773–784. [Google Scholar] [CrossRef]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.P.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [Green Version]
- Ayyaz, A.; Miao, Y.; Hannan, F.; Islam, F.; Zhang, K.; Xu, J.; Farooq, M.A.; Zhou, W. Drought tolerance in Brassica napus is accompanied with enhanced antioxidative protection, photosynthetic and hormonal regulation at seedling stage. Physiol. Plant. 2021, 172, 1133–1148. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.A.N.; Shah, T.; Cheng, Y.; Yan, L.Ü.; Zhang, X.; Zou, X. Physiological and molecular responses to cold stress in rapeseed (Brassica napus L.). J. Integr. Agric. 2019, 18, 2742–2752. [Google Scholar]
- Lohani, N.; Jain, D.; Singh, M.B.; Bhalla, P.L. Engineering multiple abiotic stress tolerance in canola, Brassica napus. Front. Plant Sci. 2020, 3. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dong, C.; Hu, M.; Bai, Z.; Tong, C.; Zuo, R.; Liu, Y.; Cheng, X.; Cheng, M.; Huang, J.; et al. Identification of flower-specific promoters through comparative transcriptome analysis in Brassica napus. Int. J. Mol. Sci. 2019, 20, 5949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, H.; Li, T.; Luo, C.; Huang, H.; Ruan, Y.; Li, X.; Niu, Y.; Fan, Y.; Sun, W.; Zhang, K.; et al. Brassicaedb: A gene expression database for brassica crops. Int. J. Mol. Sci. 2020, 21, 5831. [Google Scholar] [CrossRef]
- Cannon, S.B.; Mitra, A.; Baumgarten, A.; Young, N.D.; May, G. The roles of segmental and tandem gene duplication in the evolution of large gene families in Arabidopsis thaliana. BMC Plant Biol. 2004, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, 202–208. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van De Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ali, U.; Zhang, G.; Yu, L.; Fang, S.; Iqbal, S.; Li, H.; Lu, S.; Guo, L. Transcriptome analysis reveals genes commonly responding to multiple abiotic stresses in rapeseed. Mol. Breed. 2019, 39, 158. [Google Scholar] [CrossRef]
- Tang, M. Population Genome Variations and Subgenome Asymmetry in Brassica napus L.; Huazhong Agricultural University: Wuhan, China, 2019. [Google Scholar]
- Cho, S.K.; Ryu, M.Y.; Kim, J.H.; Hong, J.S.; Oh, T.R.; Kim, W.T.; Yang, S.W. RING E3 ligases: Key regulatory elements are involved in abiotic stress responses in plants. BMB Rep. 2017, 50, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albalat, R.; Cañestro, C. Evolution by gene loss. Nat. Rev. Genet. 2016, 17, 379–391. [Google Scholar] [CrossRef]
- Sehrish, S.; Sumbal, W.; Xie, M.; Zhao, C.; Zuo, R.; Gao, F.; Liu, S. Genome-Wide Identification and Characterization of SET Domain Family Genes in Brassica napus L. Int. J. Mol. Sci. 2022, 23, 1936. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Zuo, R.; Bai, Z.; Yang, L.; Zhao, C.; Gao, F.; Cheng, X.; Huang, J.; Liu, Y.; Li, Y. Genome-wide characterization of Serine/Arginine-Rich gene family and its genetic effects on agronomic traits of Brassica napus. Front. Plant Sci. 2022, 13, 829668. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ke, Y.; Wen, J.; Guo, P.; Ran, F.; Wang, M.; Liu, M.; Li, P.; Li, J.; Du, H. Evolution and expression analyses of the MADS-box gene family in Brassica napus. PLoS ONE 2018, 13, e0200762. [Google Scholar] [CrossRef] [PubMed]
- Oudelaar, A.M.; Higgs, D.R. The relationship between genome structure and function. Nat. Rev. Genet. 2021, 22, 154–168. [Google Scholar] [CrossRef]
- Chaudhary, B.; Flagel, L.; Stupar, R.M.; Udall, J.A.; Verma, N.; Springer, N.M.; Wendel, J.F. Reciprocal silencing, transcriptional bias and functional divergence of homeologs in polyploid cotton (Gossypium). Genetics 2009, 182, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Meena, K.K.; Sorty, A.M.; Bitla, U.M.; Choudhary, K.; Gupta, P.; Pareek, A.; Singh, D.P.; Prabha, R.; Sahu, P.K.; Gupta, V.K. Abiotic stress responses and microbe-mediated mitigation in plants: The omics strategies. Front. Plant Sci. 2017, 8, 172. [Google Scholar] [CrossRef]
- Zhu, W.; Guo, Y.; Chen, Y.; Wu, D.; Jiang, L. Genome-wide identification, phylogenetic and expression pattern analysis of GATA family genes in Brassica napus. BMC Plant Biol. 2020, 20, 543. [Google Scholar] [CrossRef]
- Borges, F.; Gomes, G.; Gardner, R.; Moreno, N.; McCormick, S.; Feijó, J.A.; Becker, J.D. Comparative transcriptomics of Arabidopsis sperm cells. Plant Physiol. 2008, 148, 1168–1181. [Google Scholar] [CrossRef] [Green Version]
- Haerizadeh, F.; Wong, C.E.; Bhalla, P.L.; Gresshoff, P.M.; Singh, M.B. Genomic expression profiling of mature soybean (Glycine max) pollen. BMC Plant Biol. 2009, 9, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavicic, M.; Mouhu, K.; Wang, F.; Bilicka, M.; Chovanček, E.; Himanen, K. Genomic and phenomic screens for flower related RING type ubiquitin E3 ligases in Arabidopsis. Front. Plant Sci. 2017, 8, 416. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Yu, C.; Chen, Y.; Lu, C.; Hwang, J. Prediction of protein subcellular localization. Proteins Struct. Funct. Bioinform. 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Chen, C.; Xia, R.; Chen, H.; He, Y. TBtools, a toolkit for biologists integrating various biological data handling tools with a user-friendly interface. bioRxiv 2018, 289660. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Zhu, Y.; Liu, R.; Zhang, A.; Zhu, M.; Xu, W.; Lin, A.; Lu, K.; Li, J. Genome wide identification and comparative analysis of glutathione transferases (GST) family genes in Brassica napus. Sci. Rep. 2019, 9, 9196. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Classification | pI | MW (kDa) | Amino acids | Chromosome | Start | End | Duplication Type | Exon Number | Subcellular Localization |
|---|---|---|---|---|---|---|---|---|---|---|
| BnaC01g04040D | subfamily A | 5.44 | 68.30577 | 598 | C01 | 2107232 | 2110597 | WGD or Segmental | 6 | Nuclear |
| BnaA01g02770D | 5.49 | 67.70018 | 592 | A01 | 1363570 | 1366772 | WGD or Segmental | 6 | Nuclear | |
| BnaA07g03810D | 5.5 | 66.84028 | 583 | A07 | 3464224 | 3466892 | WGD or Segmental | 7 | Nuclear | |
| BnaC07g05410D | 5.39 | 67.01159 | 582 | C07 | 8591383 | 8594140 | WGD or Segmental | 7 | Nuclear | |
| BnaA09g02140D | 5.38 | 61.03602 | 525 | A09 | 1051875 | 1053449 | WGD or Segmental | 1 | Nuclear | |
| BnaA02g29030D | 5.28 | 55.36126 | 481 | A02 | 21265416 | 21266987 | WGD or Segmental | 3 | Nuclear | |
| BnaC09g01530D | 5.45 | 56.70796 | 485 | C09 | 836916 | 838508 | WGD or Segmental | 3 | Nuclear | |
| BnaC02g37060D | 5.07 | 55.77174 | 485 | C02 | 40014335 | 40015918 | WGD or Segmental | 3 | Nuclear | |
| BnaC02g04010D | subfamily B | 5.53 | 58.47465 | 510 | C02 | 2040175 | 2041821 | WGD or Segmental | 2 | Nuclear |
| BnaC05g04050D | 5.12 | 63.11043 | 556 | C05 | 1976038 | 1981190 | WGD or Segmental | 15 | Nuclear | |
| BnaA02g34990D | 5.83 | 62.01988 | 541 | A02_random | 24816 | 26477 | WGD or Segmental | 2 | Nuclear | |
| BnaA10g03930D | 5.11 | 63.19455 | 559 | A10 | 2088735 | 2093862 | WGD or Segmental | 15 | Nuclear | |
| BnaA03g03500D | 5 | 56.69741 | 496 | A03 | 1688116 | 1689720 | WGD or Segmental | 2 | Nuclear | |
| BnaC03g04990D | 5.02 | 59.7861 | 526 | C03 | 2416072 | 2417649 | WGD or Segmental | 1 | Nuclear | |
| BnaA04g18230D | 4.99 | 63.39872 | 556 | A04 | 14658509 | 14662929 | WGD or Segmental | 15 | Nuclear | |
| BnaC03g17530D | 4.9 | 63.69074 | 558 | C03 | 8960359 | 8965066 | WGD or Segmental | 15 | Nuclear | |
| BnaC04g42310D | 4.92 | 63.61489 | 558 | C04 | 42886627 | 42891037 | WGD or Segmental | 15 | Nuclear | |
| BnaA08g31720D | 5.08 | 62.69513 | 554 | A08_random | 2100605 | 2104866 | WGD or Segmental | 15 | Nuclear | |
| BnaA03g14490D | 4.85 | 63.39747 | 555 | A03 | 6669544 | 6674154 | WGD or Segmental | 15 | Nuclear | |
| BnaC08g01560D | 5.03 | 62.76486 | 555 | C08 | 1211774 | 1215865 | WGD or Segmental | 15 | Nuclear | |
| BnaA02g12100D | 5.06 | 62.48365 | 548 | A02 | 6326435 | 6331272 | WGD or Segmental | 16 | Nuclear | |
| BnaC02g45230D | 5.05 | 64.28373 | 565 | C02_random | 956624 | 961488 | WGD or Segmental | 15 | Nuclear | |
| BnaAnng26970D | 4.96 | 57.65184 | 499 | Ann_random | 30922371 | 30924172 | Tandem | 2 | Nuclear | |
| BnaAnng26980D | 6.48 | 51.01917 | 440 | Ann_random | 30926298 | 30927929 | Tandem | 2 | Nuclear | |
| BnaC04g42500D | 5.09 | 63.14806 | 548 | C04 | 43066664 | 43068633 | Tandem | 2 | Nuclear | |
| BnaA03g14710D | 5.83 | 60.14362 | 522 | A03 | 6761186 | 6762872 | WGD or Segmental | 2 | Nuclear | |
| BnaC04g42490D | 5.39 | 66.50624 | 574 | C04 | 43060145 | 43062382 | WGD or Segmental | 3 | Nuclear | |
| BnaC05g04020D | 5.18 | 61.60797 | 529 | C05 | 1967024 | 1969723 | WGD or Segmental | 14 | Nuclear | |
| BnaA10g03910D | 5.28 | 60.45582 | 522 | A10 | 2073034 | 2075988 | WGD or Segmental | 15 | Nuclear | |
| BnaC03g50610D | subfamily C | 4.87 | 75.1735 | 657 | C03 | 35066895 | 35072312 | WGD or Segmental | 3 | Nuclear |
| BnaA02g33880D | 5.15 | 51.08088 | 446 | A02 | 24242394 | 24244388 | WGD or Segmental | 2 | Extracellular | |
| BnaC02g42680D | 5.31 | 51.23706 | 447 | C02 | 45189629 | 45191512 | WGD or Segmental | 2 | Extracellular | |
| BnaA06g22860D | 4.7 | 61.10942 | 532 | A06 | 16009537 | 16013955 | WGD or Segmental | 3 | Nuclear | |
| BnaC03g50620D | 4.82 | 42.64821 | 369 | C03 | 35073684 | 35074888 | Tandem | 2 | Nuclear | |
| BnaC02g01010D | 5.61 | 54.80394 | 483 | C02 | 436156 | 437604 | WGD or Segmental | 1 | Nuclear | |
| BnaA06g18370D | 4.99 | 50.6674 | 447 | A06 | 10552571 | 10553998 | WGD or Segmental | 2 | Nuclear | |
| BnaA10g22750D | 5.06 | 74.29611 | 672 | A10 | 15235470 | 15237795 | WGD or Segmental | 3 | Nuclear | |
| BnaAnng05970D | 5.38 | 54.8181 | 478 | Ann_random | 6115791 | 6117224 | WGD or Segmental | 1 | Nuclear | |
| BnaC09g47300D | 6.05 | 73.81982 | 665 | C09 | 46717181 | 46719562 | WGD or Segmental | 4 | Nuclear |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wahid, S.; Xie, M.; Sarfraz, S.; Liu, J.; Zhao, C.; Bai, Z.; Tong, C.; Cheng, X.; Gao, F.; Liu, S. Genome-Wide Identification and Analysis of Ariadne Gene Family Reveal Its Genetic Effects on Agronomic Traits of Brassica napus. Int. J. Mol. Sci. 2022, 23, 6265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116265
Wahid S, Xie M, Sarfraz S, Liu J, Zhao C, Bai Z, Tong C, Cheng X, Gao F, Liu S. Genome-Wide Identification and Analysis of Ariadne Gene Family Reveal Its Genetic Effects on Agronomic Traits of Brassica napus. International Journal of Molecular Sciences. 2022; 23(11):6265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116265
Chicago/Turabian StyleWahid, Sumbal, Meili Xie, Sehrish Sarfraz, Jie Liu, Chuanji Zhao, Zetao Bai, Chaobo Tong, Xiaohui Cheng, Feng Gao, and Shengyi Liu. 2022. "Genome-Wide Identification and Analysis of Ariadne Gene Family Reveal Its Genetic Effects on Agronomic Traits of Brassica napus" International Journal of Molecular Sciences 23, no. 11: 6265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116265