
Beyond Genetics: Metastasis as an Adaptive Response in Breast Cancer

 and
and
Abstract
:1. Breast Cancer Mortality Is Associated with Metastatic Disease
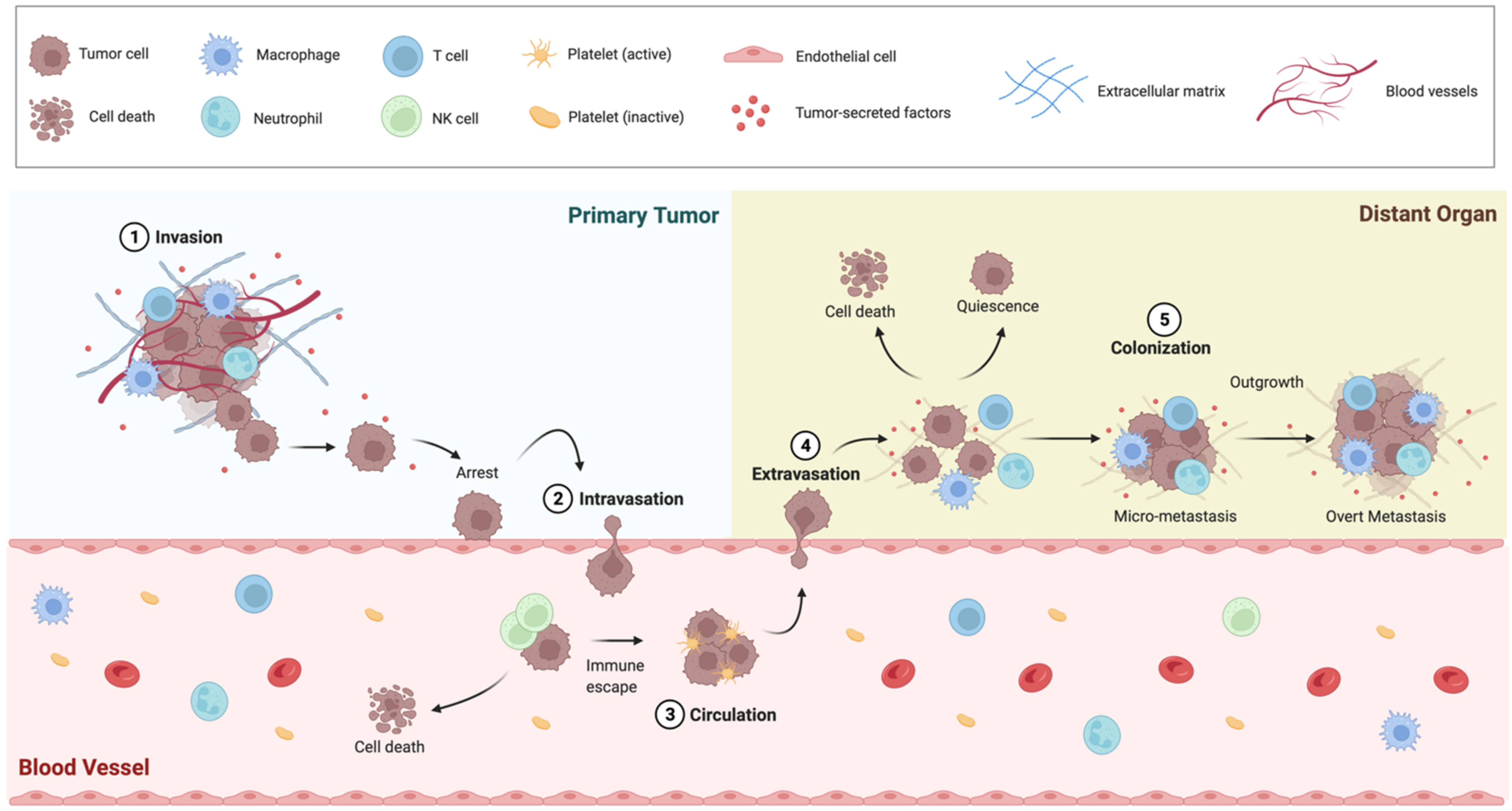
2. The BC Metastatic Progression Is a Multistep Process
2.1. Epithelial-to-Mesenchimal Transition
2.2. Intravasation and Circulating Tumor Cells
2.3. Extravasation
2.4. Metastatic Colonization
3. BC Intra-Tumor Heterogeneity and Metastasis
3.1. Genetic Heterogeneity
3.2. Transcriptional Heterogeneity
3.3. Metabolic Heterogeneity
3.4. Impacts of ITH on Patient Prognosis and Treatment
4. BC Metastatic Progression Is Not a Genetically Selected Trait
5. Adaptive Responses in BC Metastasis
5.1. Hypoxia
5.2. Metabolic Stress
5.3. ER Stress
5.4. Chemotherapy

{kind=link}
{kind=link}
| In Vitro Models | Mouse Models | Zebrafish Models |
|---|---|---|
 |  |  |
|
|
|
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Sims, A.H.; Howell, A.; Howell, S.J.; Clarke, R.B. Origins of breast cancer subtypes and therapeutic implications. Nat. Clin. Pract. Oncol. 2007, 4, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- McGuire, A.; Brown, J.A.; Malone, C.; McLaughlin, R.; Kerin, M.J. Effects of age on the detection and management of breast cancer. Cancers 2015, 7, 908–929. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, R.T.; Chen, Z.; Anderson, G.L.; Rohan, T.; Aragaki, A.; Lane, D.; Dolan, N.C.; Paskett, E.D.; McTiernan, A.; Hubbell, F.A.; et al. Ethnicity and breast cancer: Factors influencing differences in incidence and outcome. J. Natl. Cancer Inst. 2005, 97, 439–448. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, C.E.; Ma, J.; Goding Sauer, A.; Newman, L.A.; Jemal, A. Breast cancer statistics, 2017, racial Dis.parity in mortality by state. CA Cancer J. Clin. 2017, 67, 439–448. [Google Scholar] [CrossRef] [Green Version]
- Kaminska, M.; Ciszewski, T.; Lopacka-Szatan, K.; Miotla, P.; Staroslawska, E. Breast cancer risk factors. Prz. Menopauzalny 2015, 14, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Ford, D.; Easton, D.F.; Stratton, M.; Narod, S.; Goldgar, D.; Devilee, P.; Bishop, D.T.; Weber, B.; Lenoir, G.; Chang-Claude, J.; et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am. J. Hum. Genet. 1998, 62, 676–689. [Google Scholar] [CrossRef] [Green Version]
- Collaborative Group on Hormonal Factors in Breast, C. Menarche, menopause, and breast cancer risk: Individual participant meta-analysis, including 118 964 women with breast cancer from 117 epidemiological studies. Lancet Oncol. 2012, 13, 1141–1151. [Google Scholar] [CrossRef]
- James, F.R.; Wootton, S.; Jackson, A.; Wiseman, M.; Copson, E.R.; Cutress, R.I. Obesity in breast cancer--what is the risk factor? Eur. J. Cancer 2015, 51, 705–720. [Google Scholar] [CrossRef]
- Mohanty, S.S.; Mohanty, P.K. Obesity as potential breast cancer risk factor for postmenopausal women. Genes Dis. 2021, 8, 117–123. [Google Scholar] [CrossRef]
- Chen, W.Y.; Rosner, B.; Hankinson, S.E.; Colditz, G.A.; Willett, W.C. Moderate alcohol consumption during adult life, drinking patterns, and breast cancer risk. JAMA 2011, 306, 1884–1890. [Google Scholar] [CrossRef] [Green Version]
- Danaei, G.; Vander Hoorn, S.; Lopez, A.D.; Murray, C.J.; Ezzati, M.; Comparative Risk Assessment collaborating, g. Causes of cancer in the world: Comparative risk assessment of nine behavioural and environmental risk factors. Lancet 2005, 366, 1784–1793. [Google Scholar] [CrossRef] [Green Version]
- Torre, L.A.; Islami, F.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer in Women: Burden and Trends. Cancer Epidemiol. Biomark. Prev. 2017, 26, 444–457. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, G.K.; Zhao, X.; Band, H.; Band, V. Histological, molecular and functional subtypes of breast cancers. Cancer Biol. 2010, 10, 955–960. [Google Scholar] [CrossRef] [Green Version]
- Eliyatkin, N.; Yalcin, E.; Zengel, B.; Aktas, S.; Vardar, E. Molecular Classification of Breast Carcinoma: From Traditional, Old-Fashioned Way to A New Age, and A New Way. J. Breast Health 2015, 11, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Prat, A.; Pineda, E.; Adamo, B.; Galvan, P.; Fernandez, A.; Gaba, L.; Diez, M.; Viladot, M.; Arance, A.; Munoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24 (Suppl. S2), S26–S35. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yang, J.; Peng, L.; Sahin, A.A.; Huo, L.; Ward, K.C.; O′Regan, R.; Torres, M.A.; Meisel, J.L. Triple-negative breast cancer has worse overall survival and cause-specific survival than non-triple-negative breast cancer. Breast. Cancer Res. Treat. 2017, 161, 279–287. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Dillekas, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [Green Version]
- Kaskel, P.; Orth, M.; Arndt, E.; Leiter, U.; Peter, R.U.; Krahn, G. Fulminating multi-organ failure in a young woman caused by rapidly progressing melanoma metastases. Dermatology 2000, 201, 79–80. [Google Scholar] [CrossRef]
- Riggio, A.I.; Varley, K.E.; Welm, A.L. The lingering mysteries of metastatic recurrence in breast cancer. Br. J. Cancer 2021, 124, 13–26. [Google Scholar] [CrossRef]
- Lim, B.; Hortobagyi, G.N. Current challenges of metastatic breast cancer. Cancer Metastasis. Rev. 2016, 35, 495–514. [Google Scholar] [CrossRef]
- Mariotto, A.B.; Etzioni, R.; Hurlbert, M.; Penberthy, L.; Mayer, M. Estimation of the Number of Women Living with Metastatic Breast Cancer in the United States. Cancer Epidemiol. Biomark. Prev. 2017, 26, 809–815. [Google Scholar] [CrossRef] [Green Version]
- Lucci, A.; Hall, C.S.; Lodhi, A.K.; Bhattacharyya, A.; Anderson, A.E.; Xiao, L.; Bedrosian, I.; Kuerer, H.M.; Krishnamurthy, S. Circulating tumour Cells in non-metastatic breast cancer: A prospective study. Lancet Oncol. 2012, 13, 688–695. [Google Scholar] [CrossRef]
- Caswell-Jin, J.L.; Plevritis, S.K.; Tian, L.; Cadham, C.J.; Xu, C.; Stout, N.K.; Sledge, G.W.; Mandelblatt, J.S.; Kurian, A.W. Change in Survival in Metastatic Breast Cancer with Treatment Advances: Meta-Analysis and Systematic Review. JNCI Cancer Spectr. 2018, 2, pky062. [Google Scholar] [CrossRef] [Green Version]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Pantel, K. Tumor Cell Dissemination: Emerging Biological insights from animal models and cancer patients. Cancer Cell 2013, 23, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, C.A. Selection and adaptation during metastatic cancer progression. Nature 2013, 501, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Husemann, Y.; Geigl, J.B.; Schubert, F.; Musiani, P.; Meyer, M.; Burghart, E.; Forni, G.; Eils, R.; Fehm, T.; Riethmuller, G.; et al. Systemic spread is an early step in breast cancer. Cancer Cell 2008, 13, 58–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, H.; Obradovic, M.M.S.; Hoffmann, M.; Harper, K.L.; Sosa, M.S.; Werner-Klein, M.; Nanduri, L.K.; Werno, C.; Ehrl, C.; Maneck, M.; et al. Early Dis.semination seeds metastasis in breast cancer. Nature 2016, 540, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of early Dis.semination and metastasis in Her2(+) mammary cancer. Nature 2016, 540, 588–592. [Google Scholar] [CrossRef]
- Ofri, A.; Moore, K. Occult breast cancer: Where are we at? Breast 2020, 54, 211–215. [Google Scholar] [CrossRef]
- Weigelt, B.; Peterse, J.L.; Van ′t Veer, L.J. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602. [Google Scholar] [CrossRef]
- Soni, A.; Ren, Z.; Hameed, O.; Chanda, D.; Morgan, C.J.; Siegal, G.P.; Wei, S. Breast cancer subtypes predispose the site of distant metastases. Am. J. Clin. Pathol. 2015, 143, 471–478. [Google Scholar] [CrossRef]
- Press, D.J.; Miller, M.E.; Liederbach, E.; Yao, K.; Huo, D. De novo metastasis in breast cancer: Occurrence and overall survival stratified by molecular subtype. Clin. Exp. Metastasis 2017, 34, 457–465. [Google Scholar] [CrossRef]
- Arshad, F.; Wang, L.; Sy, C.; Avraham, S.; Avraham, H.K. Blood-brain barrier Int.egrity and breast cancer metastasis to the brain. Pathol. Res. Int. 2010, 2011, 920509. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Yang, J.; Liu, Y.; You, H.; Dong, Y.; Lyu, J. Prognostic factors and survival outcomes according to tumor subtype in patients with breast cancer lung metastases. PeerJ 2019, 7, e8298. [Google Scholar] [CrossRef] [Green Version]
- Gasparini, G.; Weidner, N.; Bevilacqua, P.; Maluta, S.; Dalla Palma, P.; Caffo, O.; Barbareschi, M.; Boracchi, P.; Marubini, E.; Pozza, F. Tumor microvessel density, p53 expression, tumor size, and peritumoral lymphatic vessel invasion are relevant prognostic markers in node-negative breast carcinoma. J. Clin. Oncol. 1994, 12, 454–466. [Google Scholar] [CrossRef]
- Fitzpatrick, D.J.; Lai, C.S.; Parkyn, R.F.; Walters, D.; Humeniuk, V.; Walsh, D.C. Time to breast cancer relapse predicted by primary tumour characteristics, not lymph node involvement. World J. Surg. 2014, 38, 1668–1675. [Google Scholar] [CrossRef]
- Roda, N.; Blandano, G.; Pelicci, P.G. Blood Vessels and Peripheral Nerves as Key Players in Cancer Progression and Therapy Resistance. Cancers 2021, 13, 4471. [Google Scholar] [CrossRef] [PubMed]
- Fazilaty, H.; Rago, L.; Kass Youssef, K.; Ocana, O.H.; Garcia-Asencio, F.; Arcas, A.; Galceran, J.; Nieto, M.A. A gene regulatory network to control EMT programs in development and Disease. Nat. Commun. 2019, 10, 5115. [Google Scholar] [CrossRef] [PubMed]
- Aharonov, A.; Shakked, A.; Umansky, K.B.; Savidor, A.; Genzelinakh, A.; Kain, D.; Lendengolts, D.; Revach, O.Y.; Morikawa, Y.; Dong, J.; et al. ERBB2 drives YAP activation and EMT-like processes during cardiac regeneration. Nat. Cell Biol. 2020, 22, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Sheng, G. Defining epithelial-mesenchymal transitions in animal development. Development 2021, 148, 198036. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Meng, F.; Liu, G.; Zhang, B.; Zhu, J.; Wu, F.; Ethier, S.P.; Miller, F.; Wu, G. Forkhead transcription factor foxq1 promotes epithelial-mesenchymal transition and breast cancer metastasis. Cancer Res. 2011, 71, 1292–1301. [Google Scholar] [CrossRef] [Green Version]
- Horiguchi, K.; Sakamoto, K.; Koinuma, D.; Semba, K.; Inoue, A.; Inoue, S.; Fujii, H.; Yamaguchi, A.; Miyazawa, K.; Miyazono, K.; et al. TGF-beta drives epithelial-mesenchymal transition through deltaEF1-mediated downregulation of ESRP. Oncogene 2012, 31, 3190–3201. [Google Scholar] [CrossRef]
- Stankic, M.; Pavlovic, S.; Chin, Y.; Brogi, E.; Padua, D.; Norton, L.; Massague, J.; Benezra, R. TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013, 5, 1228–1242. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer Cells through paracrine TGF-beta signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Park, J.H.; Oh, S.M. Activation of NF-kappaB by TOPK upregulates Snail/Slug expression in TGF-beta1 signaling to induce epithelial-mesenchymal transition and invasion of breast cancer Cells. Biochem. Biophys. Res. Commun. 2020, 530, 122–129. [Google Scholar] [CrossRef]
- Han, D.; Wang, L.; Chen, B.; Zhao, W.; Liang, Y.; Li, Y.; Zhang, H.; Liu, Y.; Wang, X.; Chen, T.; et al. USP1-WDR48 deubiquitinase complex enhances TGF-beta induced epithelial-mesenchymal transition of TNBC Cells via stabilizing TAK1. Cell Cycle 2021, 20, 320–331. [Google Scholar] [CrossRef]
- Incassati, A.; Pinderhughes, A.; Eelkema, R.; Cowin, P. Links between transforming growth factor-beta and canonical Wnt signaling yield new insights into breast cancer susceptibility, suppression and tumor heterogeneity. Breast. Cancer Res. 2009, 11, 103. [Google Scholar] [CrossRef] [Green Version]
- Serra, R.; Easter, S.L.; Jiang, W.; Baxley, S.E. Wnt5a as an effector of TGFbeta in mammary development and cancer. J. Mammary Gland. Biol. Neoplasia 2011, 16, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.W.; Merkel, A.R.; Page, J.M.; Ruppender, N.S.; Guelcher, S.A.; Sterling, J.A. Wnt signaling induces gene expression of factors associated with bone destruction in lung and breast cancer. Clin. Exp. Metastasis 2014, 31, 945–959. [Google Scholar] [CrossRef]
- Ma, F.; Li, W.; Liu, C.; Li, W.; Yu, H.; Lei, B.; Ren, Y.; Li, Z.; Pang, D.; Qian, C. MiR-23a promotes TGF-beta1-induced EMT and tumor metastasis in breast cancer Cells by directly targeting CDH1 and activating Wnt/beta-catenin signaling. Oncotarget 2017, 8, 69538–69550. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, X.; Zhang, H.; Li, X.; Li, X.; Cong, M.; Peng, F.; Yu, J.; Zhang, X.; Yang, Q.; Hu, G. Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor DKK1. Nat. Cell Biol. 2017, 19, 1274–1285. [Google Scholar] [CrossRef]
- Buechel, D.; Sugiyama, N.; Rubinstein, N.; Saxena, M.; Kalathur, R.K.R.; Luond, F.; Vafaizadeh, V.; Valenta, T.; Hausmann, G.; Cantu, C.; et al. Parsing beta-catenin′s cell adhesion and Wnt signaling functions in malignant mammary tumor progression. Proc. Natl. Acad. Sci. USA 2021, 118, e2020227118. [Google Scholar] [CrossRef]
- Esposito, M.; Fang, C.; Cook, K.C.; Park, N.; Wei, Y.; Spadazzi, C.; Bracha, D.; Gunaratna, R.T.; Laevsky, G.; DeCoste, C.J.; et al. TGF-beta-induced DACT1 biomolecular condensates repress Wnt signalling to promote bone metastasis. Nat. Cell Biol. 2021, 23, 257–267. [Google Scholar] [CrossRef]
- Sun, Y.; Lowther, W.; Kato, K.; Bianco, C.; Kenney, N.; Strizzi, L.; Raafat, D.; Hirota, M.; Khan, N.I.; Bargo, S.; et al. Notch4 intracellular domain binding to Smad3 and inhibition of the TGF-beta signaling. Oncogene 2005, 24, 5365–5374. [Google Scholar] [CrossRef] [Green Version]
- Leong, K.G.; Niessen, K.; Kulic, I.; Raouf, A.; Eaves, C.; Pollet, I.; Karsan, A. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J. Exp. Med. 2007, 204, 2935–2948. [Google Scholar] [CrossRef] [Green Version]
- Sethi, N.; Dai, X.; Winter, C.G.; Kang, Y. Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone Cells. Cancer Cell 2011, 19, 192–205. [Google Scholar] [CrossRef] [Green Version]
- Bendre, M.S.; Gaddy-Kurten, D.; Mon-Foote, T.; Akel, N.S.; Skinner, R.A.; Nicholas, R.W.; Suva, L.J. Expression of interleukin 8 and not parathyroid hormone-related protein by human breast cancer Cells correlates with bone metastasis in vivo. Cancer Res. 2002, 62, 5571–5579. [Google Scholar]
- Studebaker, A.W.; Storci, G.; Werbeck, J.L.; Sansone, P.; Sasser, A.K.; Tavolari, S.; Huang, T.; Chan, M.W.; Marini, F.C.; Rosol, T.J.; et al. Fibroblasts isolated from common sites of breast cancer metastasis enhance cancer cell growth rates and invasiveness in an interleukin-6-dependent manner. Cancer Res. 2008, 68, 9087–9095. [Google Scholar] [CrossRef] [Green Version]
- Oh, K.; Ko, E.; Kim, H.S.; Park, A.K.; Moon, H.G.; Noh, D.Y.; Lee, D.S. Transglutaminase 2 facilitates the distant hematogenous metastasis of breast cancer by modulating interleukin-6 in cancer Cells. Breast. Cancer Res. 2011, 13, R96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Zha, Z.; Lang, B.; Liu, J.; Yao, X. Heregulin-beta1 promotes metastasis of breast cancer cell line SKBR3 through upregulation of Snail and induction of epithelial-mesenchymal transition. Cancer Lett. 2009, 280, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Neve, E.P.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Yuen, H.F.; Chan, Y.K.; Grills, C.; McCrudden, C.M.; Gunasekharan, V.; Shi, Z.; Wong, A.S.; Lappin, T.R.; Chan, K.W.; Fennell, D.A.; et al. Polyomavirus enhancer activator 3 protein promotes breast cancer metastatic progression through Snail-induced epithelial-mesenchymal transition. J. Pathol. 2011, 224, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Chimge, N.O.; Baniwal, S.K.; Little, G.H.; Chen, Y.B.; Kahn, M.; Tripathy, D.; Borok, Z.; Frenkel, B. Regulation of breast cancer metastasis by Runx2 and estrogen signaling: The role of SNAI2. Breast. Cancer Res. 2011, 13, R127. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Srivastava, S.K. HER2 mediated de novo production of TGFbeta leads to SNAIL driven epithelial-to-mesenchymal transition and metastasis of breast cancer. Mol. Oncol. 2014, 8, 1532–1547. [Google Scholar] [CrossRef]
- Wu, Z.Q.; Li, X.Y.; Hu, C.Y.; Ford, M.; Kleer, C.G.; Weiss, S.J. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc. Natl. Acad. Sci. USA 2012, 109, 16654–16659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Zhao, X.; Xiao, Q.; Liu, Q.; Ding, K.; Yu, F.; Zhang, R.; Zhu, T.; Ge, G. Snail and Slug mediate tamoxifen resistance in breast cancer cells through activation of EGFR-ERK independent of epithelial-mesenchymal transition. J. Mol. Cell Biol. 2014, 6, 352–354. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Zhao, X.; Zhang, X.; Luo, M.; Zuo, X.; Huang, S.; Wang, Y.; Gu, S.; Zhao, X. Notch1 signaling regulates the epithelial-mesenchymal transition and invasion of breast cancer in a Slug-dependent manner. Mol. Cancer 2015, 14, 28. [Google Scholar] [CrossRef] [Green Version]
- Li, N.Y.; Weber, C.E.; Wai, P.Y.; Cuevas, B.D.; Zhang, J.; Kuo, P.C.; Mi, Z. An MAPK-dependent pathway induces epithelial-mesenchymal transition via Twist activation in human breast cancer cell lines. Surgery 2013, 154, 404–410. [Google Scholar] [CrossRef]
- Lim, J.C.; Koh, V.C.; Tan, J.S.; Tan, W.J.; Thike, A.A.; Tan, P.H. Prognostic significance of epithelial-mesenchymal transition proteins Twist and Foxc2 in phyllodes tumours of the breast. Breast. Cancer Res. Treat. 2015, 150, 19–29. [Google Scholar] [CrossRef]
- Yang, J.; Hou, Y.; Zhou, M.; Wen, S.; Zhou, J.; Xu, L.; Tang, X.; Du, Y.E.; Hu, P.; Liu, M. Twist induces epithelial-mesenchymal transition and cell motility in breast cancer via ITGB1-FAK/ILK signaling axis and its associated downstream network. Int. J. Biochem. Cell Biol. 2016, 71, 62–71. [Google Scholar] [CrossRef]
- Cieply, B.; Farris, J.; Denvir, J.; Ford, H.L.; Frisch, S.M. Epithelial-mesenchymal transition and tumor suppression are controlled by a reciprocal feedback loop between ZEB1 and Grainyhead-like-2. Cancer Res. 2013, 73, 6299–6309. [Google Scholar] [CrossRef] [Green Version]
- Hugo, H.J.; Pereira, L.; Suryadinata, R.; Drabsch, Y.; Gonda, T.J.; Gunasinghe, N.P.; PInto, C.; Soo, E.T.; Van Denderen, B.J.; Hill, P.; et al. Direct repression of MYB by ZEB1 suppresses proliferation and epithelial gene expression during epithelial-to-mesenchymal transition of breast cancer cells. Breast Cancer Res. 2013, 15, R113. [Google Scholar] [CrossRef] [Green Version]
- Avtanski, D.B.; Nagalingam, A.; Bonner, M.Y.; Arbiser, J.L.; Saxena, N.K.; Sharma, D. Honokiol inhibits epithelial-mesenchymal transition in breast cancer cells by targeting signal transducer and activator of transcription 3/Zeb1/E-cadherin axis. Mol. Oncol. 2014, 8, 565–580. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, M.K.; Park, J.H.; Lee, H.J.; Shin, D.H.; Kang, Y.; Lee, C.H.; Kong, G. Loss of the polycomb protein Mel-18 enhances the epithelial-mesenchymal transition by ZEB1 and ZEB2 expression through the downregulation of miR-205 in breast cancer. Oncogene 2014, 33, 1325–1335. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.; Song, S.; Xu, Y.; Li, H.; Liu, H. Knockdown of ZEB1 suppressed the formation of vasculogenic mimicry and epithelial-mesenchymal transition in the human breast cancer cell line MDA-MB-231. Mol. Med. Rep. 2018, 17, 6711–6716. [Google Scholar] [CrossRef] [Green Version]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef] [Green Version]
- Nieman, M.T.; Prudoff, R.S.; Johnson, K.R.; Wheelock, M.J. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J. Cell Biol. 1999, 147, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Garcia, B.; Eiro, N.; Marin, L.; Gonzalez-Reyes, S.; Gonzalez, L.O.; Lamelas, M.L.; Vizoso, F.J. Expression and prognostic significance of fibronectin and matrix metalloproteases in breast cancer metastasis. HistoPathology 2014, 64, 512–522. [Google Scholar] [CrossRef]
- Korsching, E.; Packeisen, J.; Liedtke, C.; Hungermann, D.; Wulfing, P.; Van Diest, P.J.; Brandt, B.; Boecker, W.; Buerger, H. The origin of vimentin expression in invasive breast cancer: Epithelial-mesenchymal transition, myoepithelial histogenesis or histogenesis from progenitor cells with bilinear differentiation potential? J. Pathol. 2005, 206, 451–457. [Google Scholar] [CrossRef]
- Vuoriluoto, K.; Haugen, H.; Kiviluoto, S.; Mpindi, J.P.; Nevo, J.; Gjerdrum, C.; Tiron, C.; Lorens, J.B.; Ivaska, J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 2011, 30, 1436–1448. [Google Scholar] [CrossRef] [Green Version]
- Whipple, R.A.; Matrone, M.A.; Cho, E.H.; Balzer, E.M.; Vitolo, M.I.; Yoon, J.R.; Ioffe, O.B.; Tuttle, K.C.; Yang, J.; Martin, S.S. Epithelial-to-mesenchymal transition promotes tubulin detyrosination and microtentacles that enhance endothelial engagement. Cancer Res. 2010, 70, 8127–8137. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yang, M.; Chen, R.; Su, W.; Li, P.; Chen, S.; Chen, Z.; Chen, A.; Li, S.; Hu, C. IBP regulates epithelial-to-mesenchymal transition and the motility of breast cancer cells via Rac1, RhoA and Cdc42 signaling pathways. Oncogene 2014, 33, 3374–3382. [Google Scholar] [CrossRef] [Green Version]
- Pereira De Carvalho, B.; Chern, Y.J.; He, J.; Chan, C.H. The ubiquitin ligase RNF8 regulates Rho GTPases and promotes cytoskeletal changes and motility in triple-negative breast cancer cells. FEBS Lett. 2021, 595, 241–252. [Google Scholar] [CrossRef]
- Wang, X.; Lu, H.; Urvalek, A.M.; Li, T.; Yu, L.; Lamar, J.; DiPersio, C.M.; Feustel, P.J.; Zhao, J. KLF8 promotes human breast cancer cell invasion and metastasis by transcriptional activation of MMP9. Oncogene 2011, 30, 1901–1911. [Google Scholar] [CrossRef] [Green Version]
- Eckert, M.A.; Santiago-Medina, M.; Lwin, T.M.; Kim, J.; Courtneidge, S.A.; Yang, J. ADAM12 induction by Twist1 promotes tumor invasion and metastasis via regulation of invadopodia and focal adhesions. J. Cell Sci. 2017, 130, 2036–2048. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Fontemaggi, G.; Gurtner, A.; Strano, S.; Higashi, Y.; Sacchi, A.; Piaggio, G.; Blandino, G. The transcriptional repressor ZEB regulates p73 expression at the crossroad between proliferation and differentiation. Mol. Cell. Biol. 2001, 21, 8461–8470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iseri, O.D.; Kars, M.D.; Arpaci, F.; Atalay, C.; Pak, I.; Gunduz, U. Drug resistant MCF-7 Cells exhibit epithelial-mesenchymal transition gene expression pattern. Biomed. Pharm. 2011, 65, 40–45. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, L.; He, X.; Zhang, P.; Sun, C.; Xu, X.; Lu, Y.; Li, F. TGF-beta plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem. Biophys. Res. Commun. 2018, 502, 160–165. [Google Scholar] [CrossRef]
- Soundararajan, R.; Fradette, J.J.; Konen, J.M.; Moulder, S.; Zhang, X.; Gibbons, D.L.; Varadarajan, N.; Wistuba, I.I.; Tripathy, D.; Bernatchez, C.; et al. Targeting the Interplay between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers 2019, 11, 714. [Google Scholar] [CrossRef] [Green Version]
- Gunasinghe, N.P.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis. Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef]
- Maguire, A.; Brogi, E. Sentinel lymph nodes for breast carcinoma: An update on current practice. HistoPathology 2016, 68, 152–167. [Google Scholar] [CrossRef] [Green Version]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [Green Version]
- Hanna, N.; Fidler, I.J. Role of natural killer cells in the destruction of circulating tumor emboli. J. Natl. Cancer Inst. 1980, 65, 801–809. [Google Scholar] [CrossRef]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef]
- Smit, M.A.; Geiger, T.R.; Song, J.Y.; Gitelman, I.; Peeper, D.S. A Twist-Snail axis critical for TrkB-induced epithelial-mesenchymal transition-like transformation, anoikis resistance, and metastasis. Mol. Cell. Biol. 2009, 29, 3722–3737. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Hu, Z.; Barney, K.A.; Degen, J.L. Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell-dependent and-independent mechanisms. Blood 2007, 110, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, A.; Brooks, M.W.; Houshyar, S.; Reinhardt, F.; Ardolino, M.; Fessler, E.; Chen, M.B.; Krall, J.A.; DeCock, J.; Zervantonakis, I.K.; et al. Neutrophils Suppress Intraluminal NK Cell.-Mediated Tumor Cell Clearance and Enhance Extravasation of disseminated Carcinoma cells. Cancer Discov. 2016, 6, 630–649. [Google Scholar] [CrossRef] [Green Version]
- Alix-Panabieres, C.; Pantel, K. Challenges in circulating tumour cell research. Nat. Rev. Cancer 2014, 14, 623–631. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Reymond, N.; D′Agua, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef]
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 2013, 24, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Weis, S.; Cui, J.; Barnes, L.; Cheresh, D. Endothelial barrier Disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J. Cell Biol. 2004, 167, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Gupta, G.P.; Nguyen, D.X.; Chiang, A.C.; Bos, P.D.; Kim, J.Y.; Nadal, C.; Gomis, R.R.; Manova-Todorova, K.; Massague, J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 2007, 446, 765–770. [Google Scholar] [CrossRef]
- Padua, D.; Zhang, X.H.; Wang, Q.; Nadal, C.; Gerald, W.L.; Gomis, R.R.; Massague, J. TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008, 133, 66–77. [Google Scholar] [CrossRef] [Green Version]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Gao, H.; Chakraborty, G.; Lee-Lim, A.P.; Mo, Q.; Decker, M.; Vonica, A.; Shen, R.; Brogi, E.; Brivanlou, A.H.; Giancotti, F.G. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 2012, 150, 764–779. [Google Scholar] [CrossRef] [Green Version]
- Skibinski, A.; Kuperwasser, C. The origin of breast tumor heterogeneity. Oncogene 2015, 34, 5309–5316. [Google Scholar] [CrossRef] [Green Version]
- Symmans, W.F.; Liu, J.; Knowles, D.M.; Inghirami, G. Breast cancer heterogeneity: Evaluation of clonality in primary and metastatic lesions. Hum. Pathol. 1995, 26, 210–216. [Google Scholar] [CrossRef]
- Teixeira, M.R.; Tsarouha, H.; Kraggerud, S.M.; Pandis, N.; Dimitriadis, E.; Andersen, J.A.; Lothe, R.A.; Heim, S. Evaluation of breast cancer polyclonality by combined chromosome banding and comparative genomic hybridization analysis. Neoplasia 2001, 3, 204–214. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Lee, A.V.; Rosen, J.M. The Cellular Origin and Evolution of Breast Cancer. Cold. Spring Harb. Perspect. Med. 2017, 7, a027128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewer, M.; Lee, L.; Leibovitch, M.; Zhang, G.; Liu, J.; Findlay, S.D.; Vincent, K.M.; Tandoc, K.; Dieters-Castator, D.; Quail, D.F.; et al. Translational control of breast cancer plasticity. Nat. Commun. 2020, 11, 2498. [Google Scholar] [CrossRef] [PubMed]
- Roda, N.; Gambino, V.; Giorgio, M. Metabolic Constrains Rule Metastasis Progression. Cells 2020, 9, 2081. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Yates, L.R.; Gerstung, M.; Knappskog, S.; Desmedt, C.; Gundem, G.; Van Loo, P.; Aas, T.; Alexandrov, L.B.; Larsimont, D.; Davies, H.; et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 2015, 21, 751–759. [Google Scholar] [CrossRef]
- Geyer, F.C.; Weigelt, B.; Natrajan, R.; Lambros, M.B.; De Biase, D.; Vatcheva, R.; Savage, K.; Mackay, A.; Ashworth, A.; Reis-Filho, J.S. Molecular analysis reveals a genetic basis for the phenotypic diversity of metaplastic breast carcinomas. J. Pathol. 2010, 220, 562–573. [Google Scholar] [CrossRef]
- Patani, N.; Barbashina, V.; Lambros, M.B.; Gauthier, A.; Mansour, M.; Mackay, A.; Reis-Filho, J.S. Direct evidence for concurrent morphological and genetic heterogeneity in an invasive ductal carcinoma of triple-negative phenotype. J. Clin. Pathol. 2011, 64, 822–828. [Google Scholar] [CrossRef]
- Lee, H.J.; Seo, A.N.; Kim, E.J.; Jang, M.H.; Suh, K.J.; Ryu, H.S.; Kim, Y.J.; Kim, J.H.; Im, S.A.; Gong, G.; et al. HER2 heterogeneity affects trastuzumab responses and survival in patients with HER2-positive metastatic breast cancer. Am. J. Clin. Pathol. 2014, 142, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.P.; Morin, R.D.; Khattra, J.; Prentice, L.; Pugh, T.; Burleigh, A.; Delaney, A.; Gelmon, K.; Guliany, R.; Senz, J.; et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 2009, 461, 809–813. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef]
- Eirew, P.; Steif, A.; Khattra, J.; Ha, G.; Yap, D.; Farahani, H.; Gelmon, K.; Chia, S.; Mar, C.; Wan, A.; et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 2015, 518, 422–426. [Google Scholar] [CrossRef]
- Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schuffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef]
- Jackson, H.W.; Fischer, J.R.; Zanotelli, V.R.T.; Ali, H.R.; Mechera, R.; Soysal, S.D.; Moch, H.; Muenst, S.; Varga, Z.; Weber, W.P.; et al. The single-cell Pathology landscape of breast cancer. Nature 2020, 578, 615–620. [Google Scholar] [CrossRef]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive Disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef] [Green Version]
- Rios, A.C.; Capaldo, B.D.; Vaillant, F.; Pal, B.; Van Ineveld, R.; Dawson, C.A.; Chen, Y.; Nolan, E.; Fu, N.Y.; Group, D.; et al. Intraclonal Plasticity in Mammary Tumors Revealed through Large-Scale Single-Cell Resolution 3D Imaging. Cancer Cell 2019, 35, 618–632.e616. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Ding, K.; Priedigkeit, N.; Elangovan, A.; Levine, K.M.; Carleton, N.; Savariau, L.; Atkinson, J.M.; Oesterreich, S.; Lee, A.V. Single-Cell Transcriptomic Heterogeneity in Invasive Ductal and Lobular Breast Cancer Cells. Cancer Res. 2021, 81, 268–281. [Google Scholar] [CrossRef]
- Wu, S.Z.; Al-Eryani, G.; Roden, D.L.; Junankar, S.; Harvey, K.; Andersson, A.; Thennavan, A.; Wang, C.; Torpy, J.R.; Bartonicek, N.; et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 2021, 53, 1334–1347. [Google Scholar] [CrossRef]
- Yeo, S.K.; Zhu, X.; Okamoto, T.; Hao, M.; Wang, C.; Lu, P.; Lu, L.J.; Guan, J.L. Single-Cell RNA-sequencing reveals Dis.tinct patterns of cell state heterogeneity in mouse models of breast cancer. Elife 2020, 9, e58810. [Google Scholar] [CrossRef]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor′s dilemma? Biochim. Biophys. Acta 2011, 1807, 552–561. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.N.; Zheng, G.; Tchou, J.; Nioka, S.; Li, L.Z. Characterizing the metabolic heterogeneity in human breast cancer xenografts by 3D high resolution fluorescence imaging. Springerplus 2013, 2, 73. [Google Scholar] [CrossRef] [Green Version]
- Farnie, G.; Sotgia, F.; Lisanti, M.P. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015, 6, 30472–30486. [Google Scholar] [CrossRef] [Green Version]
- Lamb, R.; Ozsvari, B.; Bonucelli, G.; Smith, D.L.; Pestell, R.G.; Martinez-Outschoorn, U.E.; Clarke, R.B.; Sotgia, F.; Lisanti, M.P. Dissecting tumor metabolic heterogeneity: Telomerase and large cell size metabolically define a sub-population of stem-like, mitochondrial-rich, cancer cells. Oncotarget 2015, 6, 21892–21905. [Google Scholar] [CrossRef] [Green Version]
- Komaki, K.; Sano, N.; Tangoku, A. Problems in histological grading of malignancy and its clinical significance in patients with operable breast cancer. Breast. Cancer 2006, 13, 249–253. [Google Scholar] [CrossRef]
- Bhang, H.E.; Ruddy, D.A.; Krishnamurthy Radhakrishna, V.; Caushi, J.X.; Zhao, R.; Hims, M.M.; Singh, A.P.; Kao, I.; Rakiec, D.; Shaw, P.; et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat. Med. 2015, 21, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Bentires-Alj, M. Breast Tumor Heterogeneity: Source of Fitness, Hurdle for Therapy. Mol. Cell 2015, 60, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramon, Y.C.S.; Sese, M.; Capdevila, C.; Aasen, T.; De Mattos-Arruda, L.; Diaz-Cano, S.J.; Hernandez-Losa, J.; Castellvi, J. Clinical implications of intratumor heterogeneity: Challenges and opportunities. J. Mol. Med. 2020, 98, 161–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Wang, Y.; Li, Q.; Cao, L.; Sun, Z.; Jin, J.; Fang, H.; Zhu, A.; Li, Y.; Zhang, W.; et al. Intratumor heterogeneity predicts metastasis of triple-negative breast cancer. Carcinogenesis 2017, 38, 900–909. [Google Scholar] [CrossRef]
- Mroz, E.A.; Tward, A.D.; Hammon, R.J.; Ren, Y.; Rocco, J.W. Int.ra-tumor genetic heterogeneity and mortality in head and neck cancer: Analysis of data from the Cancer Genome Atlas. PLoS Med. 2015, 12, e1001786. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Jiang, Y.Z.; Liu, X.Y.; Liu, Y.R.; Shao, Z.M. Clinical and molecular relevance of mutant-allele tumor heterogeneity in breast cancer. Breast. Cancer Res. Treat. 2017, 162, 39–48. [Google Scholar] [CrossRef]
- Saha, A.; Harowicz, M.R.; Cain, E.H.; Hall, A.H.; Hwang, E.S.; Marks, J.R.; Marcom, P.K.; Mazurowski, M.A. Intra-tumor molecular heterogeneity in breast cancer: Definitions of measures and association with distant recurrence-free survival. Breast. Cancer Res. Treat. 2018, 172, 123–132. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Neelakantan, D.; Ford, H.L. Clonal cooperativity in heterogenous cancers. Semin. Cell Dev. Biol. 2017, 64, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Lyons, J.G.; Siew, K.; O′Grady, R.L. Cellular Int.eractions determining the production of collagenase by a rat mammary carcinoma cell line. Int. J. Cancer 1989, 43, 119–125. [Google Scholar] [CrossRef]
- Janiszewska, M.; Tabassum, D.P.; Castano, Z.; Cristea, S.; Yamamoto, K.N.; Kingston, N.L.; Murphy, K.C.; Shu, S.; Harper, N.W.; Del Alcazar, C.G.; et al. Subclonal cooperation drives metastasis by modulating local and systemic immune microenvironments. Nat. Cell Biol. 2019, 21, 879–888. [Google Scholar] [CrossRef]
- Angus, L.; Smid, M.; Wilting, S.M.; Van Riet, J.; Van Hoeck, A.; Nguyen, L.; Nik-Zainal, S.; Steenbruggen, T.G.; Tjan-Heijnen, V.C.G.; Labots, M.; et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat. Genet. 2019, 51, 1450–1458. [Google Scholar] [CrossRef]
- Ng, C.K.Y.; Bidard, F.C.; Piscuoglio, S.; Geyer, F.C.; Lim, R.S.; De Bruijn, I.; Shen, R.; Pareja, F.; Berman, S.H.; Wang, L.; et al. Genetic Heterogeneity in Therapy-Naive Synchronous Primary Breast Cancers and Their Metastases. Clin. Cancer Res. 2017, 23, 4402–4415. [Google Scholar] [CrossRef] [Green Version]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Ellis, M.J.; Li, S.; Larson, D.E.; Chen, K.; Wallis, J.W.; Harris, C.C.; McLellan, M.D.; Fulton, R.S.; Fulton, L.L.; et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010, 464, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Park, K.; Lim, S.H.; Kim, H.S.; Yoo, K.H.; Jung, K.S.; Song, H.N.; Hong, M.; Do, I.G.; Ahn, T.; et al. Mutational profiling of brain metastasis from breast cancer: Matched pair analysis of targeted sequencing between brain metastasis and primary breast cancer. Oncotarget 2015, 6, 43731–43742. [Google Scholar] [CrossRef] [Green Version]
- Moelans, C.B.; Van der Groep, P.; Hoefnagel, L.D.C.; Van de Vijver, M.J.; Wesseling, P.; Wesseling, J.; Van der Wall, E.; Van Diest, P.J. Genomic evolution from primary breast carcinoma to Distant metastasis: Few copy number changes of breast cancer related genes. Cancer Lett. 2014, 344, 138–146. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Siegel, M.B.; Kanchi, K.L.; Miller, C.A.; Ding, L.; Zhao, W.; He, X.; Parker, J.S.; Wendl, M.C.; Fulton, R.S.; et al. Tumor Evolution in Two Patients with Basal-like Breast Cancer: A Retrospective Genomics Study of Multiple Metastases. PLoS Med. 2016, 13, e1002174. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Guille, A.; Adelaide, J.; Garnier, S.; Carbuccia, N.; Monneur, A.; Charafe-Jauffret, E.; Goncalves, A.; Viens, P.; et al. Comparative genomic analysis of primary tumors and metastases in breast cancer. Oncotarget 2016, 7, 27208–27219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aftimos, P.; Oliveira, M.; Irrthum, A.; Fumagalli, D.; Sotiriou, C.; Gal-Yam, E.N.; Robson, M.E.; Ndozeng, J.; Di Leo, A.; Ciruelos, E.M.; et al. Genomic and Transcriptomic Analyses of Breast Cancer Primaries and Matched Metastases in AURORA, the Breast International Group (BIG) Molecular Screening Initiative. Cancer Discov. 2021, 11, 2796–2811. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Li, Z.; Ma, Z.; Curtis, C. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat. Genet. 2020, 52, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Casasent, A.K.; Schalck, A.; Gao, R.; Sei, E.; Long, A.; Pangburn, W.; Casasent, T.; Meric-Bernstam, F.; Edgerton, M.E.; Navin, N.E. Multiclonal Invasion in Breast Tumors Identified by Topographic Single Cell Sequencing. Cell 2018, 172, 205–217 e212. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective Dis.semination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [Green Version]
- Tiede, S.; Kalathur, R.K.R.; Luond, F.; Von Allmen, L.; Szczerba, B.M.; Hess, M.; Vlajnic, T.; Muller, B.; Canales Murillo, J.; Aceto, N.; et al. Multi-color clonal tracking reveals intra-stage proliferative heterogeneity during mammary tumor progression. Oncogene 2021, 40, 12–27. [Google Scholar] [CrossRef]
- Paul, M.R.; Pan, T.C.; Pant, D.K.; Shih, N.N.; Chen, Y.; Harvey, K.L.; Solomon, A.; Lieberman, D.; Morrissette, J.J.; Soucier-Ernst, D.; et al. Genomic landscape of metastatic breast cancer identifies preferentially dysregulated pathways and targets. J. Clin. Invest 2020, 130, 4252–4265. [Google Scholar] [CrossRef]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184 e167. [Google Scholar] [CrossRef] [Green Version]
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; Van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015, 5, 1164–1177. [Google Scholar] [CrossRef] [Green Version]
- Diossy, M.; Reiniger, L.; Sztupinszki, Z.; Krzystanek, M.; Timms, K.M.; Neff, C.; Solimeno, C.; Pruss, D.; Eklund, A.C.; Toth, E.; et al. Breast cancer brain metastases show increased levels of genomic aberration-based homologous recombination deficiency scores relative to their corresponding primary tumors. Ann. Oncol. 2018, 29, 1948–1954. [Google Scholar] [CrossRef]
- Schrijver, W.; Selenica, P.; Lee, J.Y.; Ng, C.K.Y.; Burke, K.A.; Piscuoglio, S.; Berman, S.H.; Reis-Filho, J.S.; Weigelt, B.; Van Diest, P.J.; et al. Mutation Profiling of Key Cancer Genes in Primary Breast Cancers and Their Distant Metastases. Cancer Res. 2018, 78, 3112–3121. [Google Scholar] [CrossRef] [Green Version]
- Marjon, P.L.; Bobrovnikova-Marjon, E.V.; Abcouwer, S.F. Expression of the pro-angiogenic factors vascular endothelial growth factor and interleukin-8/CXCL8 by human breast carcinomas is responsive to nutrient deprivation and endoplasmic reticulum stress. Mol. Cancer 2004, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Quintavalle, M.; Elia, L.; Price, J.H.; Heynen-Genel, S.; Courtneidge, S.A. A cell-based high-content screening assay reveals activators and inhibitors of cancer cell invasion. Sci Signal. 2011, 4, ra49. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Xue, Y.; Si, Y.; Wang, Q.; Wang, Z.; Yuan, J.; Zhang, X. The unfolded protein response potentiates epithelial-to-mesenchymal transition (EMT) of gastric cancer cells under severe hypoxic conditions. Med. Oncol. 2015, 32, 447. [Google Scholar] [CrossRef]
- Semenza, G.L. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim. Biophys. Acta 2016, 1863, 382–391. [Google Scholar] [CrossRef]
- Semenza, G.L. Molecular mechanisms mediating metastasis of hypoxic breast cancer cells. Trends Mol. Med. 2012, 18, 534–543. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Sceneay, J.; Godde, N.; Kinwel, T.; Ham, S.; Thompson, E.W.; Humbert, P.O.; Moller, A. Intermittent hypoxia induces a metastatic phenotype in breast cancer. Oncogene 2018, 37, 4214–4225. [Google Scholar] [CrossRef]
- Weidner, N.; Semple, J.P.; Welch, W.R.; Folkman, J. Tumor angiogenesis and metastasis--correlation in invasive breast carcinoma. N. Engl. J. Med. 1991, 324, 1–8. [Google Scholar] [CrossRef]
- Horak, E.R.; Leek, R.; Klenk, N.; LeJeune, S.; Smith, K.; Stuart, N.; Greenall, M.; Stepniewska, K.; Harris, A.L. Angiogenesis, assessed by platelet/endothelial Cell. adhesion molecule antibodies, as indicator of node metastases and survival in breast cancer. Lancet 1992, 340, 1120–1124. [Google Scholar] [CrossRef]
- Vaupel, P.; Hockel, M.; Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox. Signal. 2007, 9, 1221–1235. [Google Scholar] [CrossRef]
- Gao, T.; Li, J.Z.; Lu, Y.; Zhang, C.Y.; Li, Q.; Mao, J.; Li, L.H. The mechanism between epithelial mesenchymal transition in breast cancer and hypoxia microenvironment. Biomed. Pharm. 2016, 80, 393–405. [Google Scholar] [CrossRef]
- Montagner, M.; Enzo, E.; Forcato, M.; Zanconato, F.; Parenti, A.; Rampazzo, E.; Basso, G.; Leo, G.; Rosato, A.; Bicciato, S.; et al. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nature 2012, 487, 380–384. [Google Scholar] [CrossRef]
- Gameiro, P.A.; Struhl, K. Nutrient Deprivation Elicits a Transcriptional and Translational Inflammatory Response Coupled to Decreased Protein Synthesis. Cell Rep. 2018, 24, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Hui, S.; Ghergurovich, J.M.; Fan, J.; Intlekofer, A.M.; White, R.M.; Rabinowitz, J.D.; Thompson, C.B.; Zhang, J. As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell. Metab. 2018, 27, 428–438 e425. [Google Scholar] [CrossRef] [Green Version]
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381. [Google Scholar] [CrossRef]
- Lee, Y.J.; Galoforo, S.S.; Berns, C.M.; Chen, J.C.; Davis, B.H.; Sim, J.E.; Corry, P.M.; Spitz, D.R. Glucose deprivation-induced cytotoxicity and alterations in mitogen-activated protein kinase activation are mediated by oxidative stress in multidrug-resistant human breast carcinoma Cells. J. Biol. Chem. 1998, 273, 5294–5299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, N.S.; Bicknell, R. Hypoxia and oxidative stress in breast cancer. Oxidative stress: Its effects on the growth, metastatic potential and response to therapy of breast cancer. Breast Cancer Res. 2001, 3, 323–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahalingaiah, P.K.; Singh, K.P. Chronic oxidative stress increases growth and tumorigenic potential of MCF-7 breast cancer cells. PLoS ONE 2014, 9, e87371. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Ordway, B.; Rafiei, I.; Borad, P.; Fang, B.; Koomen, J.L.; Zhang, C.; Yoder, S.; Johnson, J.; Damaghi, M. Int.egrative Analysis of Breast Cancer Cells Reveals an Epithelial-Mesenchymal Transition Role in Adaptation to Acidic Microenvironment. Front. Oncol. 2020, 10, 304. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Prisco, M.; Ertel, A.; Tsirigos, A.; Lin, Z.; Pavlides, S.; Wang, C.; Flomenberg, N.; Knudsen, E.S.; Howell, A.; et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: Achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011, 10, 1271–1286. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. The role of the unfolded protein response in tumour development: Friend or foe? Nat. Rev. Cancer 2004, 4, 966–977. [Google Scholar] [CrossRef]
- Avril, T.; Vauleon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Weissman, A.M. The Unfolded Protein Response, Degradation from Endoplasmic Reticulum and Cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer and neurological disorders. Antioxid. Redox. Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- McGrath, E.P.; Logue, S.E.; Mnich, K.; Deegan, S.; Jager, R.; Gorman, A.M.; Samali, A. The Unfolded Protein Response in Breast Cancer. Cancers 2018, 10, 344. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Liu, H.; Song, Z.; Jiang, Y.; Kim, H.; Samavati, L.; Nguyen, H.M.; Yang, Z.Q. The UPR Transducer IRE1 Promotes Breast Cancer Malignancy by Degrading Tumor Suppressor microRNAs. iScience 2020, 23, 101503. [Google Scholar] [CrossRef]
- Sicari, D.; Fantuz, M.; Bellazzo, A.; Valentino, E.; Apollonio, M.; Pontisso, I.; Di Cristino, F.; Dal Ferro, M.; Bicciato, S.; Del Sal, G.; et al. Mutant p53 improves cancer Cells′ resistance to endoplasmic reticulum stress by sustaining activation of the UPR regulator ATF6. Oncogene 2019, 38, 6184–6195. [Google Scholar] [CrossRef]
- Abba, M.C.; Lacunza, E.; Nunez, M.I.; Colussi, A.; Isla-Larrain, M.; Segal-Eiras, A.; Croce, M.V.; Aldaz, C.M. Rhomboid domain containing 2 (RHBDD2): A novel cancer-related gene over-expressed in breast cancer. Biochim. Biophys. Acta 2009, 1792, 988–997. [Google Scholar] [CrossRef] [Green Version]
- Dery, M.A.; Jodoin, J.; Ursini-Siegel, J.; Aleynikova, O.; Ferrario, C.; Hassan, S.; Basik, M.; LeBlanc, A.C. Endoplasmic reticulum stress induces PRNP prion protein gene expression in breast cancer. Breast Cancer Res. 2013, 15, R22. [Google Scholar] [CrossRef] [Green Version]
- Lacunza, E.; Rabassa, M.E.; Canzoneri, R.; Pellon-Maison, M.; Croce, M.V.; Aldaz, C.M.; Abba, M.C. Identification of signaling pathways modulated by RHBDD2 in breast cancer Cells: A link to the unfolded protein response. Cell. Stress Chaperones 2014, 19, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Nagelkerke, A.; Bussink, J.; Mujcic, H.; Wouters, B.G.; Lehmann, S.; Sweep, F.C.; Span, P.N. Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast. Cancer Res. 2013, 15, R2. [Google Scholar] [CrossRef] [Green Version]
- Varone, E.; Decio, A.; Chernorudskiy, A.; Minoli, L.; Brunelli, L.; Ioli, F.; Piotti, A.; Pastorelli, R.; Fratelli, M.; Gobbi, M.; et al. The ER stress response mediator ERO1 triggers cancer metastasis by favoring the angiogenic switch in hypoxic conditions. Oncogene 2021, 40, 1721–1736. [Google Scholar] [CrossRef]
- Piccart-Gebhart, M.J.; Procter, M.; Leyland-Jones, B.; Goldhirsch, A.; Untch, M.; Smith, I.; Gianni, L.; Baselga, J.; Bell, R.; Jackisch, C.; et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1659–1672. [Google Scholar] [CrossRef] [Green Version]
- O’Shaughnessy, J.; Osborne, C.; Pippen, J.E.; Yoffe, M.; Patt, D.; Rocha, C.; Koo, I.C.; Sherman, B.M.; Bradley, C. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N. Engl. J. Med. 2011, 364, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Burstein, H.J. Systemic Therapy for Estrogen Receptor-Positive, HER2-Negative Breast Cancer. N. Engl. J. Med. 2020, 383, 2557–2570. [Google Scholar] [CrossRef]
- Middleton, J.D.; Stover, D.G.; Hai, T. Chemotherapy-Exacerbated Breast Cancer Metastasis: A Paradox Explainable by Dysregulated Adaptive-Response. Int. J. Mol. Sci 2018, 19, 3333. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Volk-Draper, L.; Hall, K.; Griggs, C.; Rajput, S.; Kohio, P.; DeNardo, D.; Ran, S. Paclitaxel therapy promotes breast cancer metastasis in a TLR4-dependent manner. Cancer Res. 2014, 74, 5421–5434. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, G.S.; Pastoriza, J.M.; Wang, Y.; Harney, A.S.; Entenberg, D.; Pignatelli, J.; Sharma, V.P.; Xue, E.A.; Cheng, E.; D′Alfonso, T.M.; et al. Neoadjuvant chemotherapy induces breast cancer metastasis through a TMEM-mediated mechanism. Sci. Transl. Med. 2017, 9, eaan0026. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Zhou, X.; Yang, J.J.; Liu, X.; Zhao, X.H.; Wang, Q.X.; Han, L.; Song, X.; Zhu, Z.Y.; Tian, W.P.; et al. AC1MMYR2 impairs high dose paclitaxel-induced tumor metastasis by targeting miR-21/CDK5 axis. Cancer Lett. 2015, 362, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Chen, I.; Shimoda, L.A.; Park, Y.; Zhang, C.; Tran, L.; Zhang, H.; Semenza, G.L. Chemotherapy-Induced Ca(2+) Release Stimulates Breast Cancer Stem Cell Enrichment. Cell Rep. 2017, 18, 1946–1957. [Google Scholar] [CrossRef]
- Keklikoglou, I.; Cianciaruso, C.; Guc, E.; Squadrito, M.L.; Spring, L.M.; Tazzyman, S.; Lambein, L.; Poissonnier, A.; Ferraro, G.B.; Baer, C.; et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat. Cell Biol. 2019, 21, 190–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wills, C.A.; Liu, X.; Chen, L.; Zhao, Y.; Dower, C.M.; Sundstrom, J.; Wang, H.G. Chemotherapy-Induced Upregulation of Small Extracellular Vesicle-Associated PTX3 Accelerates Breast Cancer Metastasis. Cancer Res. 2021, 81, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Cory, G. Scratch-wound assay. Methods Mol. Biol. 2011, 769, 25–30. [Google Scholar] [CrossRef]
- Hulkower, K.I.; Herber, R.L. Cell migration and invasion assays as tools for drug discovery. Pharmaceutics 2011, 3, 107–124. [Google Scholar] [CrossRef] [Green Version]
- Khanna, C.; Hunter, K. Modeling metastasis in vivo. Carcinogenesis 2005, 26, 513–523. [Google Scholar] [CrossRef] [Green Version]
- Guy, C.T.; Cardiff, R.D.; Muller, W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic Disease. Mol. Cell. Biol. 1992, 12, 954–961. [Google Scholar] [CrossRef]
- Bibby, M.C. Orthotopic models of cancer for preclinical drug evaluation: Advantages and disadvantages. Eur. J. Cancer 2004, 40, 852–857. [Google Scholar] [CrossRef]
- Kim, J.B.; Urban, K.; Cochran, E.; Lee, S.; Ang, A.; Rice, B.; Bata, A.; Campbell, K.; Coffee, R.; Gorodinsky, A.; et al. Non-invasive detection of a small number of Bioluminescent cancer Cells in vivo. PLoS ONE 2010, 5, e9364. [Google Scholar] [CrossRef]
- Ritsma, L.; Steller, E.J.; Beerling, E.; Loomans, C.J.; Zomer, A.; Gerlach, C.; Vrisekoop, N.; Seinstra, D.; Van Gurp, L.; Schafer, R.; et al. Intravital microscopy through an abdominal imaging window reveals a pre-micrometastasis stage during liver metastasis. Sci. Transl. Med. 2012, 4, 158ra145. [Google Scholar] [CrossRef]
- Hason, M.; Bartunek, P. Zebrafish Models of Cancer-New Insights on Modeling Human Cancer in a Non-Mammalian Vertebrate. Genes 2019, 10, 935. [Google Scholar] [CrossRef] [Green Version]
- Fior, R.; Povoa, V.; Mendes, R.V.; Carvalho, T.; Gomes, A.; Figueiredo, N.; Ferreira, M.G. Single-Cell. functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proc. Natl. Acad. Sci. USA 2017, 114, E8234–E8243. [Google Scholar] [CrossRef] [Green Version]
- Follain, G.; Osmani, N.; Fuchs, C.; Allio, G.; Harlepp, S.; Goetz, J.G. Using the Zebrafish Embryo to Dissect the Early Steps of the Metastasis Cascade. Methods Mol. Biol. 2018, 1749, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Follain, G.; Osmani, N.; Azevedo, A.S.; Allio, G.; Mercier, L.; Karreman, M.A.; Solecki, G.; Garcia Leon, M.J.; Lefebvre, O.; Fekonja, N.; et al. Hemodynamic Forces Tune the Arrest, Adhesion, and Extravasation of Circulating Tumor Cells. Dev. Cell. 2018, 45, 33–52.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asokan, N.; Daetwyler, S.; Bernas, S.N.; Schmied, C.; Vogler, S.; Lambert, K.; Wobus, M.; Wermke, M.; Kempermann, G.; Huisken, J.; et al. Long-term in vivo imaging reveals tumor-specific Dissemination and captures host tumor Interaction in zebrafish xenografts. Sci. Rep. 2020, 10, 13254. [Google Scholar] [CrossRef] [PubMed]
- Nicoli, S.; Ribatti, D.; Cotelli, F.; Presta, M. Mammalian tumor xenografts induce neovascularization in zebrafish embryos. Cancer Res. 2007, 67, 2927–2931. [Google Scholar] [CrossRef] [Green Version]
- Roh-Johnson, M.; Shah, A.N.; Stonick, J.A.; Poudel, K.R.; Kargl, J.; Yang, G.H.; Di Martino, J.; Hernandez, R.E.; Gast, C.E.; Zarour, L.R.; et al. Macrophage-Dependent Cytoplasmic Transfer during Melanoma Invasion In Vivo. Dev. Cell. 2017, 43, 549–562.e546. [Google Scholar] [CrossRef]
- Britto, D.D.; Wyroba, B.; Chen, W.; Lockwood, R.A.; Tran, K.B.; Shepherd, P.R.; Hall, C.J.; Crosier, K.E.; Crosier, P.S.; Astin, J.W. Macrophages enhance Vegfa-driven angiogenesis in an embryonic zebrafish tumour xenograft model. Dis. Model. Mech. 2018, 11, dmm.035998. [Google Scholar] [CrossRef] [Green Version]
- Povoa, V.; Rebelo de Almeida, C.; Maia-Gil, M.; Sobral, D.; Domingues, M.; Martinez-Lopez, M.; De Almeida Fuzeta, M.; Silva, C.; Grosso, A.R.; Fior, R. Innate immune evasion revealed in a colorectal zebrafish xenograft model. Nat. Commun. 2021, 12, 1156. [Google Scholar] [CrossRef]
- Costa, B.; Estrada, M.F.; Mendes, R.V.; Fior, R. Zebrafish Avatars towards Personalized Medicine-A Comparative Review between Avatar Models. Cells 2020, 9, 293. [Google Scholar] [CrossRef] [Green Version]
- Gregory, T.R. Understanding Natural Selection: Essential Concepts and Common Misconceptions. Evo. Edu. Outreach 2009, 2, 156–175. [Google Scholar] [CrossRef] [Green Version]


|
|
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruscitto, F.; Roda, N.; Priami, C.; Migliaccio, E.; Pelicci, P.G. Beyond Genetics: Metastasis as an Adaptive Response in Breast Cancer. Int. J. Mol. Sci. 2022, 23, 6271. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116271
Ruscitto F, Roda N, Priami C, Migliaccio E, Pelicci PG. Beyond Genetics: Metastasis as an Adaptive Response in Breast Cancer. International Journal of Molecular Sciences. 2022; 23(11):6271. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116271
Chicago/Turabian StyleRuscitto, Federica, Niccolò Roda, Chiara Priami, Enrica Migliaccio, and Pier Giuseppe Pelicci. 2022. "Beyond Genetics: Metastasis as an Adaptive Response in Breast Cancer" International Journal of Molecular Sciences 23, no. 11: 6271. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23116271