
Transcriptomic and Metabolomic Analysis of Seedling-Stage Soybean Responses to PEG-Simulated Drought Stress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Sequencing Quality Statistics
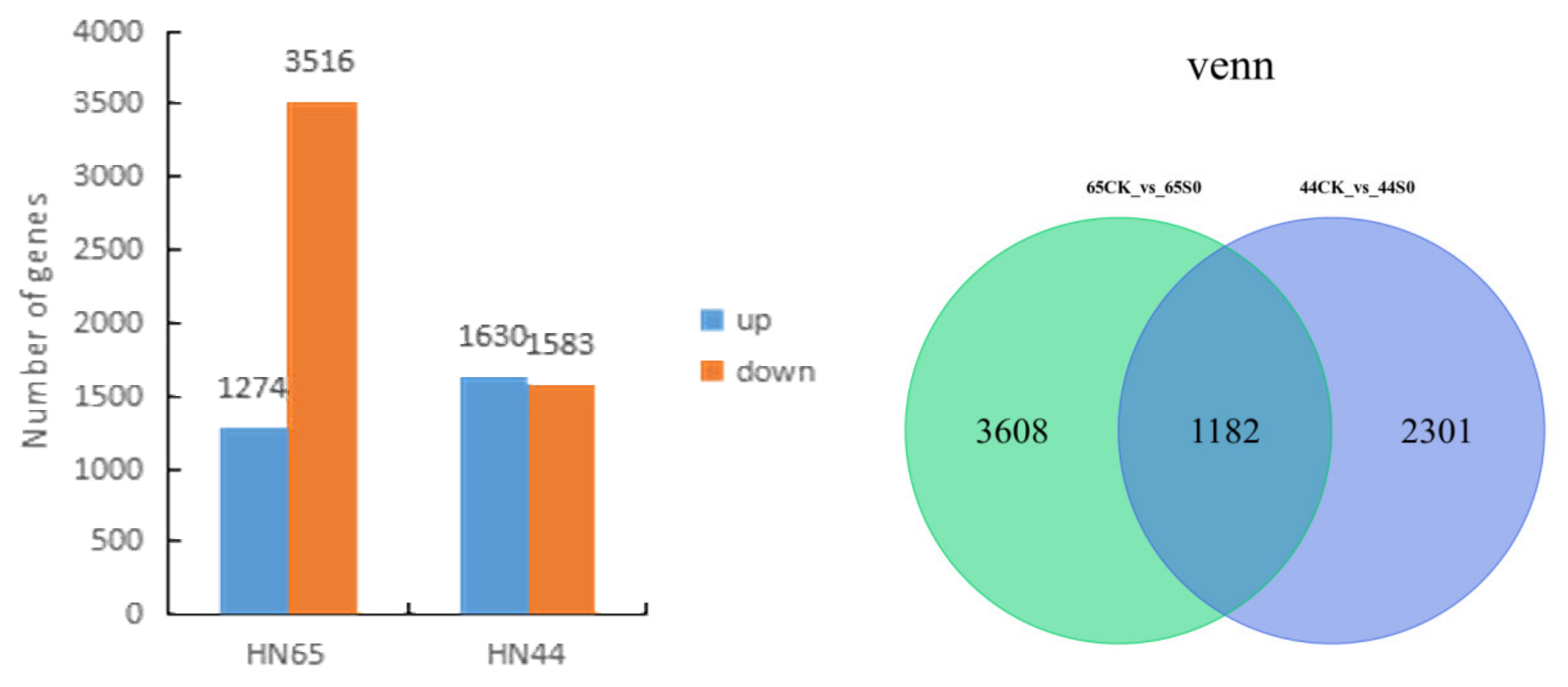
2.2. Differential Gene Screening
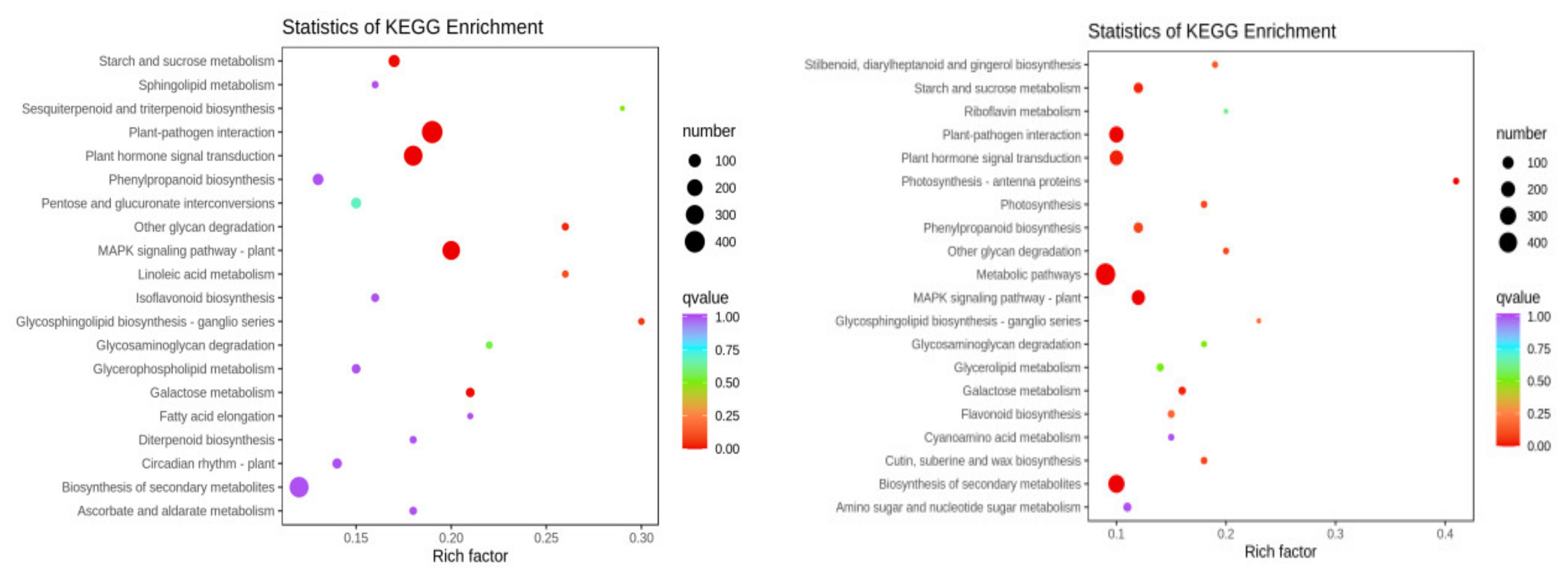
2.3. GO and KEGG Analysis of Differential Genes in Two Varieties
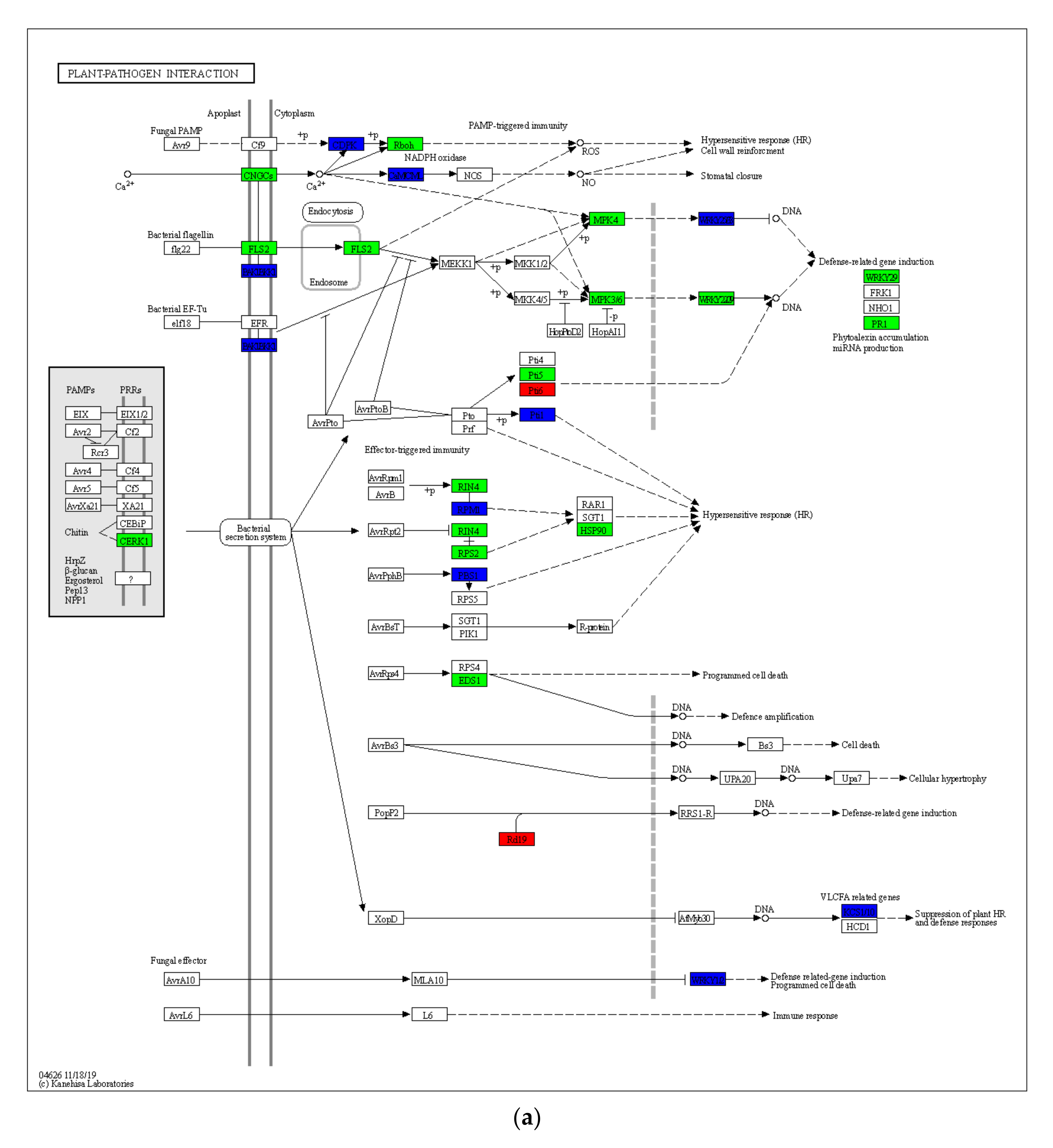
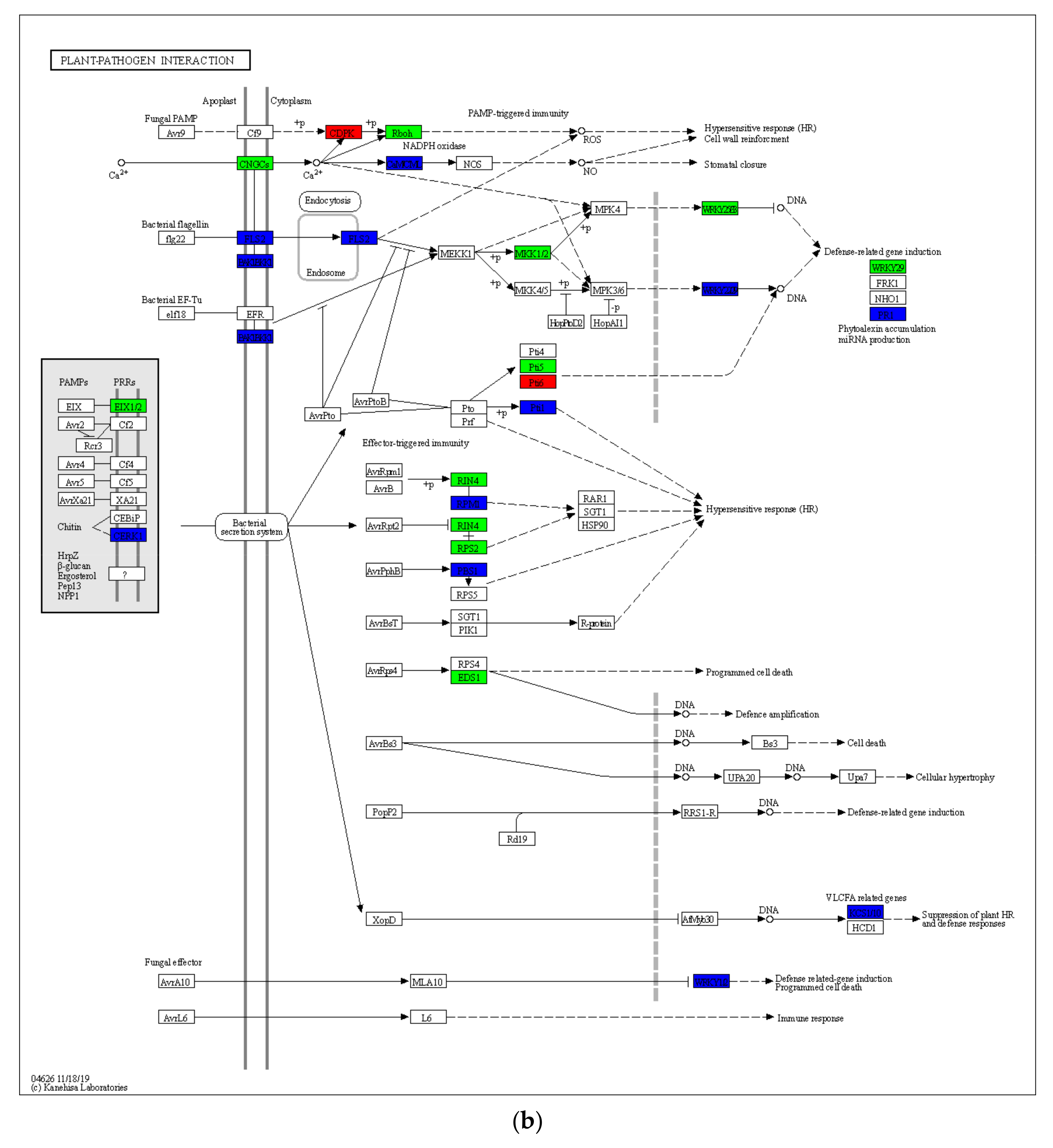
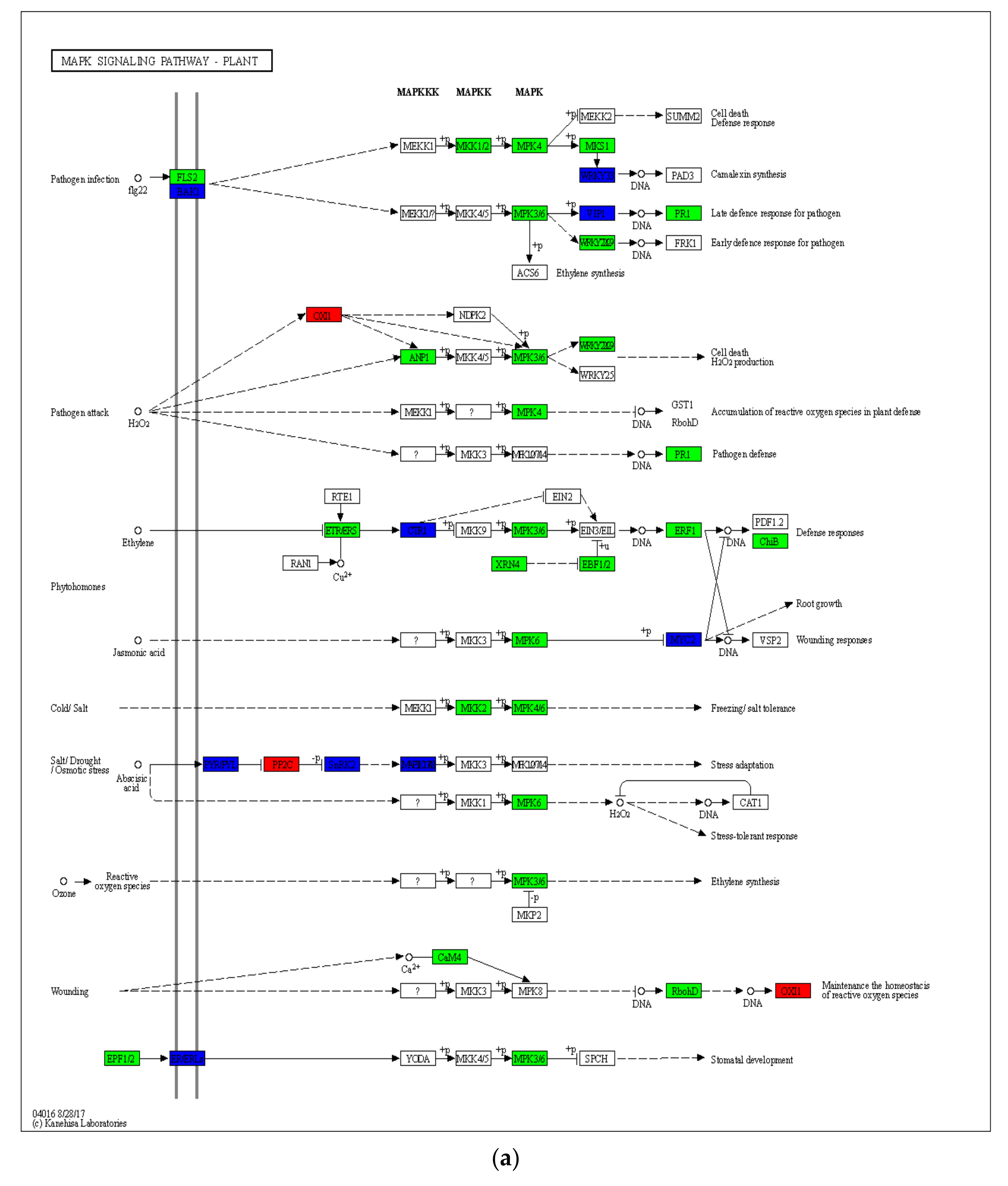
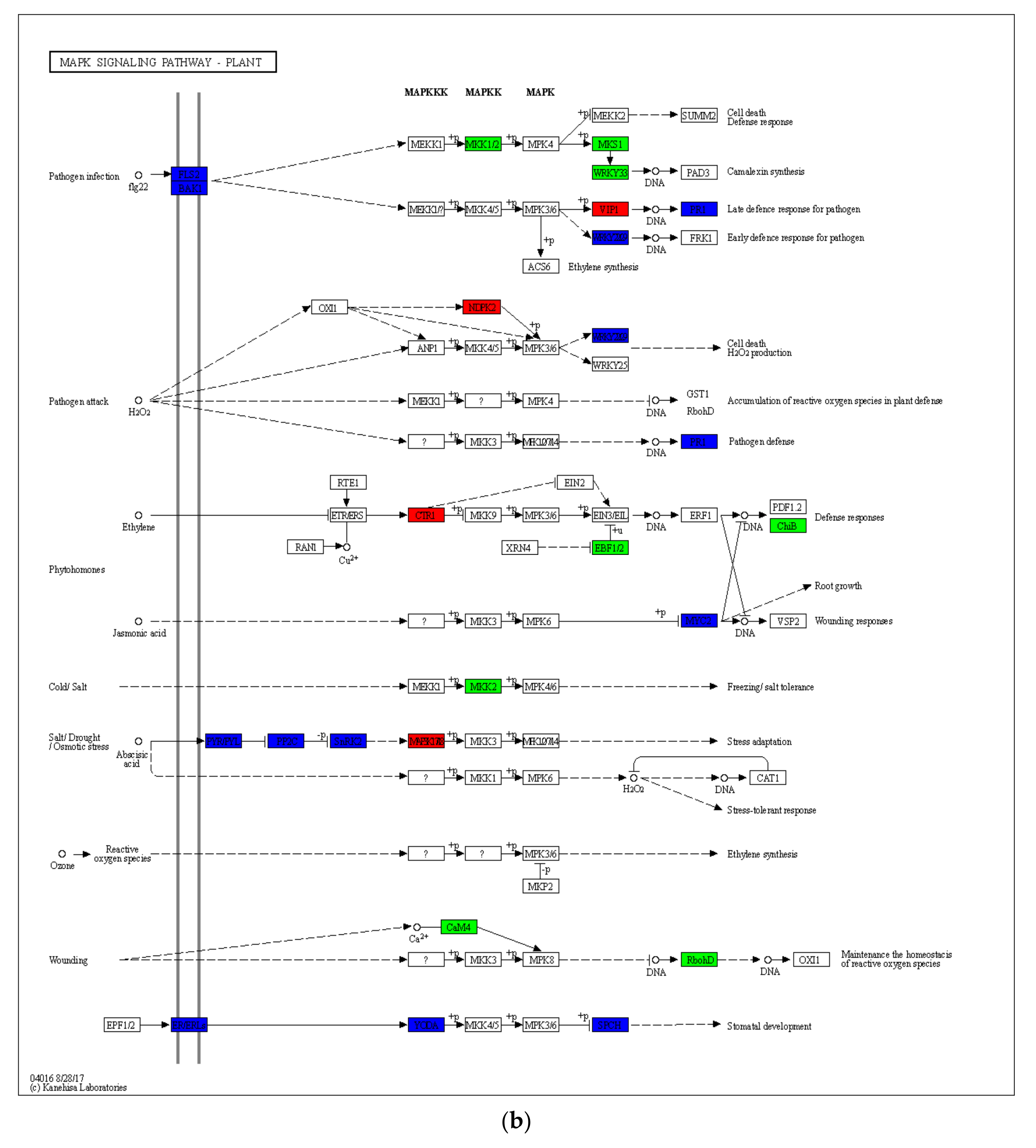
2.4. Gene Expression Differences in Drought-Induced Plant–Pathogen Interaction Pathway and MAPK Signaling Pathway of the Plant
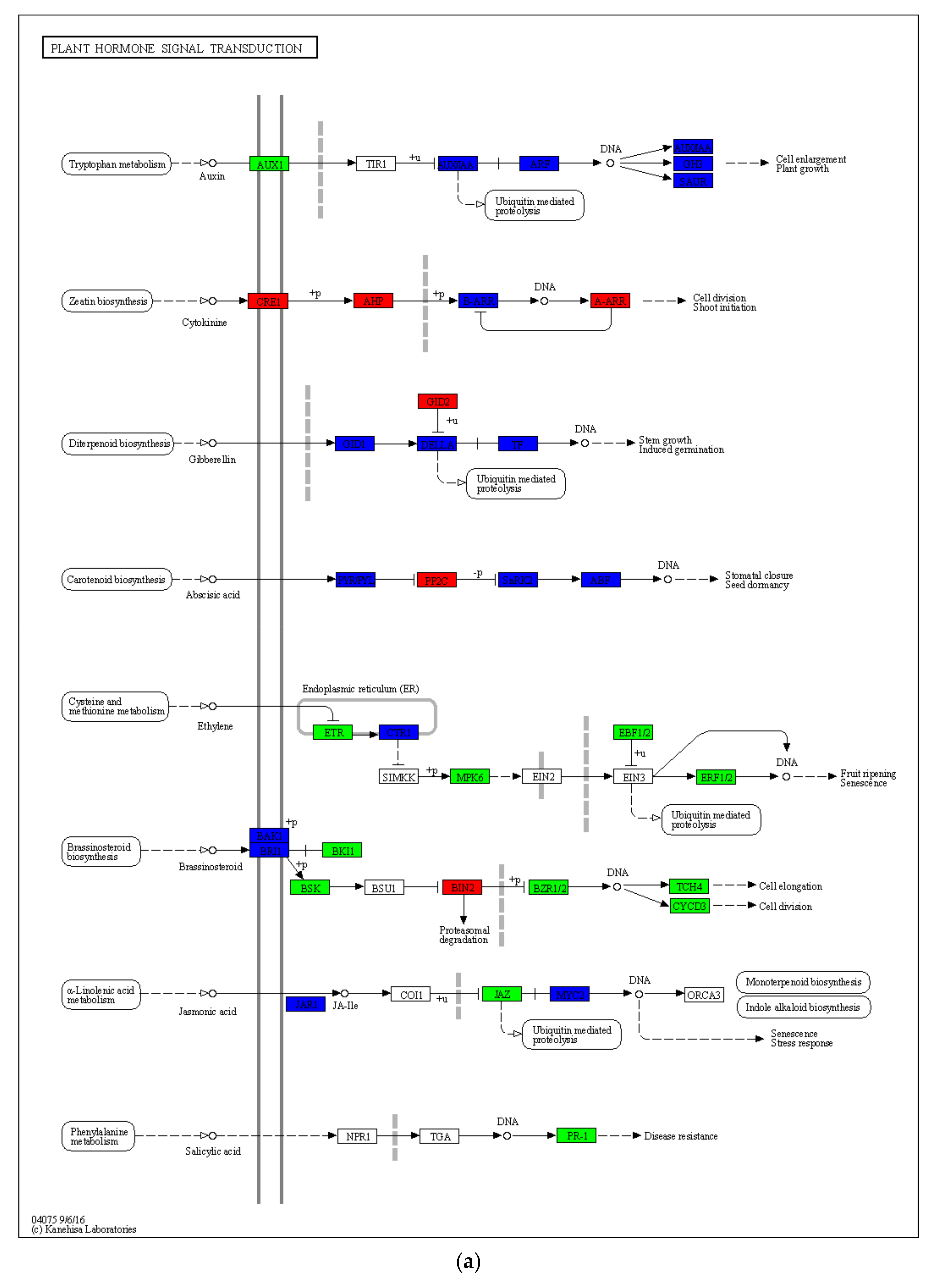
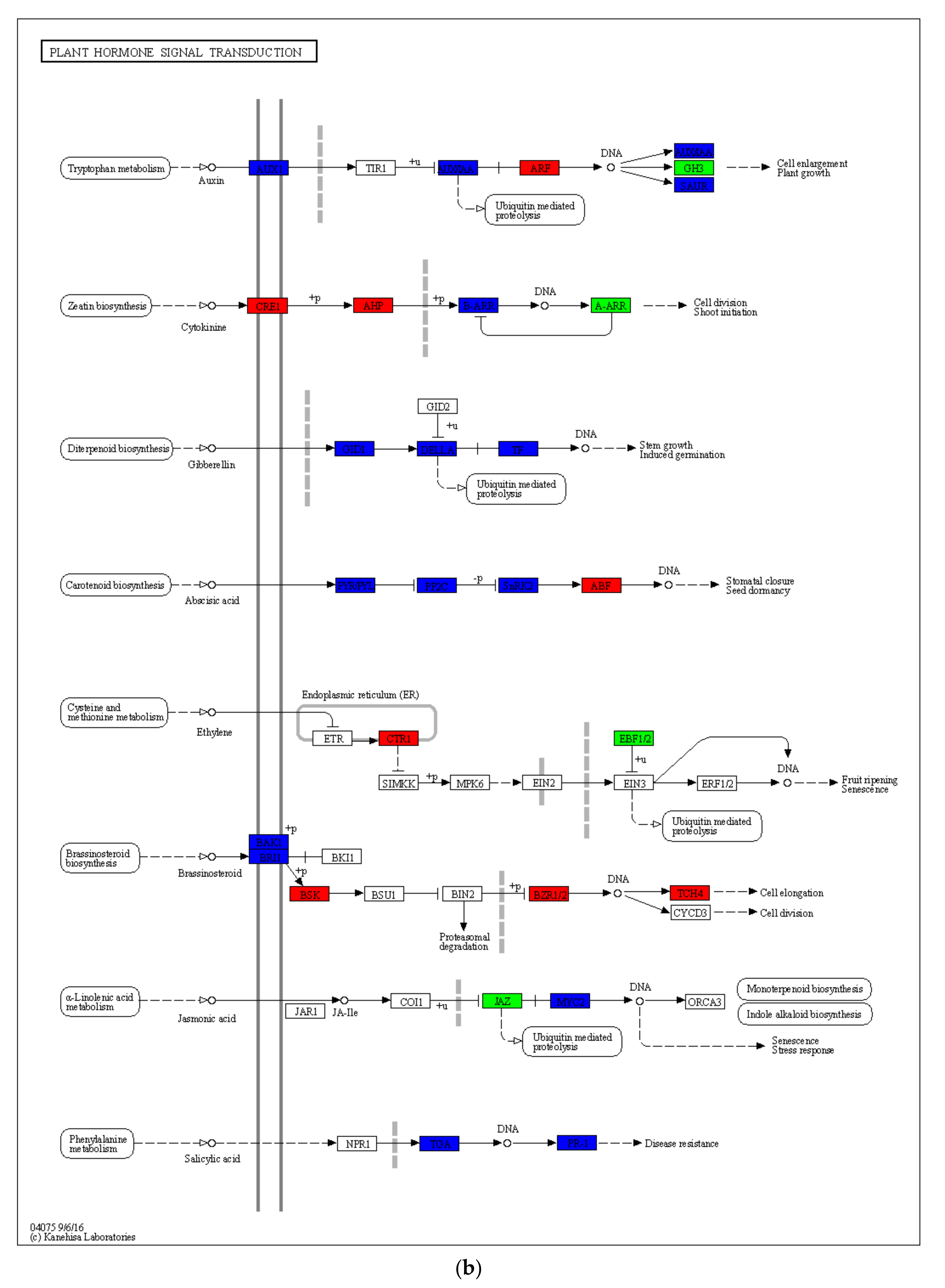
2.5. Drought-Induced Differences in Plant Hormone Signaling and Other Pathways
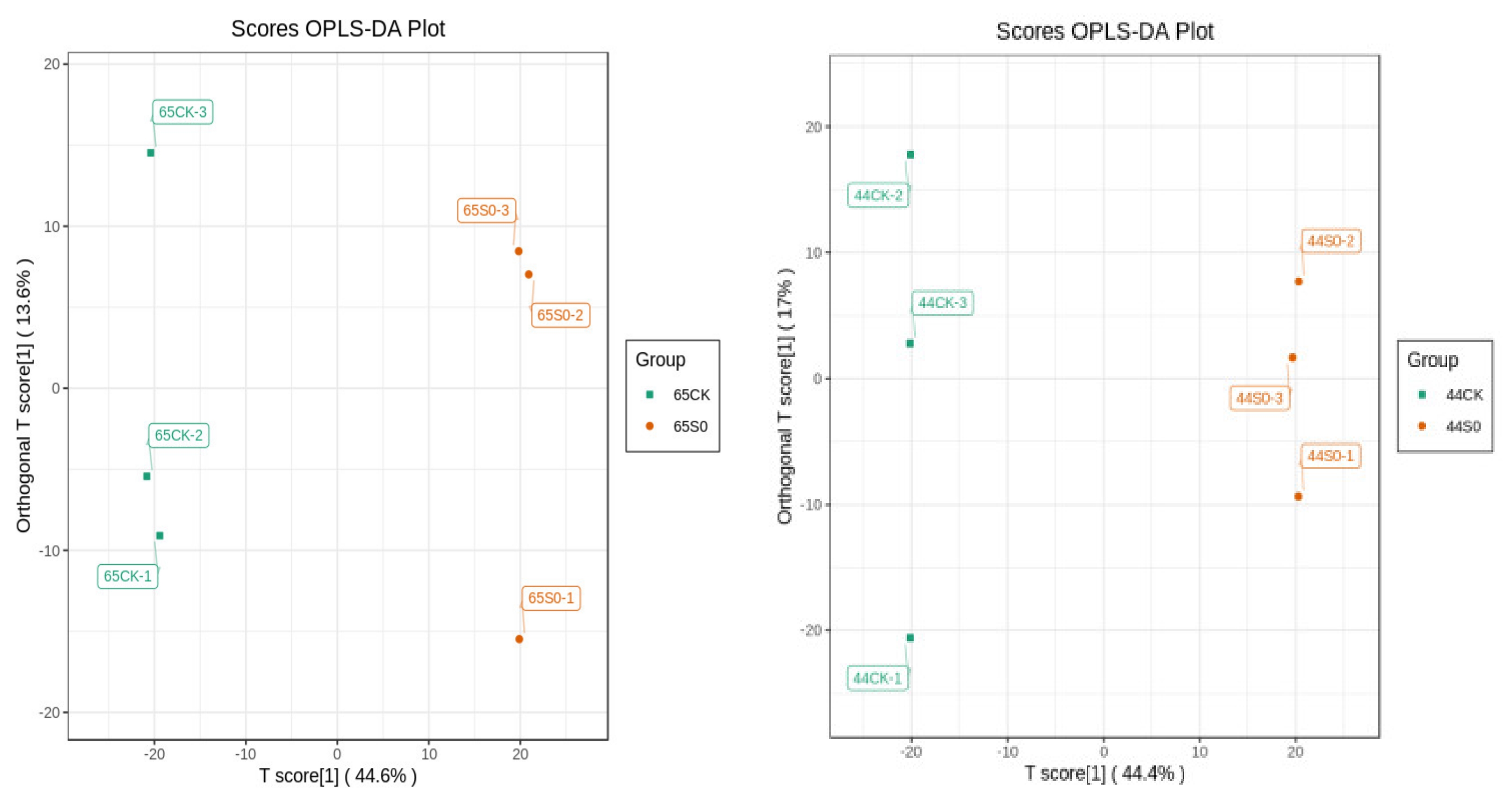
2.6. PCA and OPLS-DA Analysis
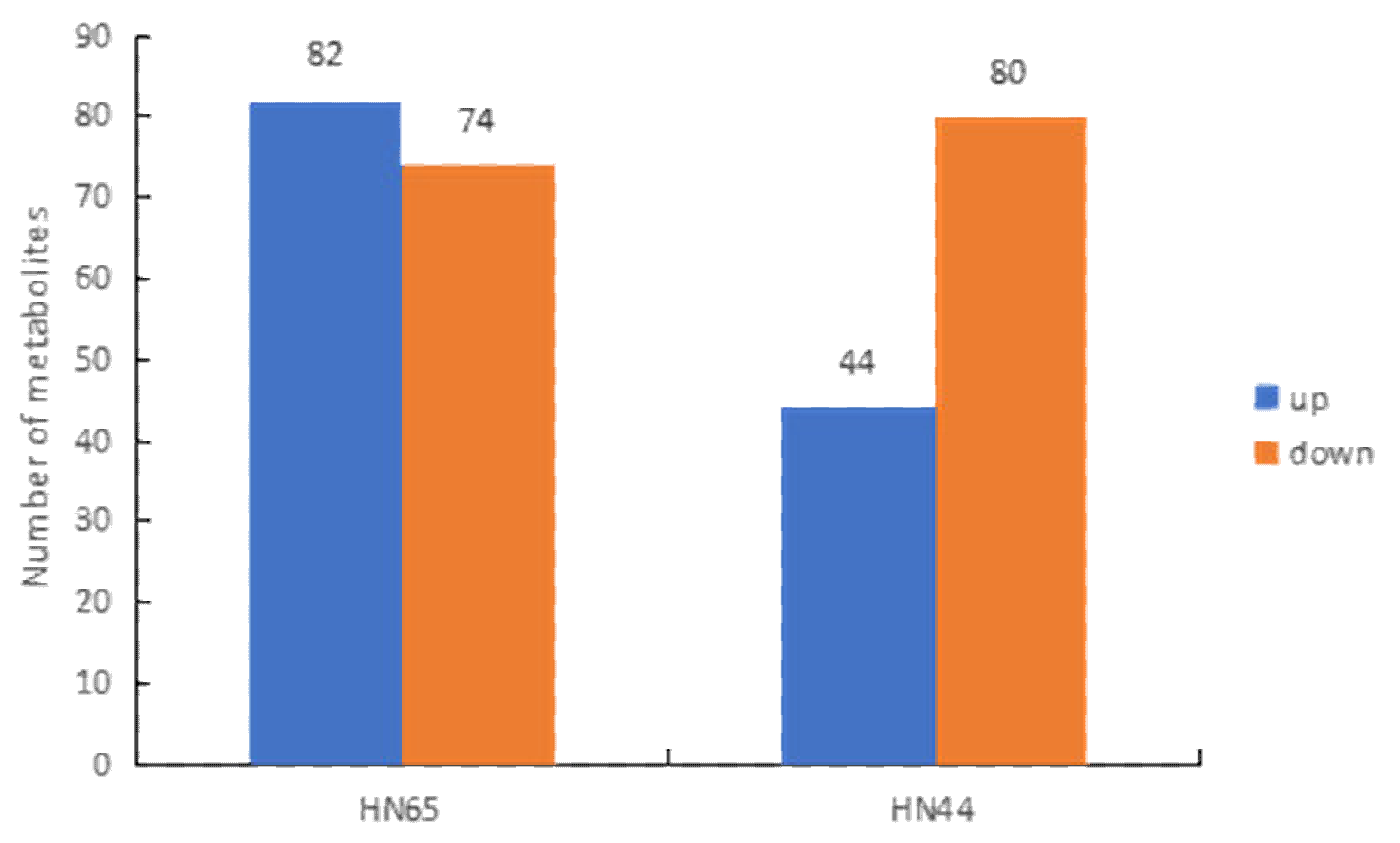
2.7. Identification and Classification of Differential Metabolites in Response to Drought in Two Varieties
2.8. Metabolism Differences of Flavonoids
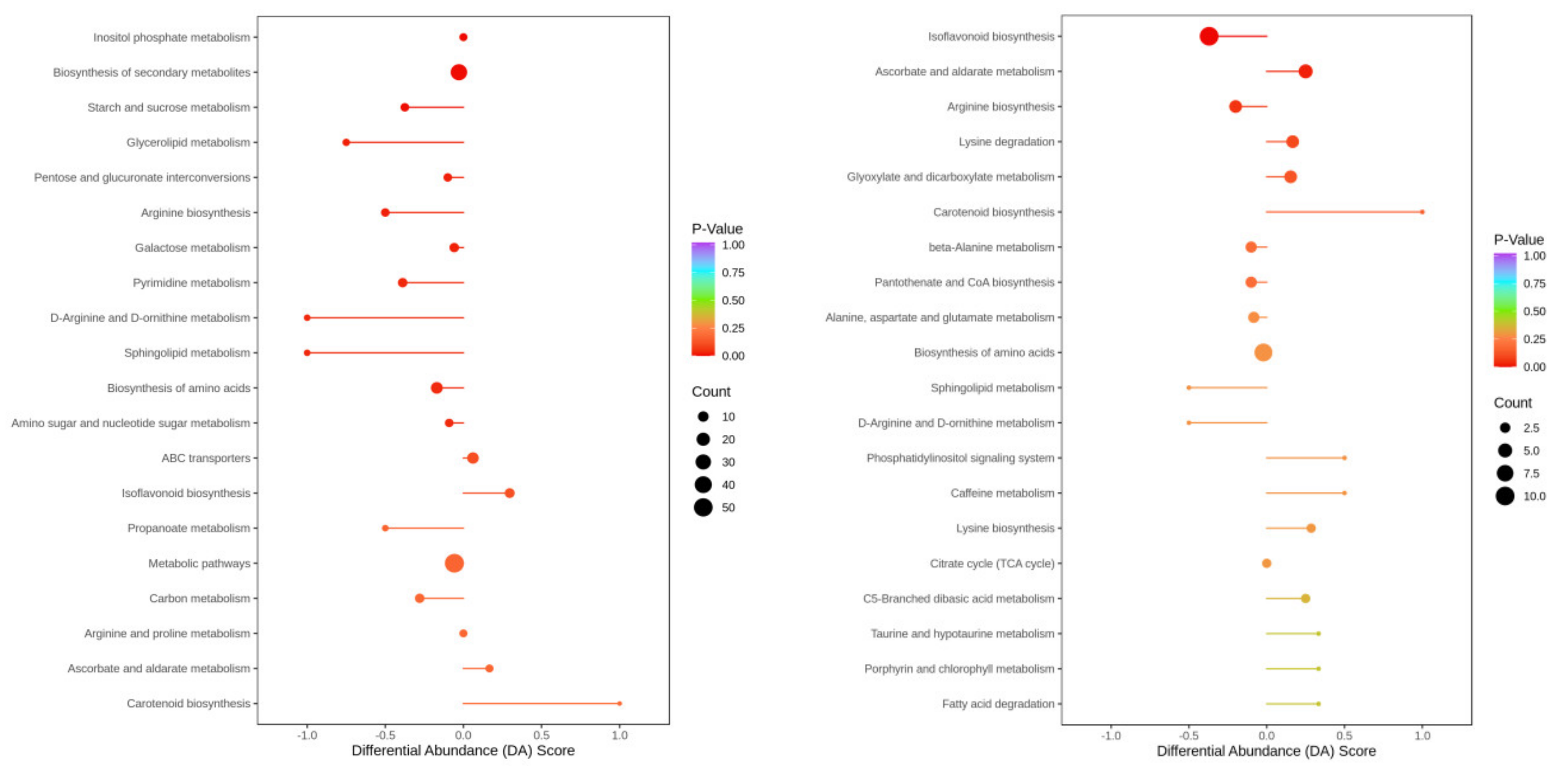
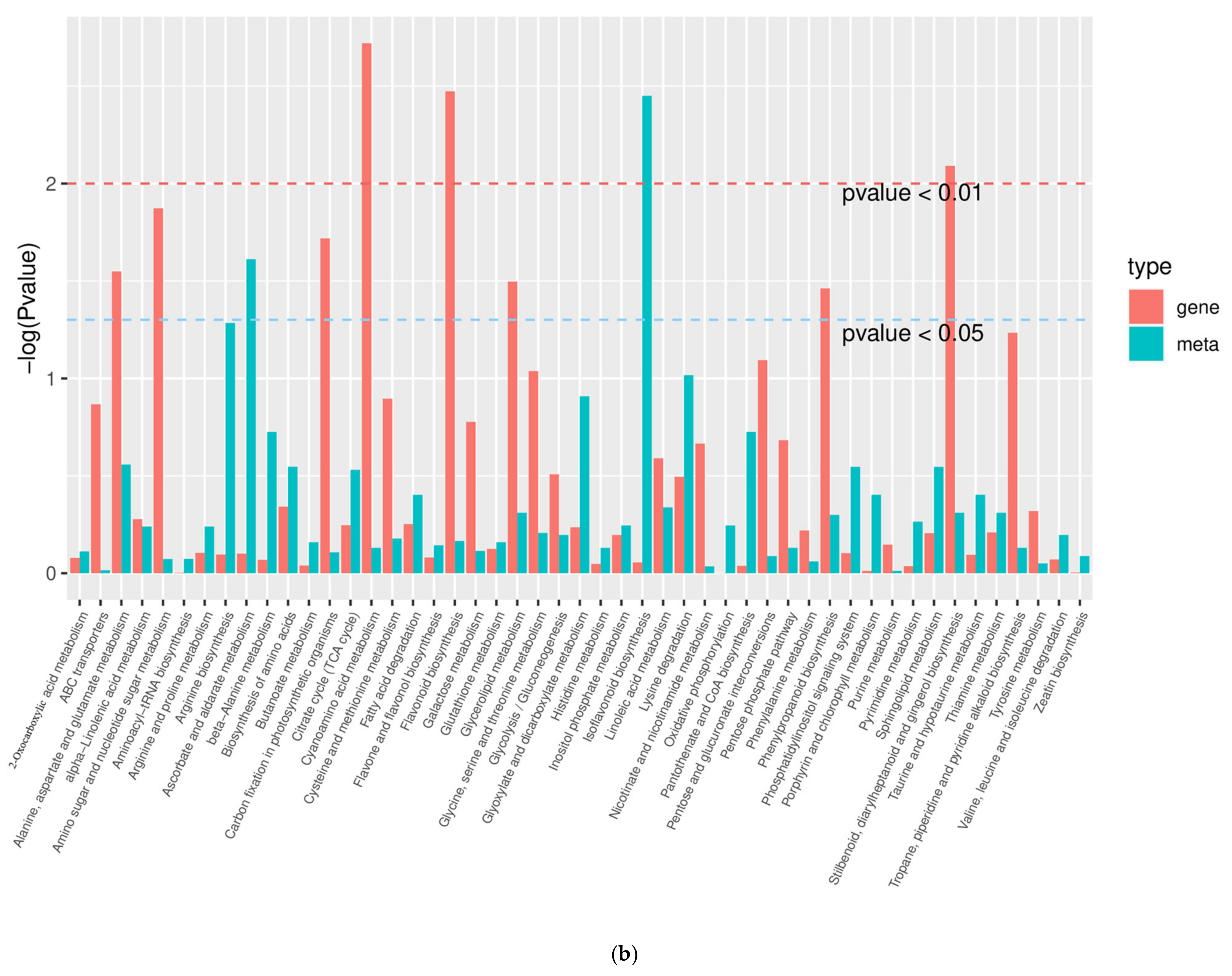
2.9. Analysis of the Overall Changes in the KEGG Metabolic Pathway
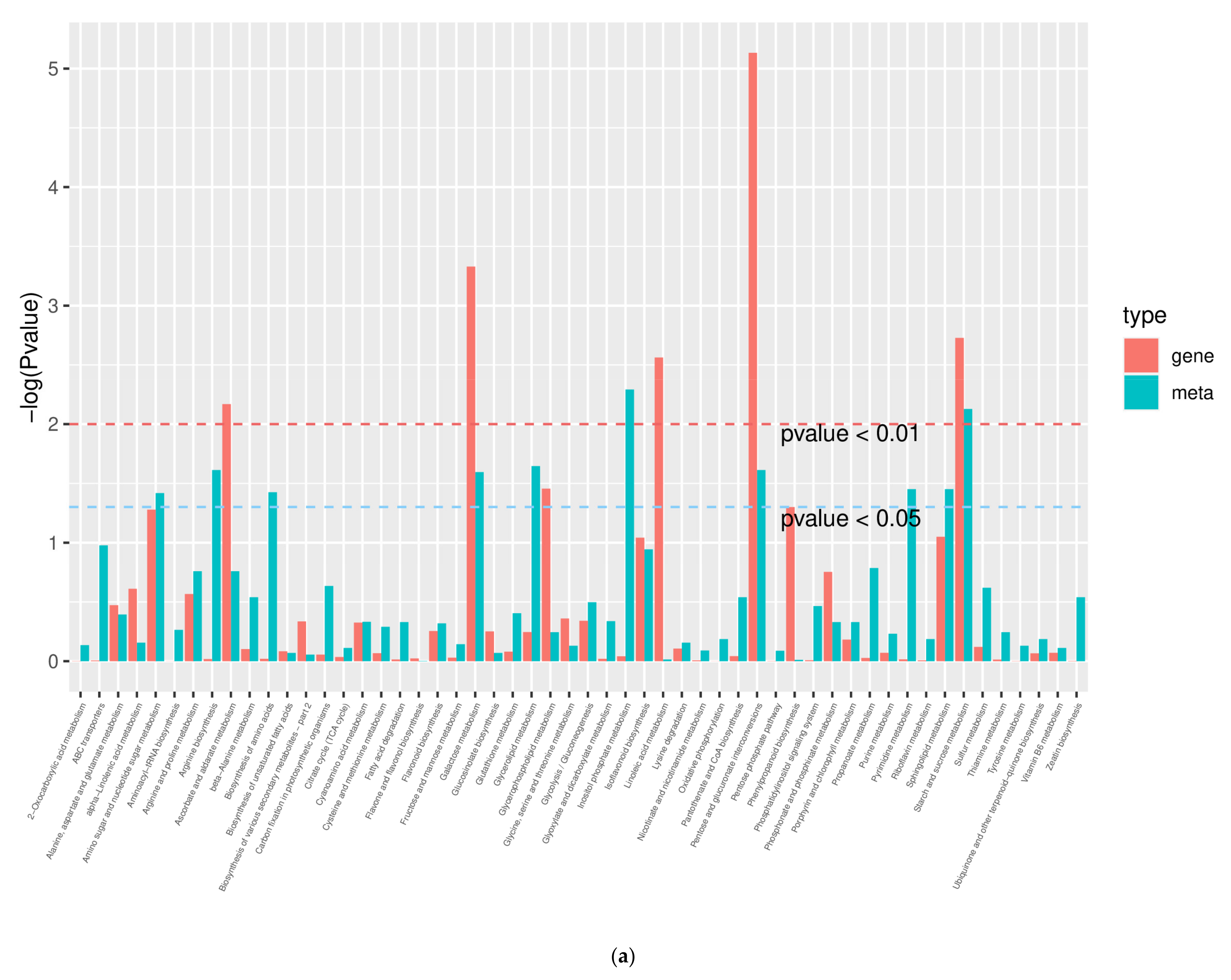
2.10. Combined Analysis of Transcriptome and Metabolome
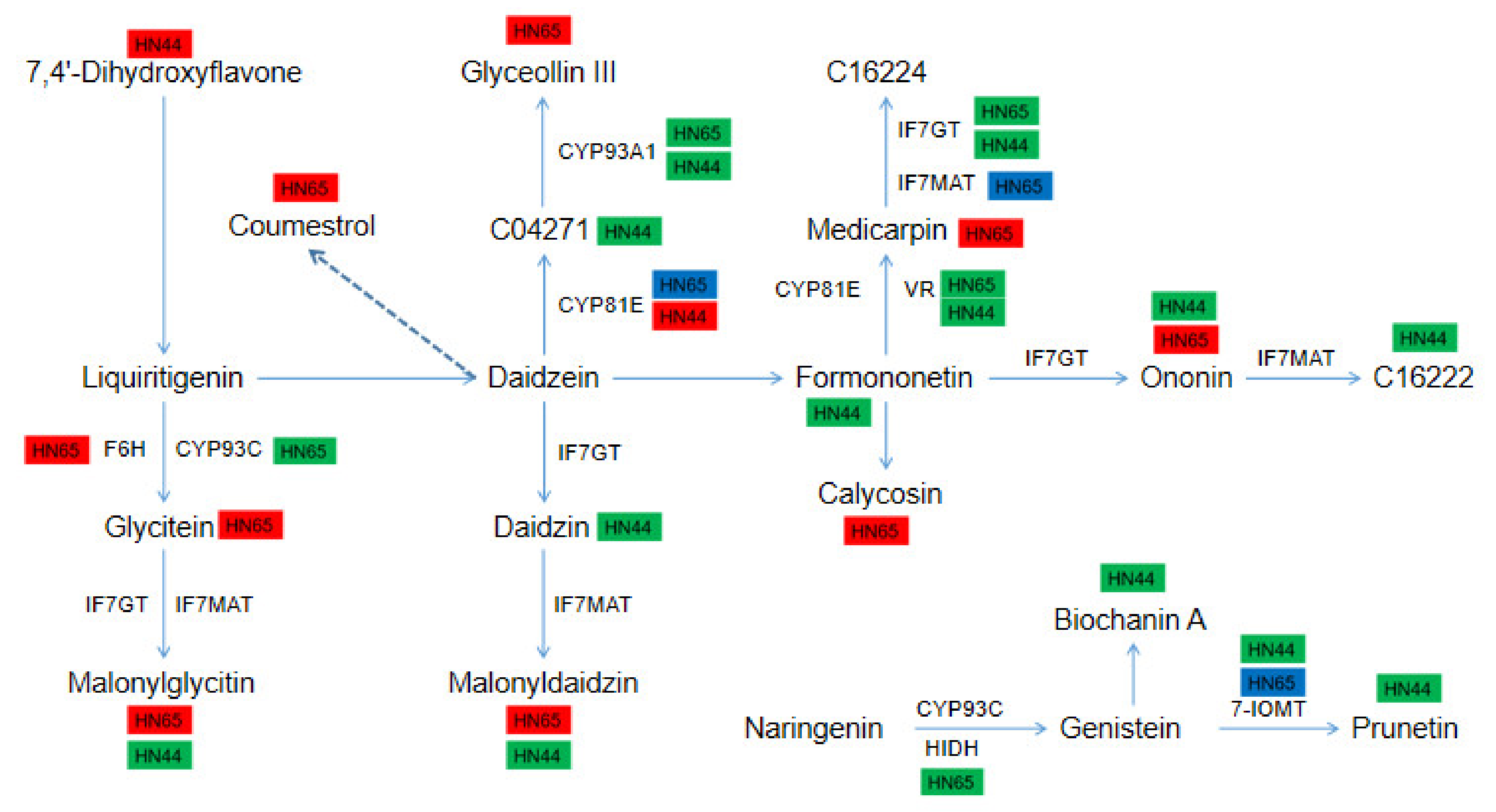
2.11. Differences in the Expression of DEGs and DAMs in the Isoflavone Biosynthesis Pathway between the Two Varieties
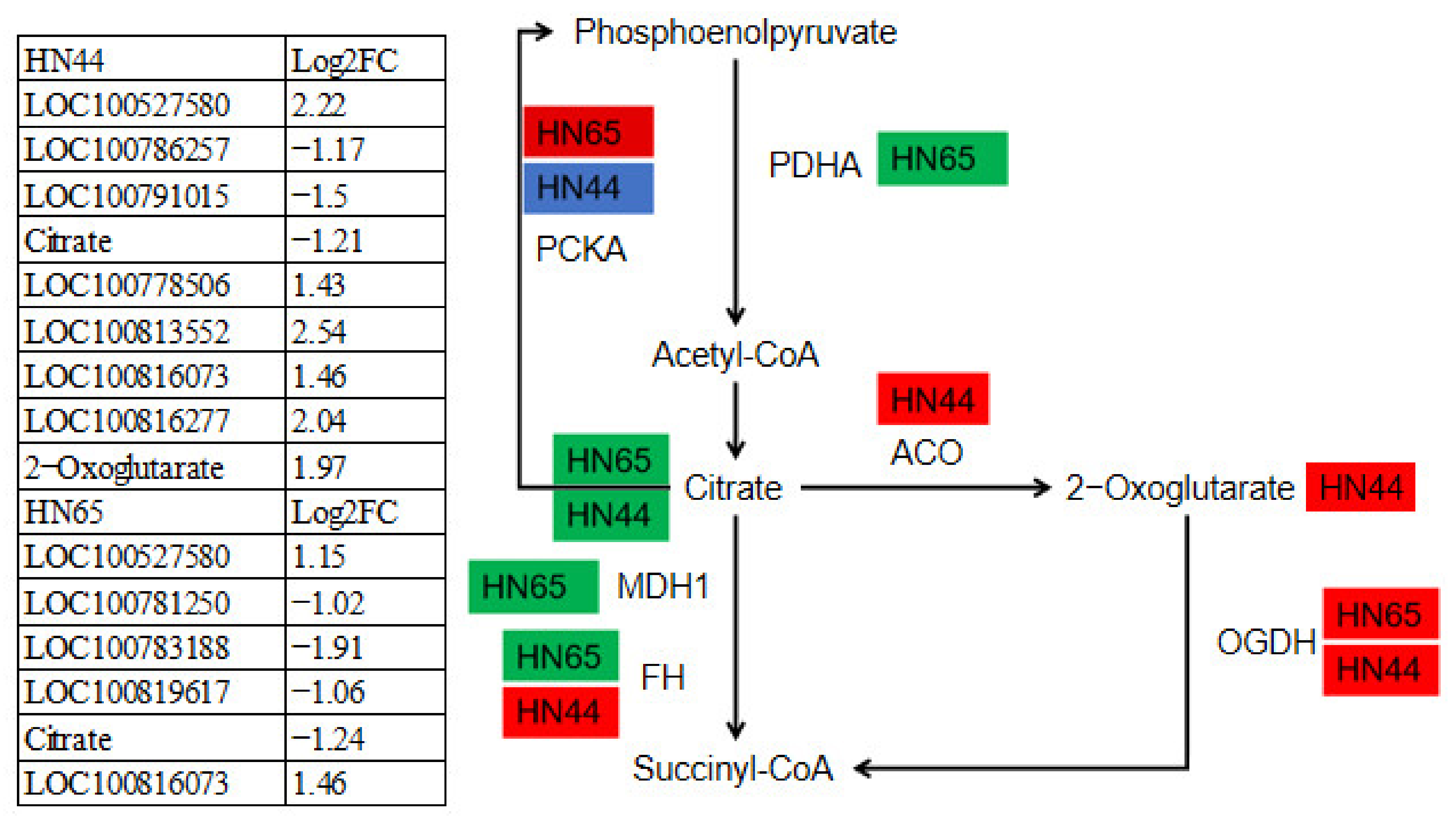
2.12. Differences in the Expression of DEGs and DAMs in the TCA Cycle between the Two Varieties
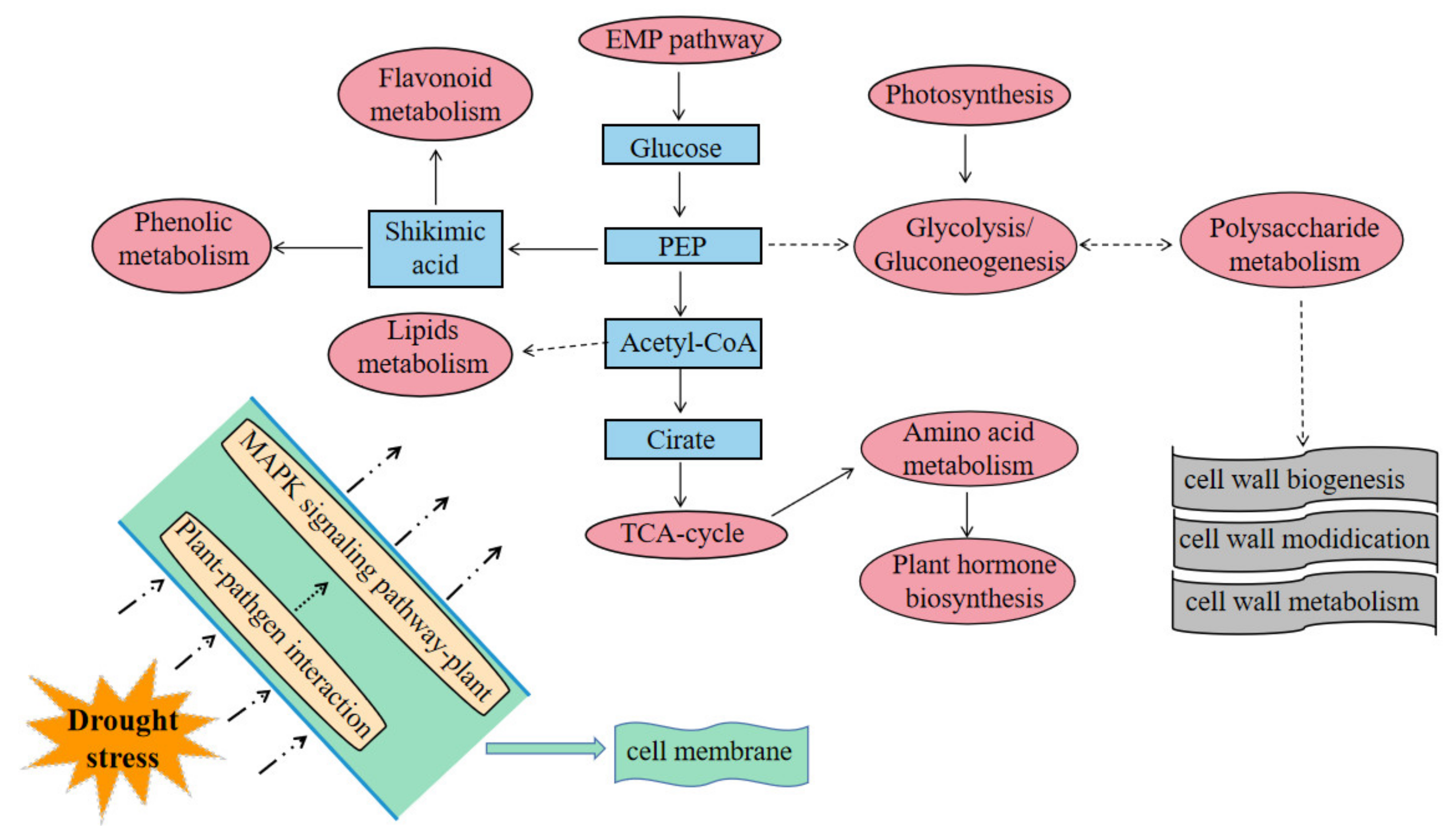
2.13. Comprehensive Understanding of Plant Drought Resistance Pathways
3. Discussion
4. Materials and Methods
4.1. Plant Material and Experimental Treatment
4.2. Transcriptomic Assay Methods
4.3. Metabolomics Assay Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tyczewska, A.; Gracz, J.; Kuczyński, J.; Twardowski, T. Deciphering the soybean molecular stress response via high-throughput approaches. Acta Biochim. Pol. 2017, 63, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Xing, Y.; Huang, J.; Xiao, X.; Liu, R. Nutritional Quality and Preliminary Investigation of Processing Characteristics for Main Soybean Cultivars. Sci. Technol. Food Ind. 2021, 42, 1–7. [Google Scholar] [CrossRef]
- Ainsworth, E.A.; Yendrek, C.R.; Skoneczka, J.A.; Long, S.P. Accelerating yield potential in soybean: Potential targets for biotechnological improvement. Plant Cell Environ. 2012, 35, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, S.; Wang, Z.; Yuan, Y.; Zhang, Z.; Liang, Q.; Yang, X.; Duan, Z.; Liu, Y.; Kong, F.; et al. Progress in soybean functional genomics over the past decade. Plant Biotechnol. J. 2021, 20, 256–282. [Google Scholar] [CrossRef]
- Feng, Z.; Ding, C.; Li, W.; Wang, D.; Cui, D. Applications of metabolomics in the research of soybean plant under abiotic stress. Food Chem. 2019, 310, 125914. [Google Scholar] [CrossRef]
- Gupta, A.; Rico-Medina, A.; Caño-Delgado, A.I. The physiology of plant responses to drought. Science 2020, 368, 266–269. [Google Scholar] [CrossRef]
- Thao, N.P.; Tran, L.-S.P. Potentials toward genetic engineering of drought-tolerant soybean. Crit. Rev. Biotechnol. 2011, 32, 349–362. [Google Scholar] [CrossRef]
- Wang, J.; Kang, L.; Liu, Z.; Lv, J.; Liu, Y.; Zou, X. Research Progress on the Impact of Drought on Plant. Hunan Agric. Sci. 2017, 7, 123–126+130. [Google Scholar] [CrossRef]
- Novaković, L.; Guo, T.; Bacic, A.; Sampathkumar, A.; Johnson, K.L. Hitting the Wall—Sensing and Signaling Pathways Involved in Plant Cell Wall Remodeling in Response to Abiotic Stress. Plants 2018, 7, 89. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wu, Z.; Zhou, Q.; Wang, X.; Song, S.; Dong, S. Physiological Response of Soybean Plants to Water Deficit. Front. Plant Sci. 2022, 12, 809692. [Google Scholar] [CrossRef]
- Shen, N.; Wang, T.; Gan, Q.; Liu, S.; Wang, L.; Jin, B. Plant flavonoids: Classification, distribution, biosynthesis, and antioxidant activity. Food Chem. 2022, 383, 132531. [Google Scholar] [CrossRef]
- Baldoni, E.; Genga, A.; Cominelli, E. Plant MYB Transcription Factors: Their Role in Drought Response Mechanisms. Int. J. Mol. Sci. 2015, 16, 15811–15851. [Google Scholar] [CrossRef] [Green Version]
- Lata, C.; Prasad, M. Role of DREBs in regulation of abiotic stress responses in plants. J. Exp. Bot. 2011, 62, 4731–4748. [Google Scholar] [CrossRef] [Green Version]
- Joshi, R.; Wani, S.H.; Singh, B.; Bohra, A.; Dar, Z.A.; Lone, A.A.; Pareek, A.; Singla-Pareek, S.L. Transcription Factors and Plants Response to Drought Stress: Current Understanding and Future Directions. Front. Plant Sci. 2016, 7, 1029. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Yang, D.-H.; Yao, H.; Bai, G.; Zhang, Y.-H.; Xiao, B.-G. iTRAQ-based quantitative proteomic analysis reveals proteomic changes in leaves of cultivated tobacco (Nicotiana tabacum) in response to drought stress. Biochem. Biophys. Res. Commun. 2016, 469, 768–775. [Google Scholar] [CrossRef] [Green Version]
- Faghani, E.; Gharechahi, J.; Komatsu, S.; Mirzaei, M.; Khavarinejad, R.A.; Najafi, F.; Farsad, L.K.; Salekdeh, G.H. Comparative physiology and proteomic analysis of two wheat genotypes contrasting in drought tolerance. J. Proteom. 2014, 114, 1–15. [Google Scholar] [CrossRef]
- Qin, X.; Yin, Y.; Zhao, J.; An, W.; Fan, Y.; Liang, X.; Cao, Y. Metabolomic and transcriptomic analysis of Lycium chinese and L. ruthenicum under salinity stress. BMC Plant Biol. 2022, 22, 8. [Google Scholar] [CrossRef]
- Zenda, T.; Liu, S.; Dong, A.; Li, J.; Wang, Y.; Liu, X.; Wang, N.; Duan, H. Omics-Facilitated Crop Improvement for Climate Resilience and Superior Nutritive Value. Front. Plant Sci. 2021, 12, 774994. [Google Scholar] [CrossRef]
- Zhang, L.J.; Fan, J.J.; Ruan, Y.Y. Application of polyethylene glycol in the study of plant osmotic stress physiology. Plant Physiol. Commun. 2004, 40, 361–368. [Google Scholar] [CrossRef]
- Zhao, B.; Zhang, S.; Yang, W.; Li, B.; Lan, C.; Zhang, J.; Yuan, L.; Wang, Y.; Xie, Q.; Han, J.; et al. Multi-omic dissection of the drought resistance traits of soybean landrace LX. Plant Cell Environ. 2021, 44, 1379–1398. [Google Scholar] [CrossRef]
- Razi, K.; Muneer, S. Drought stress-induced physiological mechanisms, signaling pathways and molecular response of chloroplasts in common vegetable crops. Crit. Rev. Biotechnol. 2021, 41, 669–691. [Google Scholar] [CrossRef]
- Li, T.; Wang, R.; Zhao, D.; Tao, J. Effects of drought stress on physiological responses and gene expression changes in herbaceous peony (Paeonia lactiflora Pall.). Plant Signal. Behav. 2020, 15, 1746034. [Google Scholar] [CrossRef]
- Zhao, X.; Huang, L.-J.; Sun, X.-F.; Zhao, L.-L.; Wang, P.-C. Transcriptomic and Metabolomic Analyses Reveal Key Metabolites, Pathways and Candidate Genes in Sophora davidii (Franch.) Skeels Seedlings Under Drought Stress. Front. Plant Sci. 2022, 13, 785702. [Google Scholar] [CrossRef]
- Verma, V.; Ravindran, P.; Kumar, P.P. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 2016, 16, 86. [Google Scholar] [CrossRef] [Green Version]
- Xuan, H.; Huang, Y.; Zhou, L.; Deng, S.; Wang, C.; Xu, J.; Wang, H.; Zhao, J.; Guo, N.; Xing, H. Key Soybean Seedlings Drought-Responsive Genes and Pathways Revealed by Comparative Transcriptome Analyses of Two Cultivars. Int. J. Mol. Sci. 2022, 23, 2893. [Google Scholar] [CrossRef]
- Danquah, A.; de Zelicourt, A.; Colcombet, J.; Hirt, H. The role of ABA and MAPK signaling pathways in plant abiotic stress responses. Biotechnol. Adv. 2014, 32, 40–52. [Google Scholar] [CrossRef]
- Moustafa, K.; AbuQamar, S.; Jarrar, M.; Al-Rajab, A.J.; Trémouillaux-Guiller, J. MAPK cascades and major abiotic stresses. Plant. Cell Rep. 2014, 33, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, K.; Mizoguchi, T.; Yoshida, R.; Yuasa, T.; Shinozaki, K. Various abiotic stresses rapidly activate Arabidopsis MAP kinases ATMPK4 and ATMPK6. Plant J. 2000, 24, 655–665. [Google Scholar] [CrossRef]
- Teige, M.; Scheikl, E.; Eulgem, T.; Dóczi, R.; Ichimura, K.; Shinozaki, K.; Dangl, J.L.; Hirt, H. The MKK2 Pathway Mediates Cold and Salt Stress Signaling in Arabidopsis. Mol. Cell 2004, 15, 141–152. [Google Scholar] [CrossRef]
- Robatzek, S.; Wirthmueller, L. Mapping FLS2 function to structure: LRRs, kinase and its working bits. Protoplasma 2012, 250, 671–681. [Google Scholar] [CrossRef]
- Williamson, G.; Kay, C.D.; Crozier, A. The Bioavailability, Transport, and Bioactivity of Dietary Flavonoids: A Review from a Historical Perspective. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1054–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackah, M.; Shi, Y.; Wu, M.; Wang, L.; Guo, P.; Guo, L.; Jin, X.; Li, S.; Zhang, Q.; Qiu, C.; et al. Metabolomics Response to Drought Stress in Morus alba L. Variety Yu-711. Plants 2021, 10, 1636. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, Y.; Wang, X.; Li, X.; Dong, S. Physiology and metabonomics reveal differences in drought resistance among soybean varieties. Bot. Stud. 2022, 63, 8. [Google Scholar] [CrossRef] [PubMed]
- Alfano, B.; Barretta, L.; Del Giudice, A.; De Vito, S.; Di Francia, G.; Esposito, E.; Formisano, F.; Massera, E.; Miglietta, M.L.; Polichetti, T. A Review of Low-Cost Particulate Matter Sensors from the Depvelopers’ Perspectives. Sensors 2020, 20, 6819. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Shi, L.; Jiao, Y.; Li, M.; Zhong, X.; Gu, F.; Liu, Q.; Xia, X.; Li, H. Metabolic responses to drought stress in the tissues of drought-tolerant and drought-sensitive wheat genotype seedlings. AoB Plant 2018, 10, ply016. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Li, B.; Chen, Q.; Su, Y.; Wang, R.; Liu, Z.; Chen, G. Non-targeted metabolomic analysis of the variations in the metabolites of two genotypes of Glycyrrhiza uralensis Fisch. under drought stress. Ind. Crop. Prod. 2021, 176, 114402. [Google Scholar] [CrossRef]
















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Song, S.; Wang, X.; Liu, J.; Dong, S. Transcriptomic and Metabolomic Analysis of Seedling-Stage Soybean Responses to PEG-Simulated Drought Stress. Int. J. Mol. Sci. 2022, 23, 6869. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126869
Wang X, Song S, Wang X, Liu J, Dong S. Transcriptomic and Metabolomic Analysis of Seedling-Stage Soybean Responses to PEG-Simulated Drought Stress. International Journal of Molecular Sciences. 2022; 23(12):6869. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126869
Chicago/Turabian StyleWang, Xiyue, Shuang Song, Xin Wang, Jun Liu, and Shoukun Dong. 2022. "Transcriptomic and Metabolomic Analysis of Seedling-Stage Soybean Responses to PEG-Simulated Drought Stress" International Journal of Molecular Sciences 23, no. 12: 6869. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23126869