
The m6A Methyltransferase METTL3-Mediated N6-Methyladenosine Modification of DEK mRNA to Promote Gastric Cancer Cell Growth and Metastasis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. METTL3 Is Highly Expressed in GC
2.2. METTL3 Promotes the Proliferation and Migration of GC Cells
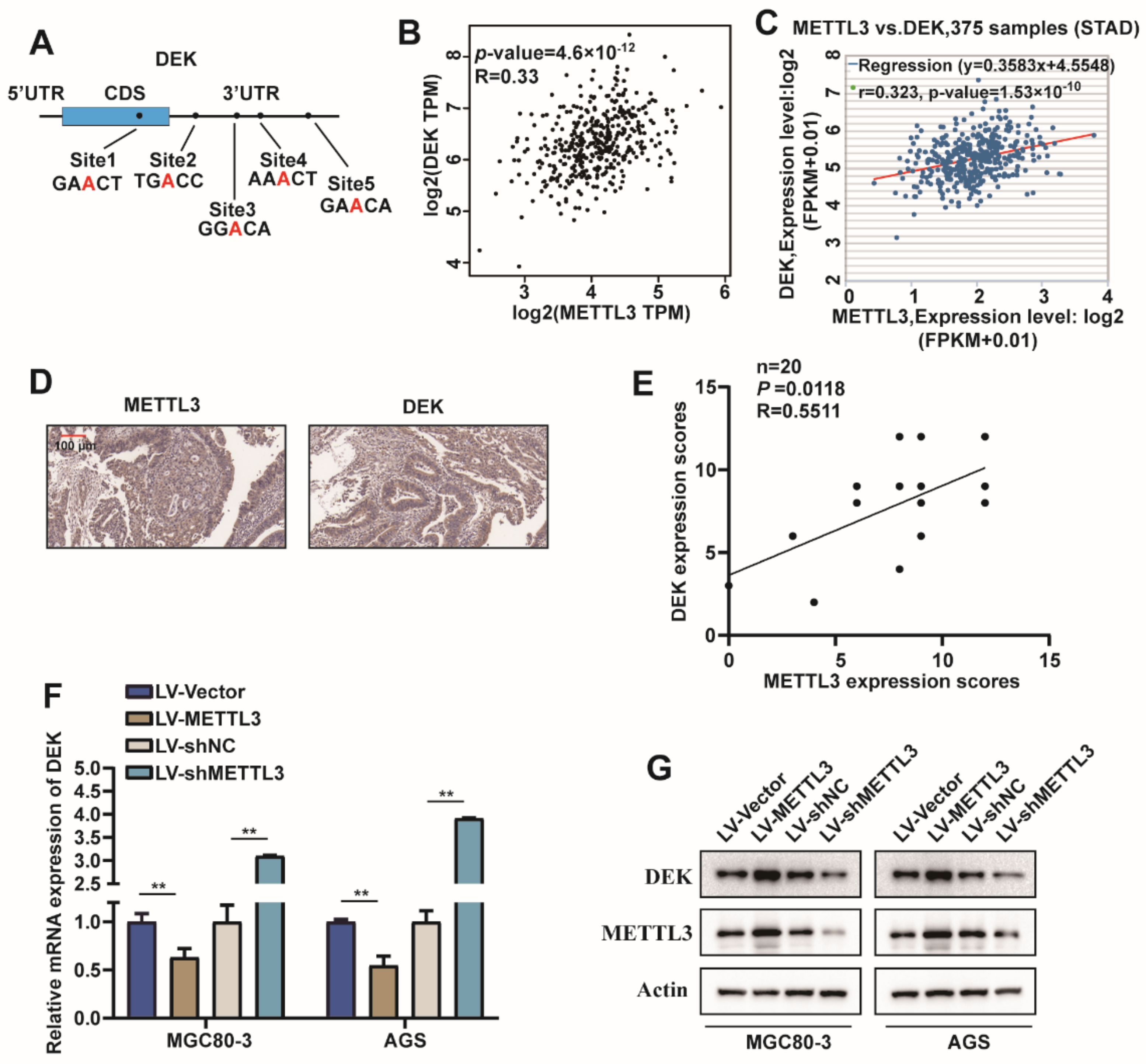
2.3. METTL3 Regulated DEK Expression in GC
2.4. METTL3 Stabilize DEK mRNA via m6A Modification in GC
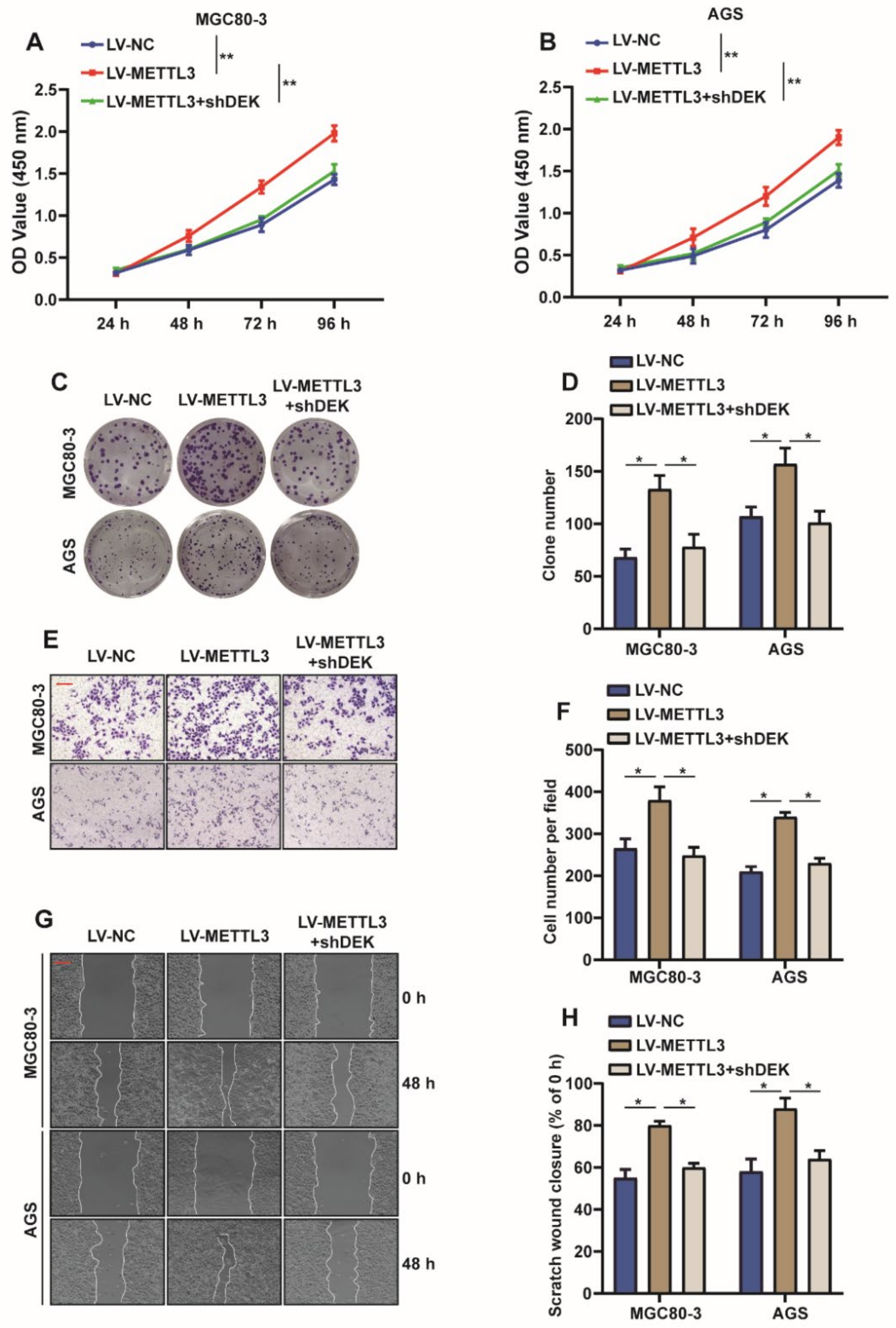
2.5. METTL3 Promotes Proliferation and Migration of GC Cells by Regulating DEK
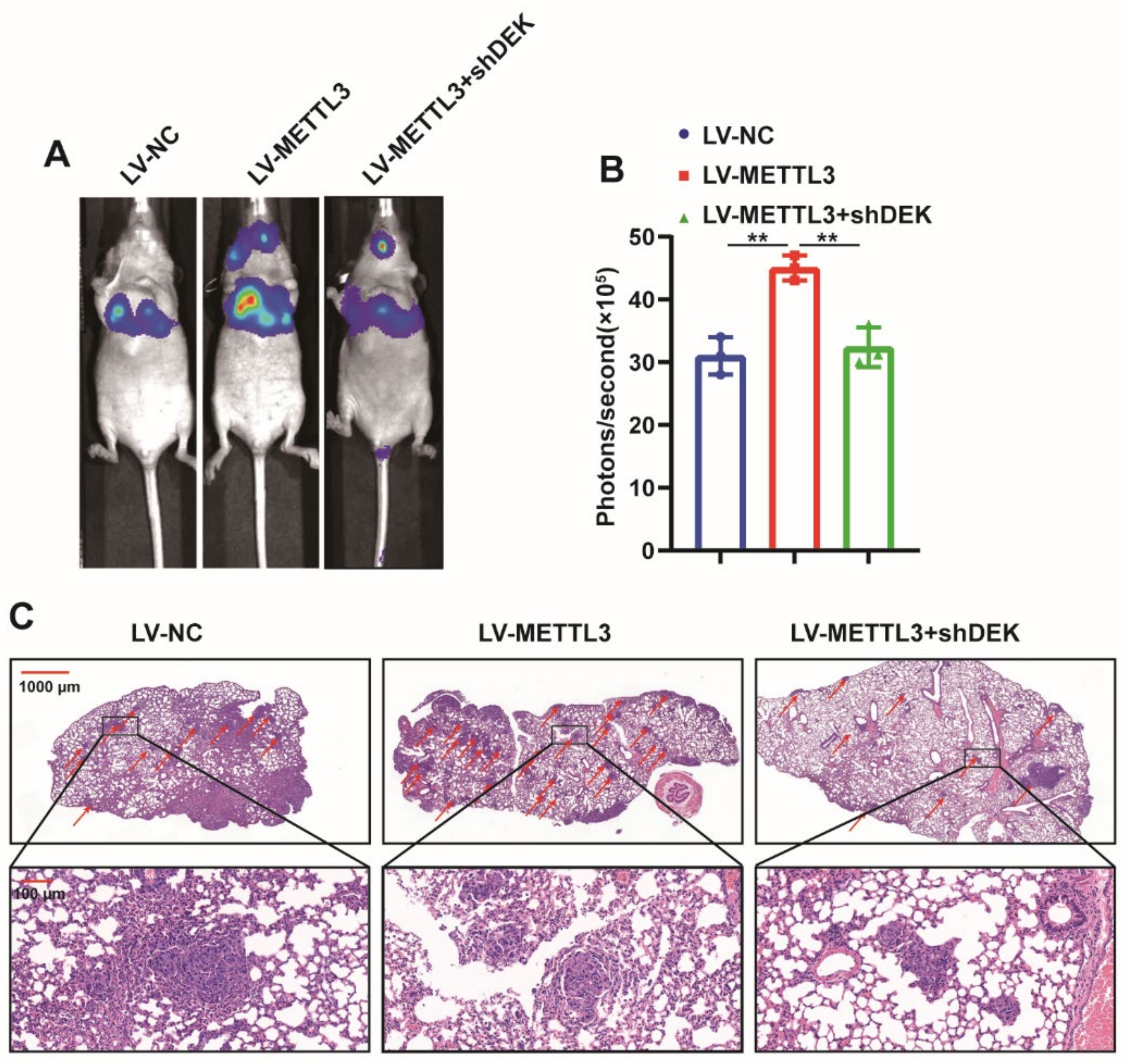
2.6. METTL3 Promotes GC Metastasis In Vivo
3. Discussion
4. Materials and Methods
4.1. Tissue Samples
4.2. Cell Lines
4.3. Quantitative Real-Time PCR (qRT-PCR)
4.4. Plasmid Construction and Cell Transfection
4.5. Western Blot
4.6. Cell Counting Kit-8 (CCK-8) Assay
4.7. Wound-Healing Assay
4.8. Colony Formation
4.9. Transwell Assay
4.10. m6A RNA Dot Blot Assay
4.11. RNA Immunoprecipitation Assay (RIP)
4.12. Methylated RNA Immunoprecipitation Assay (MeRIP)
4.13. Dual-Luciferase Reporter Assay
4.14. BALB/c Nude Mice Animal Models
4.15. IHC Analysis
4.16. Bioinformatic Analysis
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeneevassen, L.; Bessède, E.; Mégraud, F.; Lehours, P.; Dubus, P.; Varon, C. Gastric Cancer: Advances in Carcinogenesis Research and New Therapeutic Strategies. Int. J. Mol. Sci. 2021, 22, 3418. [Google Scholar] [CrossRef] [PubMed]
- Han, M.S.; Barrett, T.; Brehm, M.A.; Davis, R.J. Inflammation Mediated by JNK in Myeloid Cells Promotes the Development of Hepatitis and Hepatocellular Carcinoma. Cell Rep. 2016, 15, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef]
- Shen, H.; Lan, Y.; Zhao, Y.; Shi, Y.; Jin, J.; Xie, W. The emerging roles of N6-methyladenosine RNA methylation in human cancers. Biomark. Res. 2020, 8, 24. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Zeng, C.; Huang, W.; Li, Y.; Weng, H. Roles of METTL3 in cancer: Mechanisms and therapeutic targeting. J. Hematol. Oncol. 2020, 13, 117. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Yin, Z.; Hou, B.; Yu, M.; Chen, R.; Jin, H.; Jian, Z. Expression profiles and prognostic significance of RNA N6-methyladenosine-related genes in patients with hepatocellular carcinoma: Evidence from independent datasets. Cancer Manag. Res. 2019, 11, 3921–3931. [Google Scholar] [CrossRef] [Green Version]
- Choe, J.; Lin, S.; Zhang, W.; Liu, Q.; Wang, L.; Ramirez-Moya, J.; Du, P.; Kim, W.; Tang, S.; Sliz, P.; et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 2018, 561, 556–560. [Google Scholar] [CrossRef] [Green Version]
- Yue, B.; Song, C.; Yang, L.; Cui, R.; Cheng, X.; Zhang, Z.; Zhao, G. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol. Cancer 2019, 18, 142. [Google Scholar] [CrossRef] [Green Version]
- Riveiro-Falkenbach, E.; Soengas, M.S. Control of tumorigenesis and chemoresistance by the DEK oncogene. Clin. Cancer Res. 2010, 16, 2932–2938. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Huang, X.; Zhang, W.; Zhao, H.; Wu, G.; Lv, F.; Shi, L.; Teng, Y. Critical role of DEK and its regulation in tumorigenesis and metastasis of hepatocellular carcinoma. Oncotarget 2016, 7, 26844–26855. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhang, C.; Gan, L.; Jia, Y.; Xiong, Y.; Chen, Y.; Wang, Z.; Wang, L.; Luo, H.; Li, J.; et al. iTRAQ-Based Quantitative Proteomics Approach Identifies Novel Diagnostic Biomarkers That Were Essential for Glutamine Metabolism and Redox Homeostasis for Gastric Cancer. Proteom. Clin. Appl. 2019, 13, e1800038. [Google Scholar] [CrossRef]
- Gayosso-Gómez, L.V.; Zárraga-Granados, G.; Paredes-Garcia, P.; Falfán-Valencia, R.; Vazquez-Manríquez, M.E.; Martinez-Barrera, L.M.; Castillo-Gonzalez, P.; Rumbo-Nava, U.; Guevara-Gutierrez, R.; Rivera-Bravo, B.; et al. Identification of circulating miRNAs profiles that distinguish malignant pleural mesothelioma from lung adenocarcinoma. EXCLI J. 2014, 13, 740–750. [Google Scholar]
- Vu, L.P.; Pickering, B.F.; Cheng, Y.; Zaccara, S.; Nguyen, D.; Minuesa, G.; Chou, T.; Chow, A.; Saletore, Y.; MacKay, M.; et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017, 23, 1369–1376. [Google Scholar] [CrossRef]
- Wang, H.; Xu, B.; Shi, J. N6-methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene 2020, 722, 144076. [Google Scholar] [CrossRef]
- Wu, M.; Xia, Y.; Wang, Y.; Fan, F.; Li, X.; Song, J.; Ding, J. Development and validation of an immune-related gene prognostic model for stomach adenocarcinoma. Biosci. Rep. 2020, 40, BSR20201012. [Google Scholar] [CrossRef]
- Ye, T.; Yang, M.; Huang, D.; Wang, X.; Xue, B.; Tian, N.; Xu, X.; Bao, L.; Hu, H.; Lv, T.; et al. MicroRNA-7 as a potential therapeutic target for aberrant NF-κB-driven distant metastasis of gastric cancer. J. Exp. Clin. Cancer Res. CR 2019, 38, 55. [Google Scholar] [CrossRef] [Green Version]
- Dragomir, M.P.; Kopetz, S.; Ajani, J.A.; Calin, G.A. Non-coding RNAs in GI cancers: From cancer hallmarks to clinical utility. Gut 2020, 69, 748–763. [Google Scholar] [CrossRef]
- Canale, M.; Casadei-Gardini, A.; Ulivi, P.; Arechederra, M.; Berasain, C.; Lollini, P.L.; Fernández-Barrena, M.G.; Avila, M.A. Epigenetic Mechanisms in Gastric Cancer: Potential New Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 5500. [Google Scholar] [CrossRef]
- Saletore, Y.; Meyer, K.; Korlach, J.; Vilfan, I.D.; Jaffrey, S.; Mason, C.E. The birth of the Epitranscriptome: Deciphering the function of RNA modifications. Genome Biol. 2012, 13, 175. [Google Scholar] [CrossRef] [Green Version]
- Helm, M.; Motorin, Y. Detecting RNA modifications in the epitranscriptome: Predict and validate. Nat. Rev. Genet. 2017, 18, 275–291. [Google Scholar] [CrossRef]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.Y.; Liang, R.; Yi, Y.C.; Fan, H.N.; Chen, M.; Zhang, J.; Zhu, J.S. The m(6)A Reader YTHDF1 Facilitates the Tumorigenesis and Metastasis of Gastric Cancer via USP14 Translation in an m(6)A-Dependent Manner. Front. Cell Dev. Biol. 2021, 9, 647702. [Google Scholar] [CrossRef]
- Du, Y.; Hou, G.; Zhang, H.; Dou, J.; He, J.; Guo, Y.; Li, L.; Chen, R.; Wang, Y.; Deng, R.; et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 2018, 46, 5195–5208. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, C.; Ding, Q.; Zhao, Y.; Wang, Z.; Chen, J.; Jiang, Z.; Zhang, Y.; Xu, G.; Zhang, J.; et al. METTL3-mediated m(6)A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut 2020, 69, 1193–1205. [Google Scholar] [CrossRef]
- Liu, T.; Yang, S.; Sui, J.; Xu, S.Y.; Cheng, Y.P.; Shen, B.; Zhang, Y.; Zhang, X.M.; Yin, L.H.; Pu, Y.P.; et al. Dysregulated N6-methyladenosine methylation writer METTL3 contributes to the proliferation and migration of gastric cancer. J. Cell Physiol. 2020, 235, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, C.; Lan, L.; Yan, L.; Li, W.; Evans, I.; Ruiz, E.J.; Su, Q.; Zhao, G.; Wu, W.; et al. METTL3 promotes oxaliplatin resistance of gastric cancer CD133+ stem cells by promoting PARP1 mRNA stability. Cell Mol. Life Sci. 2022, 79, 135. [Google Scholar] [CrossRef] [PubMed]
- Huo, F.C.; Zhu, Z.M.; Zhu, W.T.; Du, Q.Y.; Liang, J.; Mou, J. METTL3-mediated m(6)A methylation of SPHK2 promotes gastric cancer progression by targeting KLF2. Oncogene 2021, 40, 2968–2981. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Xian, Q.; Wang, Q.; Wu, C.; Yan, H.; Li, X.; Lu, L.; Wu, C.; Zhu, D.; Xu, X.; et al. m6A Methyltransferase 3 Promotes the Proliferation and Migration of Gastric Cancer Cells through the m6A Modification of YAP1. J. Oncol. 2021, 2021, 8875424. [Google Scholar] [CrossRef]
- Liu, Z.F.; Yang, J.; Wei, S.P.; Luo, X.G.; Jiang, Q.S.; Chen, T.; Gong, Y.Q. Upregulated METTL3 in nasopharyngeal carcinoma enhances the motility of cancer cells. Kaohsiung J. Med. Sci. 2020, 36, 895–903. [Google Scholar] [CrossRef]
- Chen, M.; Wei, L.; Law, C.T.; Tsang, F.H.; Shen, J.; Cheng, C.L.; Tsang, L.H.; Ho, D.W.; Chiu, D.K.; Lee, J.M.; et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 2018, 67, 2254–2270. [Google Scholar] [CrossRef]
- Cheng, M.; Sheng, L.; Gao, Q.; Xiong, Q.; Zhang, H.; Wu, M.; Liang, Y.; Zhu, F.; Zhang, Y.; Zhang, X.; et al. The m(6)A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-κB/MYC signaling network. Oncogene 2019, 38, 3667–3680. [Google Scholar] [CrossRef]
- Deng, R.; Cheng, Y.; Ye, S.; Zhang, J.; Huang, R.; Li, P.; Liu, H.; Deng, Q.; Wu, X.; Lan, P.; et al. m(6)A methyltransferase METTL3 suppresses colorectal cancer proliferation and migration through p38/ERK pathways. OncoTargets Ther. 2019, 12, 4391–4402. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.Q.; Bai, L.L.; Wang, Z.; Lei, L.; Zheng, Y.W.; Li, Z.H.; Huang, W.J.; Liu, C.C.; Xu, H.T. DEK is highly expressed in breast cancer and is associated with malignant phenotype and progression. Oncol. Lett. 2021, 21, 440. [Google Scholar] [CrossRef]
- Pease, N.A.; Shephard, M.S.; Sertorio, M.; Waltz, S.E.; Vinnedge, L.M.P. DEK Expression in Breast Cancer Cells Leads to the Alternative Activation of Tumor Associated Macrophages. Cancers 2020, 12, 1936. [Google Scholar] [CrossRef]
- Hacker, K.E.; Bolland, D.E.; Tan, L.; Saha, A.K.; Niknafs, Y.S.; Markovitz, D.M.; McLean, K. The DEK Oncoprotein Functions in Ovarian Cancer Growth and Survival. Neoplasia 2018, 20, 1209–1218. [Google Scholar] [CrossRef]
- Cai, Y.; Hao, Y.; Xu, H.; Chen, K.; Ren, B. Gigantol inhibits cell proliferation and induces apoptosis by regulating DEK in non-small cell lung cancer. Exp. Ther. Med. 2021, 22, 1317. [Google Scholar] [CrossRef]
- Lee, K.F.; Tsai, M.M.; Tsai, C.Y.; Huang, C.G.; Ou, Y.H.; Hsieh, C.C.; Hsieh, H.L.; Wang, C.S.; Lin, K.H. DEK Is a Potential Biomarker Associated with Malignant Phenotype in Gastric Cancer Tissues and Plasma. Int. J. Mol. Sci. 2019, 20, 5689. [Google Scholar] [CrossRef] [Green Version]
- Fan, D.; Wang, C.; Wang, D.; Zhang, N.; Yi, T. Circular RNA circ_0000039 enhances gastric cancer progression through miR-1292-5p/DEK axis. Cancer Biomark. Sect. A Dis. Markers 2021, 30, 167–177. [Google Scholar] [CrossRef]
- Zhang, W.; Liao, K.; Liu, D. MiR-138-5p Inhibits the Proliferation of Gastric Cancer Cells by Targeting DEK. Cancer Manag. Res. 2020, 12, 8137–8147. [Google Scholar] [CrossRef]
- Hui, W.; Ma, X.; Zan, Y.; Song, L.; Zhang, S.; Dong, L. MicroRNA-1292-5p inhibits cell growth, migration and invasion of gastric carcinoma by targeting DEK. Am. J. Cancer Res. 2018, 8, 1228–1238. [Google Scholar]
- Zhang, H.; Wang, J.; Wang, Y.; Li, J.; Zhao, L.; Zhang, T.; Liao, X. Long Non-Coding LEF1-AS1 Sponge miR-5100 Regulates Apoptosis and Autophagy in Gastric Cancer Cells via the miR-5100/DEK/AMPK-mTOR Axis. Int. J. Mol. Sci. 2022, 23, 4787. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.-M.; Qi, F.-F.; Wang, J.; Duan, Y.-Y.; Zhao, L.-L.; Wang, Y.-D.; Zhang, T.-C.; Liao, X.-H. The m6A Methyltransferase METTL3-Mediated N6-Methyladenosine Modification of DEK mRNA to Promote Gastric Cancer Cell Growth and Metastasis. Int. J. Mol. Sci. 2022, 23, 6451. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126451
Zhang H-M, Qi F-F, Wang J, Duan Y-Y, Zhao L-L, Wang Y-D, Zhang T-C, Liao X-H. The m6A Methyltransferase METTL3-Mediated N6-Methyladenosine Modification of DEK mRNA to Promote Gastric Cancer Cell Growth and Metastasis. International Journal of Molecular Sciences. 2022; 23(12):6451. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126451
Chicago/Turabian StyleZhang, Hui-Min, Fei-Fei Qi, Jun Wang, Yuan-Yuan Duan, Li-Li Zhao, Yun-Dan Wang, Tong-Cun Zhang, and Xing-Hua Liao. 2022. "The m6A Methyltransferase METTL3-Mediated N6-Methyladenosine Modification of DEK mRNA to Promote Gastric Cancer Cell Growth and Metastasis" International Journal of Molecular Sciences 23, no. 12: 6451. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126451