1. Introduction
The recent expansion of tissue engineering allowed the development of innovative techniques to create tissue substitutes as a response to the organ shortage [
1]. However, the lack of an existing and efficient vascularization system prevents those substitutes from being implanted in the host body [
2,
3]. Among existing vascularization strategies, some studies mimicked angiogenesis and vasculogenesis mechanisms to induce vessels’ formation, growth, and expansion inside the host body [
4,
5,
6]. While promising, the development of a mature blood vessel network remains time-consuming and inadequately provides oxygen and nutrients to the host’s cells shortly after the surgery. Another approach enabling a more rapid vascularization of the substitutes after implantation is to directly create three-dimensional (3D) micro blood-vessel substitutes and integrate them into the tissue substitutes [
7,
8,
9]. Based on engineering techniques such as stereolithography [
10,
11], microfluidic [
6,
12], or bioprinting [
13,
14], some studies highlight the generation of 2D capillary models or 3D scaffold-embedded microchannels. However, none of these techniques allow the formation of a mature, scaffold-free, and perfusable microcapillary in 3D, conditions required to be implanted and to provide an efficient vascularization.
Here, we propose a method to create an intra-volume channel within 3D, scaffold-free, mature, and cell-covered hydrogel microfibres inspired by industrial laser processing techniques applied to transparent materials. Primarily, a microfibre was produced from a collagen hydrogel core covered by a dense and living-cell outer bilayer. Secondly, a channel was processed inside the collagen core with a near-infrared (NIR, Stockholm, Sweden), femtosecond (fs) laser source. It is well known that the use of tightly focused ultrashort laser pulses induces localized damages in the bulk of transparent materials via non-linear absorption while minimizing thermal effects in the interaction zone [
15]. Indeed, NIR laser beams allow working in the tissue biological window where the laser light is less scattered or absorbed by the material [
16].
Data on laser-hydrogel interaction have been previously reported by the same group, Verit et al. [
17]. In addition, several studies report about the generation of intra-volume channels within the volume of cell-embedded hydrogels. Applegate et al. generated 3D structures within the volume of a silk-based hydrogel with ultrashort laser pulses (100 fs) at a wavelength of 810 nm. Here, silk hydrogel was chosen for its transparency to the 810 nm wavelength enabling the creation of voids at 1 cm depth. The use of a pulse repetition rate of 80 MHz generated a strong thermal accumulation at the focal point disrupting silk fibres and leading to the apparition of a cavity inside the hydrogel [
18]. To generate channels within collagen hydrogel, Hribar et al. doped the hydrogel with gold nanorods, creating a local absorption pick at 800 nm. Using 100 fs laser pulses with only a few nJ and at a repetition rate of 80 MHz, nanorods efficiently absorbed the laser beam which was then converted into heat disrupting collagen fibres and generating channels [
19]. Using UV laser light (355 nm) and nanosecond pulses (1 ns) at a repetition rate of 100 Hz, Brandenberg et al. generated channels in the bulk of a bovine collagen hydrogel. The use of UV light and nanosecond pulses leads to channels at shallower depths and greater widths with reduced machining accuracy [
20]. The common characteristic to these three studies and the majority of studies found in the literature is the ability to preserve cellular viability right after hydrogel laser machining [
19] or up to 4 h after laser interaction [
18]. However, the good cellular viability after laser processing may be explained by the small number of embedded cells scattered throughout the material’s volume; when the beam goes through the hydrogel to generate an intra-volume channel, only a few cells happen to interact with it. In addition, Zeigler et al. have shown that a decay in cellular viability could appear up to 12 h after laser processing, whereas cellular viability tracking in the previously mentioned studies was limited to a few hours only after processing [
21].
The uniqueness of this work lies in the generation of channels within the volume of collagen hydrogel microfibres covered by a high-density bilayer of living cells, in order to closely mimic a real glomerular microfibre system. During laser processing, the laser beam directly crossed the surface cell bilayer before being focused in the hydrogel volume to create cavities. Cellular viability was evaluated using Live/Dead
® assays 24 h after laser processing to assess the influence of direct laser interaction on cell preservation. The experimental approach employed in this work is based on a previous study by the same group on intra-volume fs laser processing of cell-free gelatin hydrogel blocks where cavitation phenomena induced by a tightly focused fs laser in the hydrogel volume were investigated [
17]. It was observed that after one laser pulse, a cavitation bubble appeared at the focal point, rapidly grew to reach its maximum size, and then slowly shrank in volume affecting the gelatin hydrogel structure. Finally, it was found that channel formation was promoted when a low number of large volume cavitation bubbles characterized by a slow shrinking rate was observed in the gelatin hydrogel volume after the laser processing. This behavior was enhanced when processing at a depth of the focal point of 100 μm, at fluence higher than the cavitation threshold value of 24.5 J·cm
−2, at low repetition rate, and with a strong spatial overlap between two successive laser pulses. These results were considered as a starting point for this study, where the number of cavitation bubbles formed, their maximal size, and their shrinking rate depended on laser parameters and directly affected channel formation and cellular viability in the microfibres.
3. Discussion
This work aims to identify a narrow range of laser parameters as well as the best experimental conditions allowing the creation of a fully formed channel within the collagen core of microfibres without damage to the outer dense cell bilayer. Two new challenges were identified with respect to our previous works [
17,
24]: firstly, the identification of an efficient laser parameter window allowing channel formation in the hydrogel core and secondly, the preservation of the cellular viability of this high-density living cell bilayer.
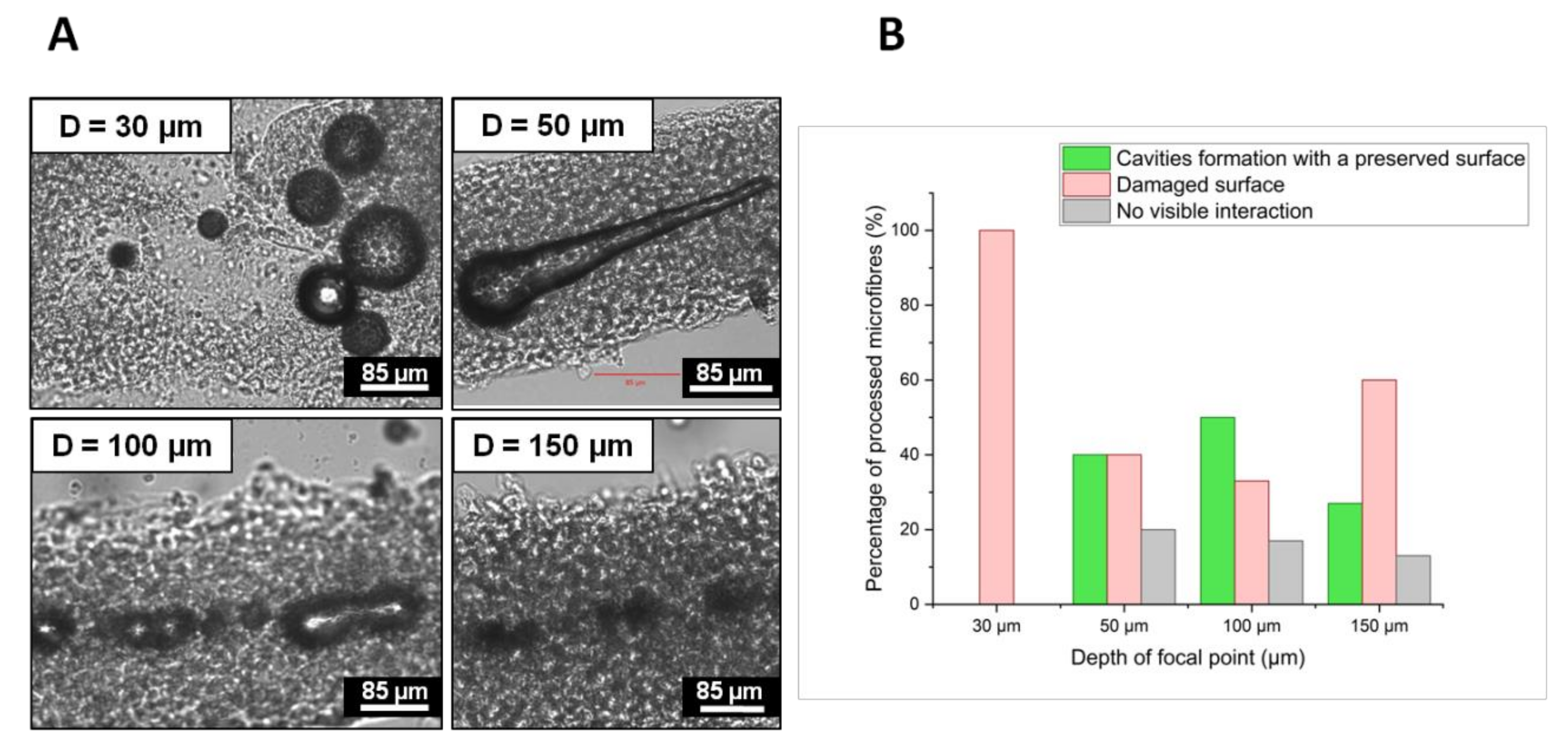
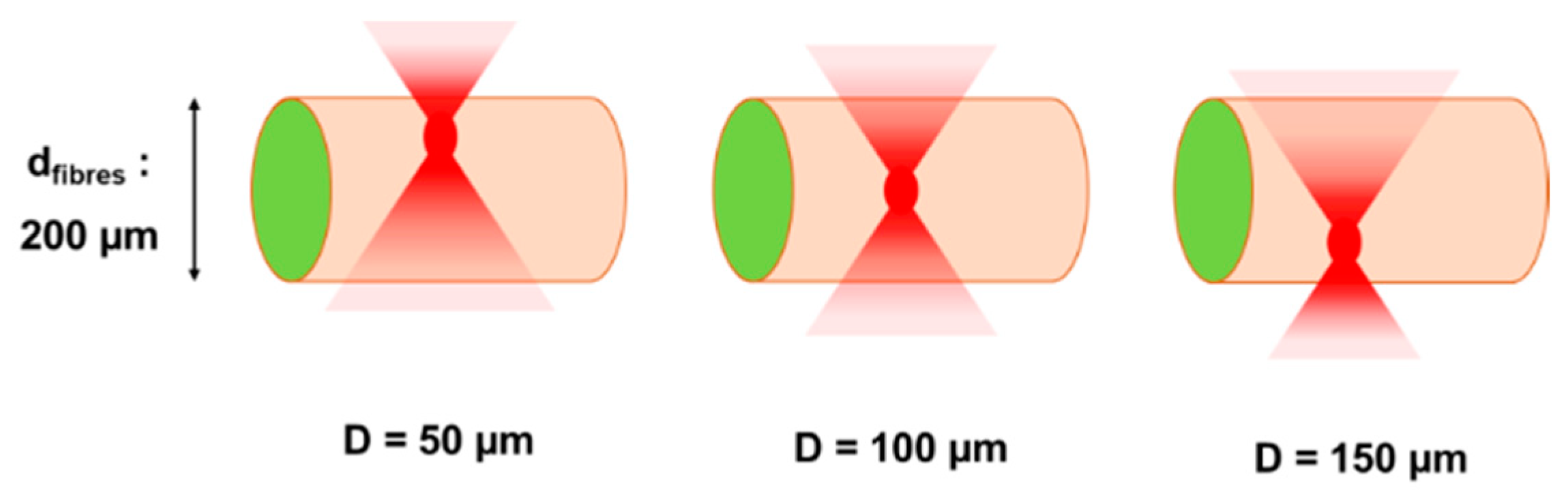
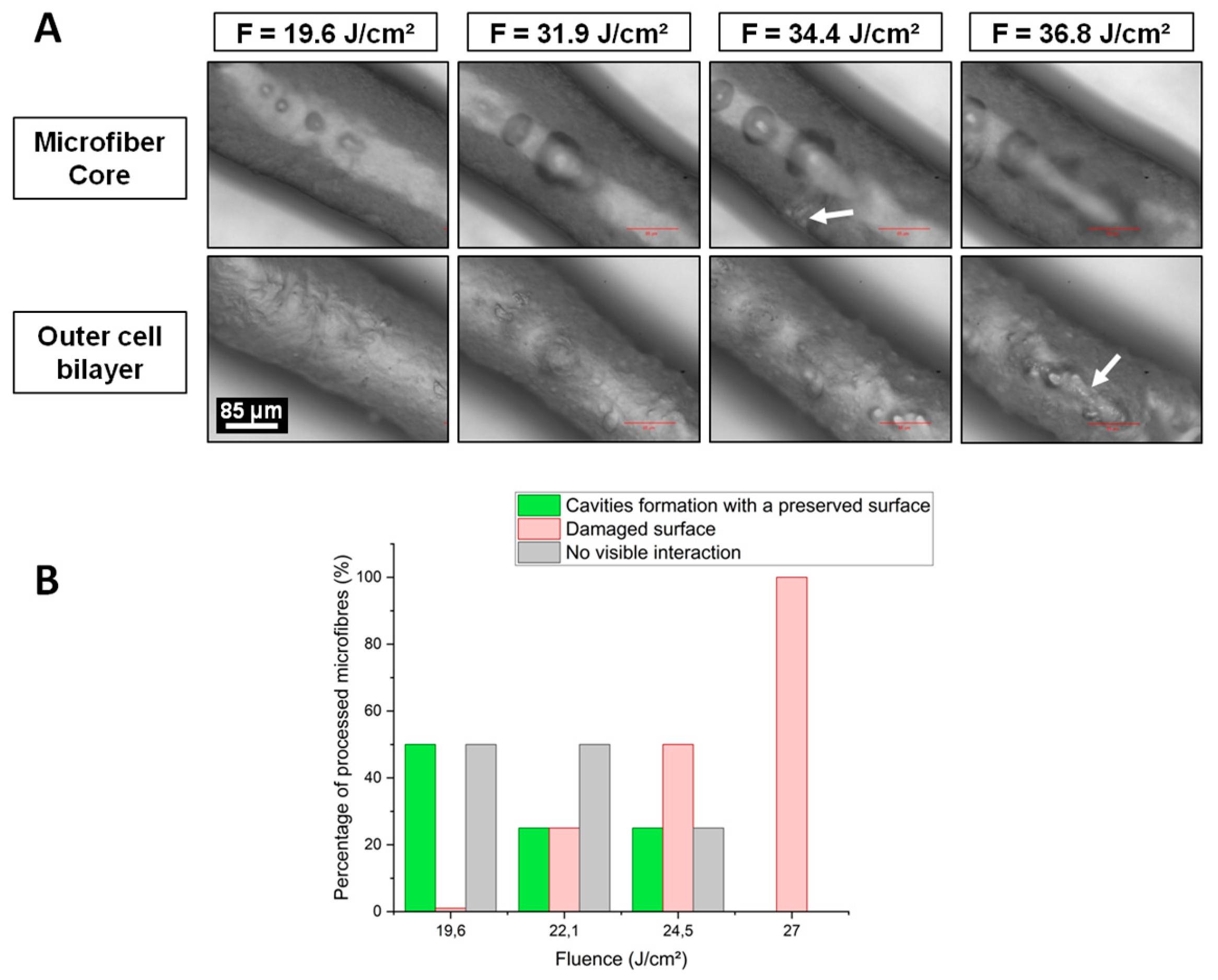
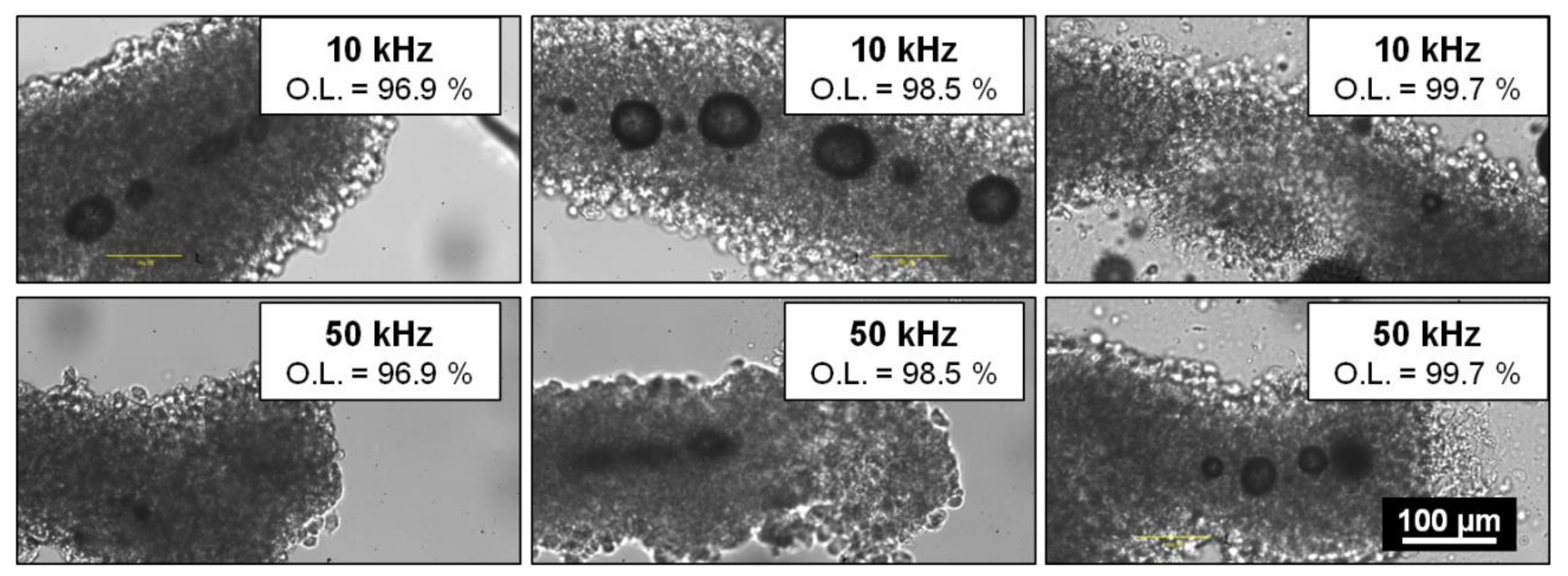
To achieve these goals, the optimal laser parameters were determined from previous observations that channel generation inside a hydrogel was enhanced by the formation of voluminous cavitation bubbles with a lifetime from a few minutes to a few hours before collapse [
17]. This type of bubbles was generated inside a gelatin hydrogel because of the similarity of its optical properties to those of collagen hydrogels [
25]. The best set of process parameters was identified at a depth D = 100 µm, a fluence F = 24.5 J·cm
−2, a repetition rate R.R. = 10 kHz and an overlap O.L. ≤ 98.5%. Under these conditions, material damages due to heat dispersion were spatially limited to the channel close vicinity. Moreover, cavitation bubbles were the farthest from the outer cell bilayer thus minimizing mechanical pressure applied on cells. If the focal point was positioned too close to the cell wall (D < 100 µm), fluence value at the microfibre surface could exceed the cell bilayer damage threshold leading to a cell viability drop or eventually the complete destruction of the microfibre structure. On the other hand, for depths greater than D = 100 µm, laser energy was likely absorbed and dispersed by the high-water content collagen, leading to a decrease of the volume of cavitation bubbles and their probability to generate channels.
By varying fluence values at D = 100 µm, the volume of cavitation bubbles and their respective lifetime could be enhanced to promote channel generation. While a fluence lower than F = 19.6 J·cm−2 did not create any cavitation bubble, highest fluence values generated non-linear effects directly on the microfibre outer cell bilayer. Above F = 36.8 J·cm−2, the fluence value at the cell wall likely exceeded the cell bilayer damage threshold, altering the laser-irradiated cells.
In literature, thermal accumulation during laser processing is a relevant factor especially when it comes to biological systems where cellular viability preservation is essential [
21,
23,
26]. The thermal load could be reduced by decreasing either the repetition rate or the spatial overlap between two successive pulses. The results presented above showed that a decrease of thermal accumulation directly influenced the behavior of cavitation bubbles during laser interaction. However even when cavitation bubbles with a diameter smaller than the average width of the collagen core were created far enough from the cell wall, no cellular viability improvement was observed. It is important to note that a strong cellular viability drop was generally observed independently of the applied laser parameters and related thermal accumulation effects.
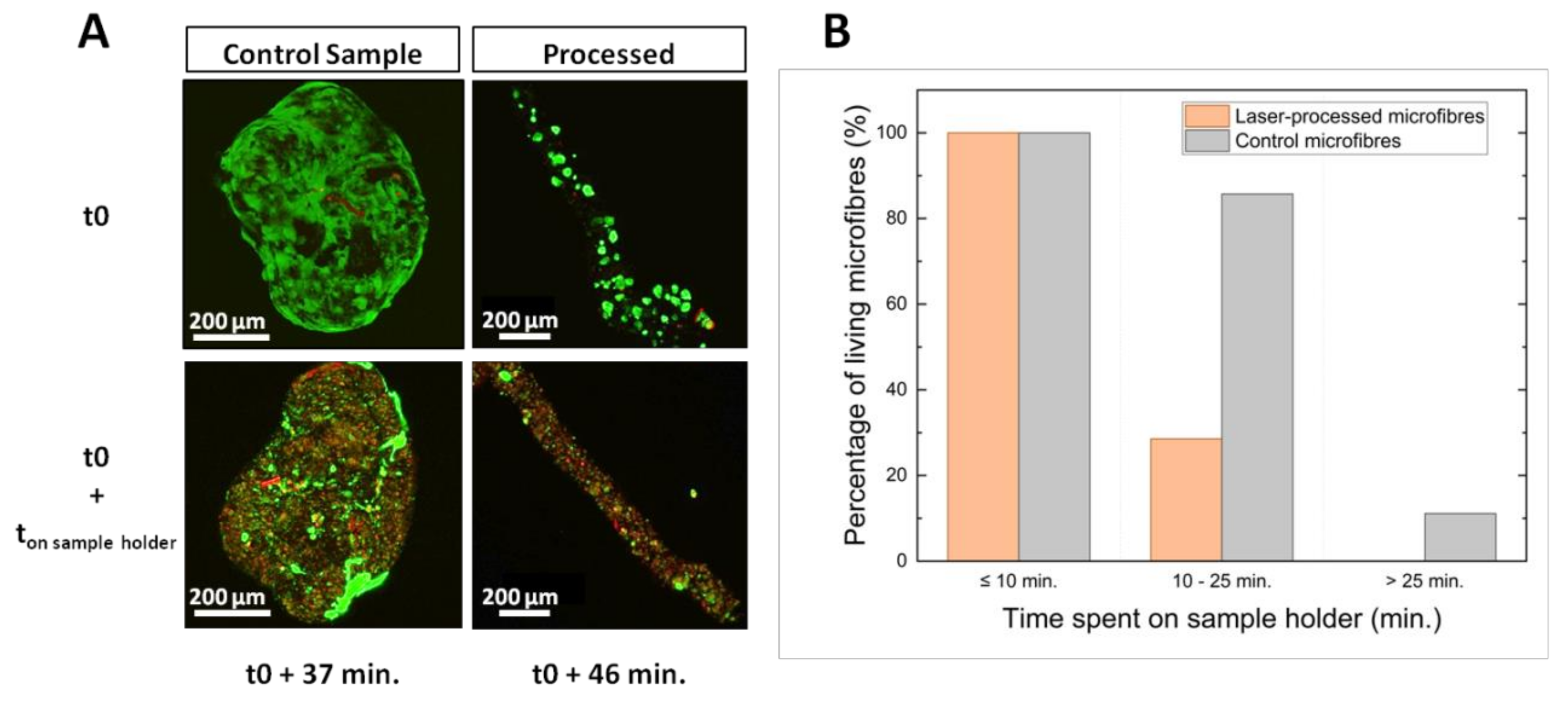
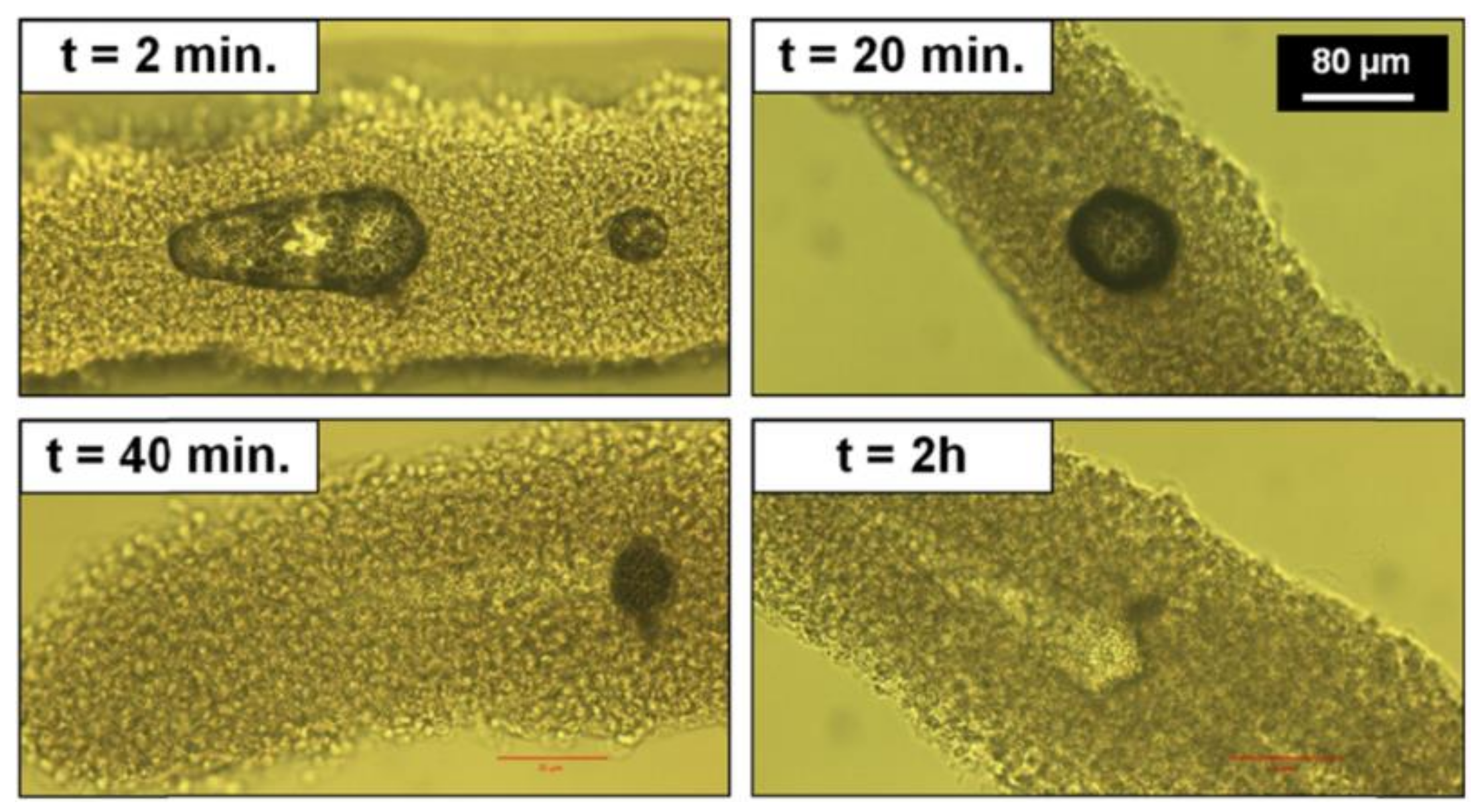
Live/Dead® assays revealed that the viability drop was accentuated by microfibre dehydration. Only a minimal amount of culture medium was used during the laser processing to prevent culture medium from dispersing the laser energy. In this environment, cell damage was negligible for about 10 min. Afterwards, cellular viability decreased quickly due to stress related to dehydration and laser processing. These results show that a good preservation of cellular viability might be possible by reducing the total processing time below 10 min. Reducing this time would be achievable by a process automation involving the detection of the microfibre center and the calculation of the laser trajectory via a synchronization of the CCD camera with the stage control software.
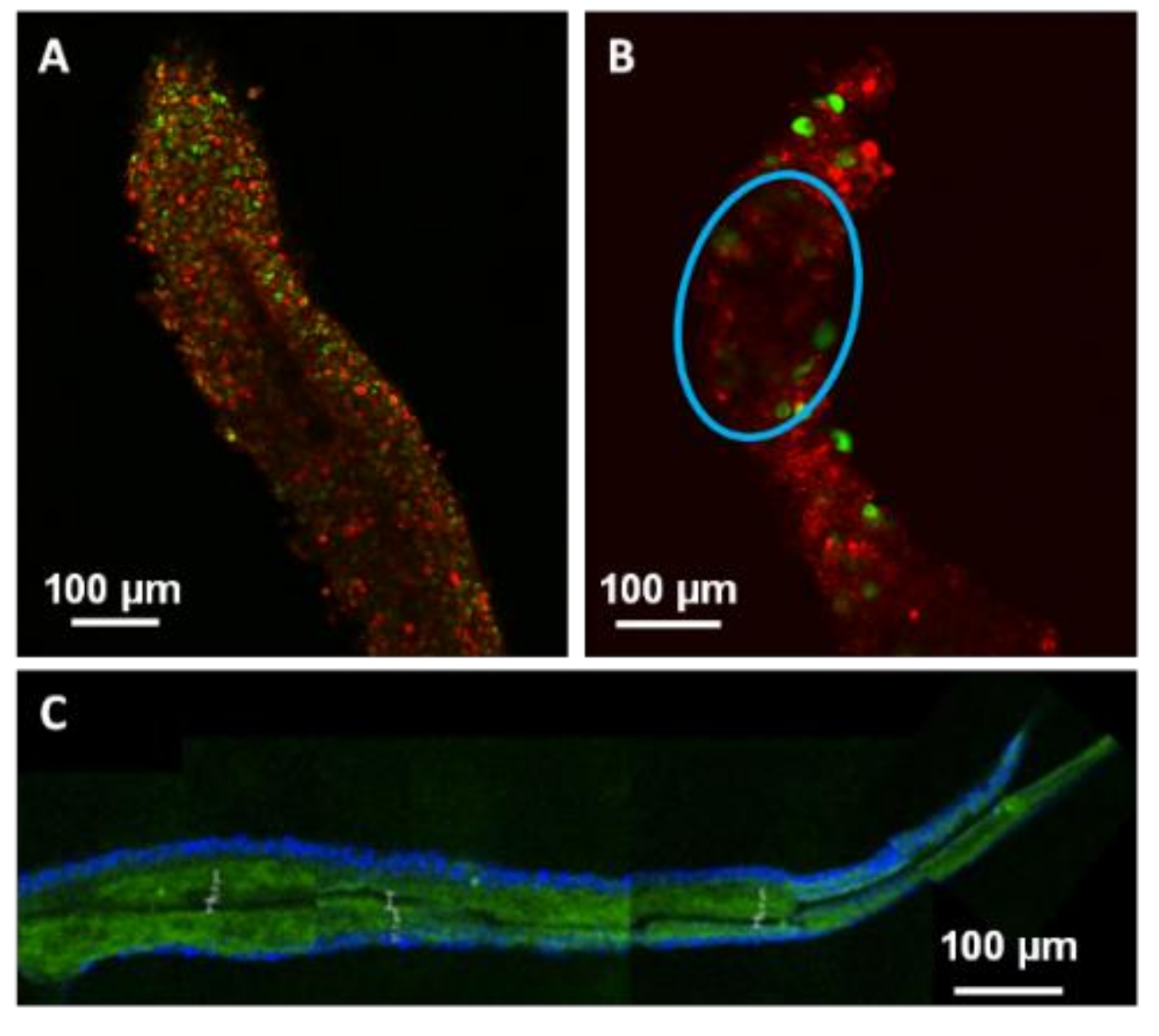
Cell hydration also affected the laser processing quality as observed by confocal microscopy 24 h after process. When microfibres were non-viable due to insufficient hydration, laser processing led to the formation of a sharp channel within the dried collagen core. These channels were generated in a similar way than the ones observed inside PFA-fixed microfibres or in synthetic hydrogel [
27]. On the other hand, when microfibres were viable due to sufficient hydration, channel detection was difficult. Either no channel or only a slight attenuation of the autofluorescence signal from the hydrogel core was detected. This observation could be explained by the fact that the collagen of hydrated fibres might be able to reabsorb water after bubble collapse due to its high water content. This way, the channel would slowly be clogged after the laser interaction. To preserve channel durability, processed microfibres should be immediately perfused after laser processing. Moreover, the presence of an internal flow might allow complete cell differentiation of the outer cell bilayer within this dynamic and 3D environment [
28,
29,
30,
31].
4. Materials and Methods
Collagen hydrogel microfibres were composed of a hydrogel core made of collagen type I and a cell bilayer with glomerular endothelial cells (GECs) on the inner side and podocytes on the outer side.
4.1. Cell Culture
Outer cell bilayer was composed of GECs and podocyte cells immortalized by temperature sensitive gene SV40. They were cultured according to the protocol by Flegeau et al. [
22]. Both cell lines proliferated at 33 °C and differentiated at 37 °C.
4.2. Living-Cell Microfibres Manufacture
Firstly, a shell of 2% reticulated alginate was made around a 350 µm diameter polycarbonate capillary tube (Paradigm Optics, Vancouver, WA, USA) by soaking it alternatively into sodium alginate (FMC BioPolymer, Philadelphia, PA, USA) and calcium chloride (Sigma Aldrich, Burlington, MA, USA) baths. The polycarbonate tube was then removed, and a solution containing type I collagen (Corning Lab, Corning, NY, USA) and GECs was simultaneously injected inside the core of the alginate shell. Since collagen solution is liquid below 4 °C, the injection was performed at cold temperature. Alginate shells were then manually removed after 18 h incubated at 33 °C. Five additional days were needed at 37 °C in order to obtain a full differentiation of GECs and their migration to the outer part of hydrogel microfibres. Secondly, podocytes were seeded over GECs, along microfibres, in order to obtain a cell bilayer covering the core of collagen hydrogel. Fourteen days after seeding, podocytes were differentiated. Cell number (35.10
6/mL) and collagen concentrations (5 mg/mL) required to create collagen hydrogel microfibres was determined in the previous study of Flegeau et al. [
22].
4.3. PFA-Fixed Microfibres Manufacture
PFA-fixed microfibres were created following the previous protocol [
22]. However, collagen type I-FITC (Sigma Aldrich, Burlington, MA, USA) was used instead of the Corning Lab collagen I. Then, microfibres were washed with PBS 1X and sank in a solution of 4% paraformaldehyde (PFA) at 4 °C. After 30 min, they were rinsed three times with PBS 1X. PFA-fixed microfibres were either immediately laser processed or stored in 12-well culture plates with PBS 1X at 4 °C.
4.4. Femtosecond Laser Processing Set-Up
Laser processing of microfibres was carried with a 350 fs laser source (Satsuma HP3 from Amplitude System, Pessac, France) running at 1030 nm central wavelength. The laser pulse energy was adjusted by sending the Gaussian linearly polarized laser beam through a polarizer and a half-wave plate. A beam expander of a factor 3 combined with a 0.4 numerical aperture microscope objective (Seiwa Optical, Seiwa Optical Europe GmbH, Frankfurt, Germany) ensured a strong focalization into the bulk of the material with a spot diameter at the focal point of about 3 µm. Sample holders were placed on computer-controlled XY translation stages (ALIO, Los Angeles, CA, USA) while the vertical position of the focusing objective was controlled by a Z translation stage (ALIO, Los Angeles, CA, USA) to precisely determine the focus position. A more detailed description of the experimental set-up used for processing and imaging the microfibres is provided in Verit et al. [
17].
4.5. Sample Conditioning before Processing
Microfibres (length in the range 0.5–1 cm) were laid on a 35 mm diameter Petri dish coated with a solution of 2% agarose with a drop of culture medium. Just before laser processing, the culture medium was removed until the microfibres adhered to the agarose coating, providing a minimal hydration for the cells during processing while avoiding possible absorption and scattering from the culture medium itself. The preparation of microfibres was estimated roughly at 1 h.
4.6. Laser Micromachining Process
Sample holders containing microfibres were placed on the XY translation stages. To generate channels within the collagen core of microfibres, straight lines were processed by the laser beam following the natural and tortuous shape of the microfibre by moving the XY translation stages at a defined translational speed, while keeping the position of the laser beam fixed. Several processing parameters were varied in order to evaluate their influence on the formation of channels within the microfibres: depth of the focal point within the microfibre (D), laser fluence (F), laser repetition rate (R.R.), and spatial overlap between two successive laser pulses (O.L.). The depth of the focal point within the material volume was ranging from D = 30 µm to D = 150 µm, where the reference D = 0 µm was considered to be the outer surface of the microfibre. The fluence ranged from F = 19.6 J·cm−2 to F = 36.8 J·cm−2. Two repetition rate values were investigated: R.R. = 10 kHz and R.R. = 50 kHz while the spatial overlap between successive laser pulses was varied by changing the speed v of the XY translation stages from v = 0.1 mm·s−1 to v = 5 mm·s−1 in order to study the influence of the following overlap values (O.L.): 96.9%, 98.5%, and 99.7%. While samples stayed on the laser processing station for just a few minutes for the treatment, the time for their preparation and handling just before and after the laser processing was estimated to up to 15 min. For each combination of laser parameters, experiments were repeated twice for the sake of repeatability. For all collected data, std <5% was obtained.
4.7. Characterization of Laser-Processed Living Cell Microfibres and Cell Viability Assessment
For each set of laser parameters, the microfibres were characterized at two different moments in time: immediately after and 24 h after laser interaction. After laser processing, processed areas were imaged via a CCD camera acA2000 (Basler, with a 0.4 N.A.) whose image plane followed the laser focal point during laser interaction. Channel formation and cellular viability were assessed 24 h later by confocal microscopy (Leica, Wetzlar, Germany) with a Live/Dead® cytotoxicity assay (Life Technologies, Carlsbad, CA, USA).
4.8. Characterization of Laser-Processed PFA-Fixed Microfibres
A DAPI solution was diluted at 1/5000 in a PBS 1X solution and applied on laser-processed PFA-fixed microfibres during 10 min in dark environment, then rinsed three times with distilled water. DAPI-labeled cell nucleus appeared in blue color in confocal microscope images (Leica) while collagen FITC appeared in green color.
5. Conclusions
The goal of this work was to develop a 3D blood microcapillary model by femtosecond laser processing. Whereas intra-volume channels are commonly created inside cell-free hydrogel or with a low density of embedded cells scattered through the whole material volume [
18,
19,
20], here, the laser processed system is upgraded to mimic more closely a real microcapillary structure, where hydrogel microfibres are enveloped by a high cellular density which strongly affects the laser processing outcomes. Moreover, the preservation of the cellular viability is assessed up to several hours after the laser processing. Channel processing was realized with a 350 fs, 1030 nm laser source strongly focused by a high numerical aperture objective. Channel formation, due to laser-induced cavitation in the hydrogel core, was optimized by processing at a focal depth of D = 100 µm, a laser fluence of F = 19.6 J·cm
−2, an overlap between O.L. = 96.9% and O.L. = 99.7%, and a repetition rate between R.R. = 10 kHz and R.R. = 50 kHz. These conditions allowed the generation of voluminous cavitation bubbles with a lifetime longer than 30 s, which was demonstrated to improve intra-volume channel durability. However, laser processing resulted in a decrease of cellular viability regardless the specific laser parameters applied for the processing. This viability drop was explained by a lack of sufficient hydration necessary to obtain a successful processing. Cellular mortality could be reduced drastically by reducing the dehydration stress through automation of the microfibre center detection and calculation of the laser trajectory. The last issue to consider concerns the sustainability of the channel created inside fully hydrated microfibres, which could be addressed by the perfusion of processed microcapillaries immediately after laser processing. To extend the exploitation of this technique to the generation of full 3D fibrous scaffolds for vascularization of thick constructs, the combination of successive manufacturing steps of laser-induced vascularization/issue generation with the employment of an automated six-axis system, which allows for laser treatment of 3D parts, could be eventually considered in a single manufacturing environment.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






