
Ferulic Acid, Pterostilbene, and Tyrosol Protect the Heart from ER-Stress-Induced Injury by Activating SIRT1-Dependent Deacetylation of eIF2α

 ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
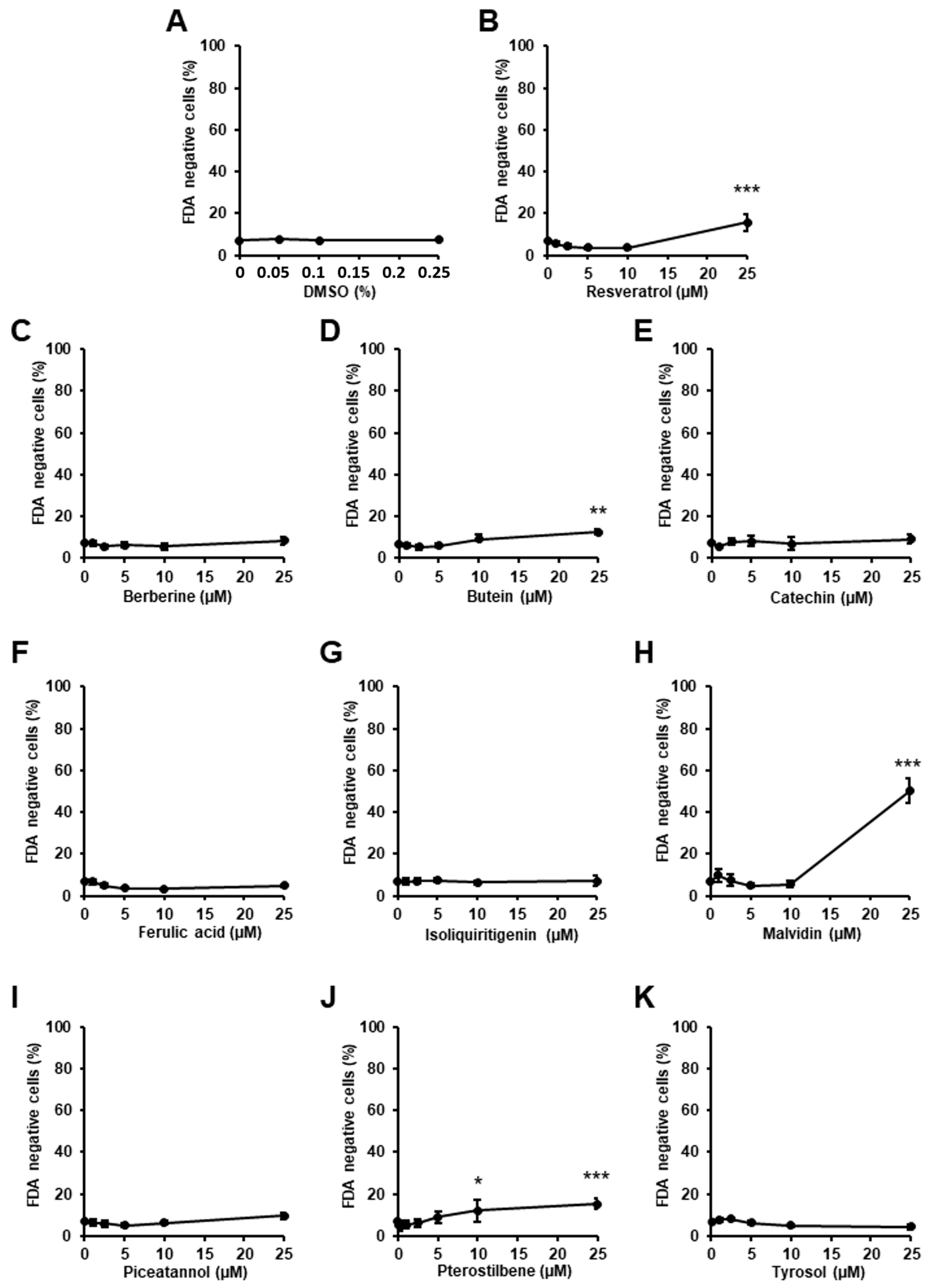
2.1. Determination of the Effect of the Phytochemicals on Cardiac Cell Viability
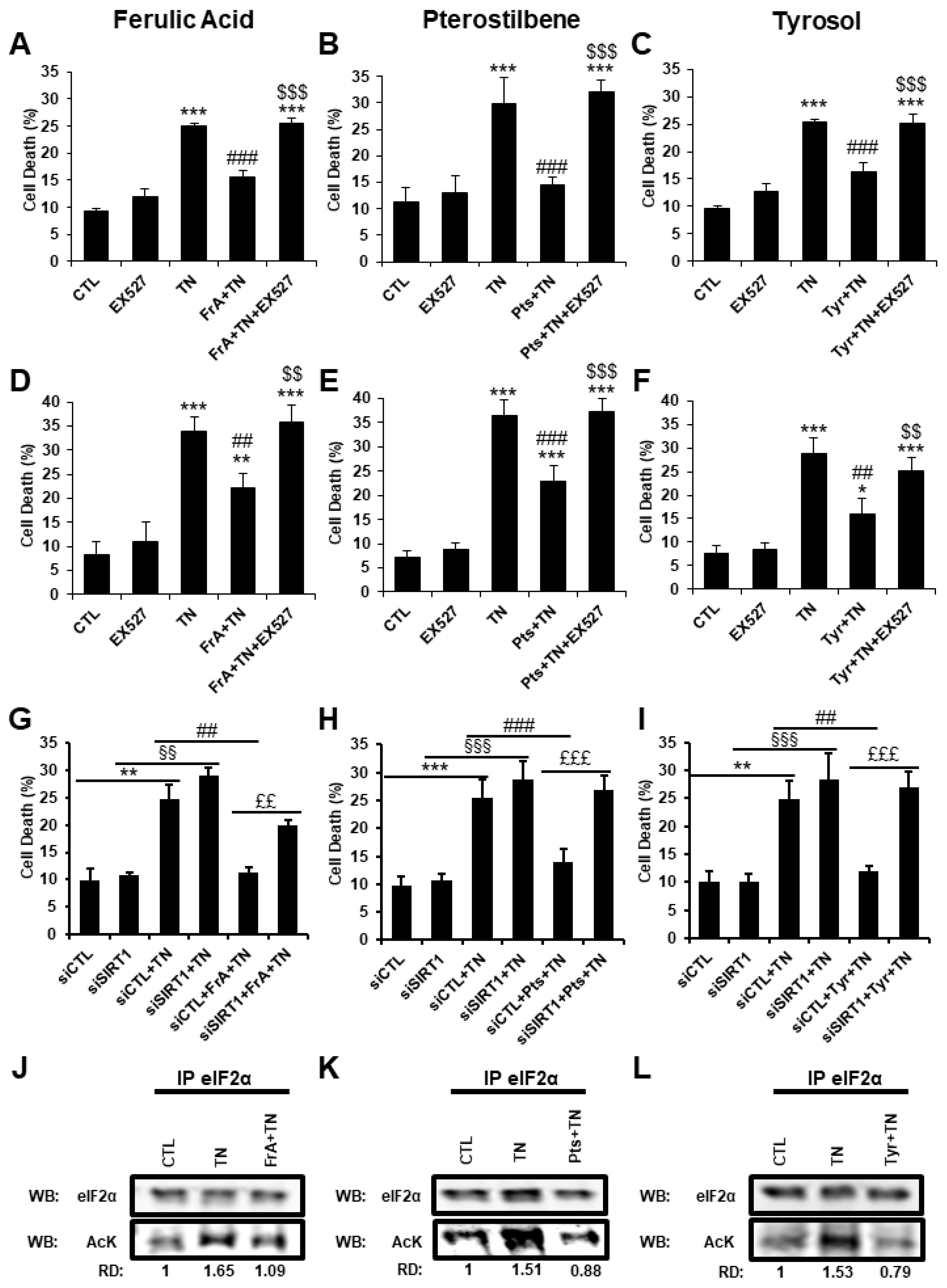
2.2. Ferulic Acid, Pterostilbene, and Tyrosol Protect Adult Cardiomyocytes from Cell Death Induced by Severe ER Stress
2.3. Ferulic Acid, Pterostilbene, and Tyrosol Protect the Heart from Damage Induced by Severe ER Stress
2.4. Ferulic Acid, Pterostilbene, and Tyrosol Protect Cardiac Cells from Apoptosis Induced by Severe ER Stress
2.5. The Cardioprotective Effect of Ferulic Acid, Pterostilbene, and Tyrosol Do Not Seem to Be Directly Related to Their Antioxidant Activity
2.6. Ferulic Acid, Pterostilbene, and Tyrosol Reduce ER-Stress-Induced Activation of the PERK Pathway of the UPR
2.7. Ferulic Acid, Pterostilbene, and Tyrosol Protect Cardiomyocytes from ER-Stress-Induced Apoptosis by Regulating the PERK Pathway through SIRT1-Mediated Deacetylation of eIF2α
3. Discussion
4. Materials and Methods
4.1. Animals and Treatments
4.2. Echocardiography
4.3. Cell Culture and Reagents
4.4. Flow Cytometry
4.5. Isolation of Adult Rat Ventricular Myocytes (ARVM)
4.6. Evaluation of Adult Cardiomyocyte Death by Fluorescence Microscopy
4.7. RNA Isolation and Quantitative RT-PCR
4.8. Western Blot Analysis
4.9. siRNA Transfection
4.10. Protein Immunoprecipitation
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Binder, P.; Fang, Q.; Wang, Z.; Xiao, W.; Liu, W.; Wang, X. Endoplasmic reticulum stress in the heart: Insights into mechanisms and drug targets. Br. J. Pharmacol. 2018, 175, 1293–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.Y.; Minamino, T.; Tsukamoto, O.; Sawada, T.; Asai, M.; Kato, H.; Asano, Y.; Fujita, M.; Takashima, S.; Hori, M.; et al. Overexpression of endoplasmic reticulum-resident chaperone attenuates cardiomyocyte death induced by proteasome inhibition. Cardiovasc. Res. 2008, 79, 600–610. [Google Scholar] [CrossRef]
- Martindale, J.J.; Fernandez, R.; Thuerauf, D.; Whittaker, R.; Gude, N.; Sussman, M.A.; Glembotski, C.C. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ. Res. 2006, 98, 1186–1193. [Google Scholar] [CrossRef] [Green Version]
- Prola, A.; Nichtova, Z.; Pires Da Silva, J.; Piquereau, J.; Monceaux, K.; Guilbert, A.; Gressette, M.; Ventura-Clapier, R.; Garnier, A.; Zahradnik, I.; et al. Endoplasmic reticulum stress induces cardiac dysfunction through architectural modifications and alteration of mitochondrial function in cardiomyocytes. Cardiovasc. Res. 2019, 115, 328–342. [Google Scholar] [CrossRef]
- Fu, H.Y.; Okada, K.; Liao, Y.; Tsukamoto, O.; Isomura, T.; Asai, M.; Sawada, T.; Okuda, K.; Asano, Y.; Sanada, S.; et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 2010, 122, 361–369. [Google Scholar] [CrossRef]
- Matsushima, S.; Sadoshima, J. The role of sirtuins in cardiac disease. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1375–H1389. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.; Islam, R. Mammalian Sirt1: Insights on its biological functions. Cell Commun. Signal. 2011, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Prola, A.; Silva, J.P.; Guilbert, A.; Lecru, L.; Piquereau, J.; Ribeiro, M.; Mateo, P.; Gressette, M.; Fortin, D.; Boursier, C.; et al. SIRT1 protects the heart from ER stress-induced cell death through eIF2alpha deacetylation. Cell Death Differ. 2017, 24, 343–356. [Google Scholar] [CrossRef] [Green Version]
- Pires Da Silva, J.; Monceaux, K.; Guilbert, A.; Gressette, M.; Piquereau, J.; Novotova, M.; Ventura-Clapier, R.; Garnier, A.; Lemaire, C. SIRT1 Protects the Heart from ER Stress-Induced Injury by Promoting eEF2K/eEF2-Dependent Autophagy. Cells 2020, 9, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhullar, K.S.; Son, M.; Kerek, E.; Cromwell, C.R.; Wingert, B.M.; Wu, K.; Jovel, J.; Camacho, C.J.; Hubbard, B.P.; Wu, J. Tripeptide IRW Upregulates NAMPT Protein Levels in Cells and Obese C57BL/6J Mice. J. Agric. Food Chem. 2021, 69, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Ashkar, F.; Wu, J. Peptides GWN and GW protect kidney cells against Dasatinib induced mitochondrial injury in a SIRT1 dependent manner. Food Chem. Mol. Sci. 2022, 4, 100069. [Google Scholar] [CrossRef] [PubMed]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Fusi, J.; Bianchi, S.; Daniele, S.; Pellegrini, S.; Martini, C.; Galetta, F.; Giovannini, L.; Franzoni, F. An in vitro comparative study of the antioxidant activity and SIRT1 modulation of natural compounds. Biomed. Pharmacother. 2018, 101, 805–819. [Google Scholar] [CrossRef]
- Gay, N.H.; Suwanjang, W.; Ruankham, W.; Songtawee, N.; Wongchitrat, P.; Prachayasittikul, V.; Prachayasittikul, S.; Phopin, K. Butein, isoliquiritigenin, and scopoletin attenuate neurodegeneration via antioxidant enzymes and SIRT1/ADAM10 signaling pathway. RSC Adv. 2020, 10, 16593–16606. [Google Scholar] [CrossRef]
- Giovannini, L.; Bianchi, S. Role of nutraceutical SIRT1 modulators in AMPK and mTOR pathway: Evidence of a synergistic effect. Nutrition 2017, 34, 82–96. [Google Scholar] [CrossRef]
- Kershaw, J.; Kim, K.H. The Therapeutic Potential of Piceatannol, a Natural Stilbene, in Metabolic Diseases: A Review. J. Med. Food 2017, 20, 427–438. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Lee, W.; Yun, J.M. Luteolin and fisetin suppress oxidative stress by modulating sirtuins and forkhead box O3a expression under in vitro diabetic conditions. Nutr. Res. Pract. 2017, 11, 430–434. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Wang, K.; Ma, Z.; Liu, D.; Yang, Y.; Sun, M.; Wen, A.; Hao, Y.; Ma, S.; Ren, F.; et al. SIRT1 activation by butein attenuates sepsis-induced brain injury in mice subjected to cecal ligation and puncture via alleviating inflammatory and oxidative stress. Toxicol. Appl. Pharmacol. 2019, 363, 34–46. [Google Scholar] [CrossRef]
- Gonzalez Arbelaez, L.F.; Ciocci Pardo, A.; Fantinelli, J.C.; Schinella, G.R.; Mosca, S.M.; Rios, J.L. Cardioprotection and natural polyphenols: An update of clinical and experimental studies. Food Funct. 2018, 9, 6129–6145. [Google Scholar] [CrossRef] [PubMed]
- Goszcz, K.; Duthie, G.G.; Stewart, D.; Leslie, S.J.; Megson, I.L. Bioactive polyphenols and cardiovascular disease: Chemical antagonists, pharmacological agents or xenobiotics that drive an adaptive response? Br. J. Pharmacol. 2017, 174, 1209–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecour, S.; Lamont, K.T. Natural polyphenols and cardioprotection. Mini Rev. Med. Chem. 2011, 11, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Iside, C.; Scafuro, M.; Nebbioso, A.; Altucci, L. SIRT1 Activation by Natural Phytochemicals: An Overview. Front. Pharmacol. 2020, 11, 1225. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Yao, H.; Caito, S.; Hwang, J.W.; Arunachalam, G.; Rahman, I. Regulation of SIRT1 in cellular functions: Role of polyphenols. Arch. Biochem. Biophys. 2010, 501, 79–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, H.; Sinclair, D.A.; Ellis, J.L.; Steegborn, C. Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharmacol. Ther. 2018, 188, 140–154. [Google Scholar] [CrossRef]
- Karaman Mayack, B.; Sippl, W.; Ntie-Kang, F. Natural Products as Modulators of Sirtuins. Molecules 2020, 25, 3287. [Google Scholar] [CrossRef]
- Cheng, Y.; Di, S.; Fan, C.; Cai, L.; Gao, C.; Jiang, P.; Hu, W.; Ma, Z.; Jiang, S.; Dong, Y.; et al. SIRT1 activation by pterostilbene attenuates the skeletal muscle oxidative stress injury and mitochondrial dysfunction induced by ischemia reperfusion injury. Apoptosis Int. J. Program. Cell Death 2016, 21, 905–916. [Google Scholar] [CrossRef]
- Belmadani, S.; Matrougui, K. Broken heart: A matter of the endoplasmic reticulum stress bad management? World J. Cardiol. 2019, 11, 159–170. [Google Scholar] [CrossRef]
- Minamino, T.; Kitakaze, M. ER stress in cardiovascular disease. J. Mol. Cell. Cardiol. 2010, 48, 1105–1110. [Google Scholar] [CrossRef]
- Hollman, P.C.; Cassidy, A.; Comte, B.; Heinonen, M.; Richelle, M.; Richling, E.; Serafini, M.; Scalbert, A.; Sies, H.; Vidry, S. The biological relevance of direct antioxidant effects of polyphenols for cardiovascular health in humans is not established. J. Nutr. 2011, 141, 989S–1009S. [Google Scholar] [CrossRef] [Green Version]
- Mladenka, P.; Zatloukalova, L.; Filipsky, T.; Hrdina, R. Cardiovascular effects of flavonoids are not caused only by direct antioxidant activity. Free. Radic. Biol. Med. 2010, 49, 963–975. [Google Scholar] [CrossRef]
- Tangney, C.C.; Rasmussen, H.E. Polyphenols, inflammation, and cardiovascular disease. Curr. Atheroscler. Rep. 2013, 15, 324. [Google Scholar] [CrossRef]
- Oak, M.H.; Auger, C.; Belcastro, E.; Park, S.H.; Lee, H.H.; Schini-Kerth, V.B. Potential mechanisms underlying cardiovascular protection by polyphenols: Role of the endothelium. Free. Radic. Biol. Med. 2018, 122, 161–170. [Google Scholar] [CrossRef]
- Dyck, G.J.B.; Raj, P.; Zieroth, S.; Dyck, J.R.B.; Ezekowitz, J.A. The Effects of Resveratrol in Patients with Cardiovascular Disease and Heart Failure: A Narrative Review. Int. J. Mol. Sci. 2019, 20, 904. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi, A.; Hayes, A.W.; Karimi, G. Resveratrol and endoplasmic reticulum stress: A review of the potential protective mechanisms of the polyphenol. Phytother. Res. PTR 2021, 35, 5564–5583. [Google Scholar] [CrossRef]
- Lou, Y.; Wang, Z.; Xu, Y.; Zhou, P.; Cao, J.; Li, Y.; Chen, Y.; Sun, J.; Fu, L. Resveratrol prevents doxorubicin-induced cardiotoxicity in H9c2 cells through the inhibition of endoplasmic reticulum stress and the activation of the Sirt1 pathway. Int. J. Mol. Med. 2015, 36, 873–880. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Zhu, J.; Zhang, X.; Wang, J.; Xiao, W.; Li, B.; Jin, L.; Lian, J.; Zhou, L.; Liu, J. Inhibition of Cardiomyocytes Hypertrophy by Resveratrol Is Associated with Amelioration of Endoplasmic Reticulum Stress. Cell. Physiol. Biochem. 2016, 39, 780–789. [Google Scholar] [CrossRef]
- He, Y.; Fu, Y.; Xi, M.; Zheng, H.; Zhang, Y.; Liu, Y.; Zhao, Y.; Xi, J.; He, Y. Zn2+ and mPTP mediate resveratrol-induced myocardial protection from endoplasmic reticulum stress. Met. Integr. Biometal Sci. 2020, 12, 290–300. [Google Scholar] [CrossRef]
- Bonnefont-Rousselot, D. Resveratrol and Cardiovascular Diseases. Nutrients 2016, 8, 250. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef] [Green Version]
- Walle, T. Bioavailability of resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 9–15. [Google Scholar] [CrossRef]
- Boocock, D.J.; Faust, G.E.; Patel, K.R.; Schinas, A.M.; Brown, V.A.; Ducharme, M.P.; Booth, T.D.; Crowell, J.A.; Perloff, M.; Gescher, A.J.; et al. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1246–1252. [Google Scholar] [CrossRef] [Green Version]
- Almeida, L.; Vaz-da-Silva, M.; Falcao, A.; Soares, E.; Costa, R.; Loureiro, A.I.; Fernandes-Lopes, C.; Rocha, J.F.; Nunes, T.; Wright, L.; et al. Pharmacokinetic and safety profile of trans-resveratrol in a rising multiple-dose study in healthy volunteers. Mol. Nutr. Food Res. 2009, 53 (Suppl. S1), S7–S15. [Google Scholar] [CrossRef]
- Cottart, C.H.; Nivet-Antoine, V.; Laguillier-Morizot, C.; Beaudeux, J.L. Resveratrol bioavailability and toxicity in humans. Mol. Nutr. Food Res. 2010, 54, 7–16. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, L.; Li, F.; Hu, C.P.; Zhang, Z. Restoration of sirt1 function by pterostilbene attenuates hypoxia-reoxygenation injury in cardiomyocytes. Eur. J. Pharmacol. 2016, 776, 26–33. [Google Scholar] [CrossRef]
- Peng, R.M.; Lin, G.R.; Ting, Y.; Hu, J.Y. Oral delivery system enhanced the bioavailability of stilbenes: Resveratrol and pterostilbene. BioFactors 2018, 44, 5–15. [Google Scholar] [CrossRef]
- Riche, D.M.; McEwen, C.L.; Riche, K.D.; Sherman, J.J.; Wofford, M.R.; Deschamp, D.; Griswold, M. Analysis of safety from a human clinical trial with pterostilbene. J. Toxicol. 2013, 2013, 463595. [Google Scholar] [CrossRef]
- Karkovic Markovic, A.; Toric, J.; Barbaric, M.; Jakobusic Brala, C. Hydroxytyrosol, Tyrosol and Derivatives and Their Potential Effects on Human Health. Molecules 2019, 24, 2001. [Google Scholar] [CrossRef] [Green Version]
- Di Benedetto, R.; Vari, R.; Scazzocchio, B.; Filesi, C.; Santangelo, C.; Giovannini, C.; Matarrese, P.; D’Archivio, M.; Masella, R. Tyrosol, the major extra virgin olive oil compound, restored intracellular antioxidant defences in spite of its weak antioxidative effectiveness. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 535–545. [Google Scholar] [CrossRef]
- Zdunska, K.; Dana, A.; Kolodziejczak, A.; Rotsztejn, H. Antioxidant Properties of Ferulic Acid and Its Possible Application. Ski. Pharmacol. Physiol. 2018, 31, 332–336. [Google Scholar] [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression during Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [Green Version]
- Otręba, M.; Kośmider, L.; Rzepecka-Stojko, A. Polyphenols’ Cardioprotective Potential: Review of Rat Fibroblasts as Well as Rat and Human Cardiomyocyte Cell Lines Research. Molecules 2021, 26, 774. [Google Scholar] [CrossRef]
- Alam, M.A. Anti-hypertensive Effect of Cereal Antioxidant Ferulic Acid and Its Mechanism of Action. Front. Nutr. 2019, 6, 121. [Google Scholar] [CrossRef]
- Lee, H.; Im, S.W.; Jung, C.H.; Jang, Y.J.; Ha, T.Y.; Ahn, J. Tyrosol, an olive oil polyphenol, inhibits ER stress-induced apoptosis in pancreatic beta-cell through JNK signaling. Biochem. Biophys. Res. Commun. 2016, 469, 748–752. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monceaux, K.; Gressette, M.; Karoui, A.; Pires Da Silva, J.; Piquereau, J.; Ventura-Clapier, R.; Garnier, A.; Mericskay, M.; Lemaire, C. Ferulic Acid, Pterostilbene, and Tyrosol Protect the Heart from ER-Stress-Induced Injury by Activating SIRT1-Dependent Deacetylation of eIF2α. Int. J. Mol. Sci. 2022, 23, 6628. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126628
Monceaux K, Gressette M, Karoui A, Pires Da Silva J, Piquereau J, Ventura-Clapier R, Garnier A, Mericskay M, Lemaire C. Ferulic Acid, Pterostilbene, and Tyrosol Protect the Heart from ER-Stress-Induced Injury by Activating SIRT1-Dependent Deacetylation of eIF2α. International Journal of Molecular Sciences. 2022; 23(12):6628. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126628
Chicago/Turabian StyleMonceaux, Kévin, Mélanie Gressette, Ahmed Karoui, Julie Pires Da Silva, Jérôme Piquereau, Renée Ventura-Clapier, Anne Garnier, Mathias Mericskay, and Christophe Lemaire. 2022. "Ferulic Acid, Pterostilbene, and Tyrosol Protect the Heart from ER-Stress-Induced Injury by Activating SIRT1-Dependent Deacetylation of eIF2α" International Journal of Molecular Sciences 23, no. 12: 6628. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126628