
Inhibition of Vascular Endothelial Cadherin Cleavage Prevents Elastic Fiber Alterations and Atherosclerosis Induced by Intermittent Hypoxia in the Mouse Aorta

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Intermittent Hypoxia Increases Transendothelium Passage of LDL and Monocytes Migration In Vitro and Blocking the Pathways Involved in VE-Cadherin Cleavage Prevents these Effects
2.2. Intermittent Hypoxia Induces VE-Cadherin Cleavage In Vivo in C57BL/6J Mice, and Inhibition of HIF-1, VEGRF Tyr-Kinases and Src-Kinases Prevents this Effect
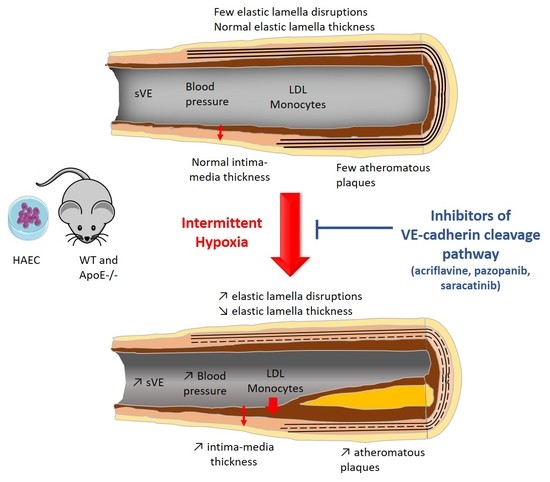
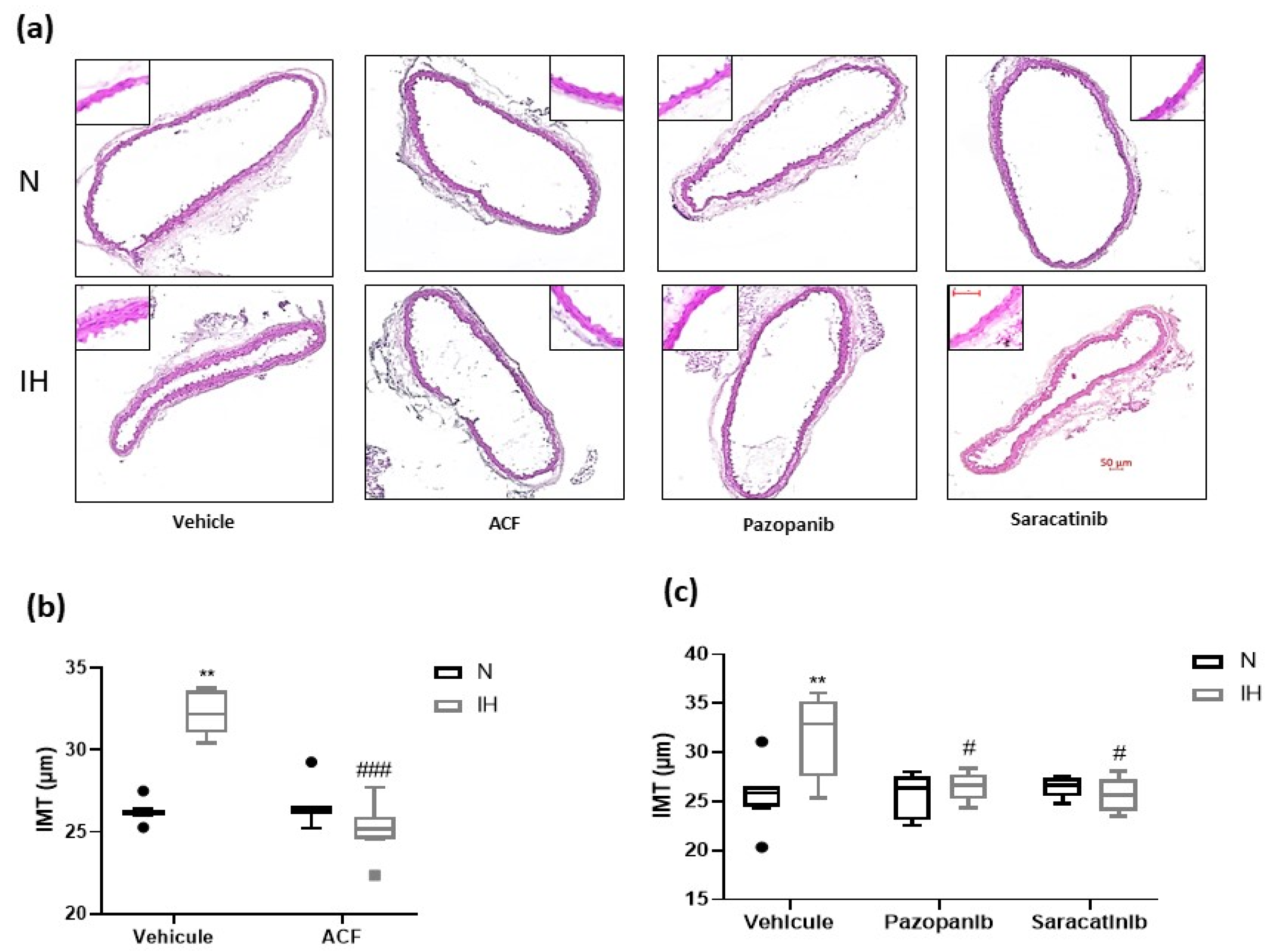
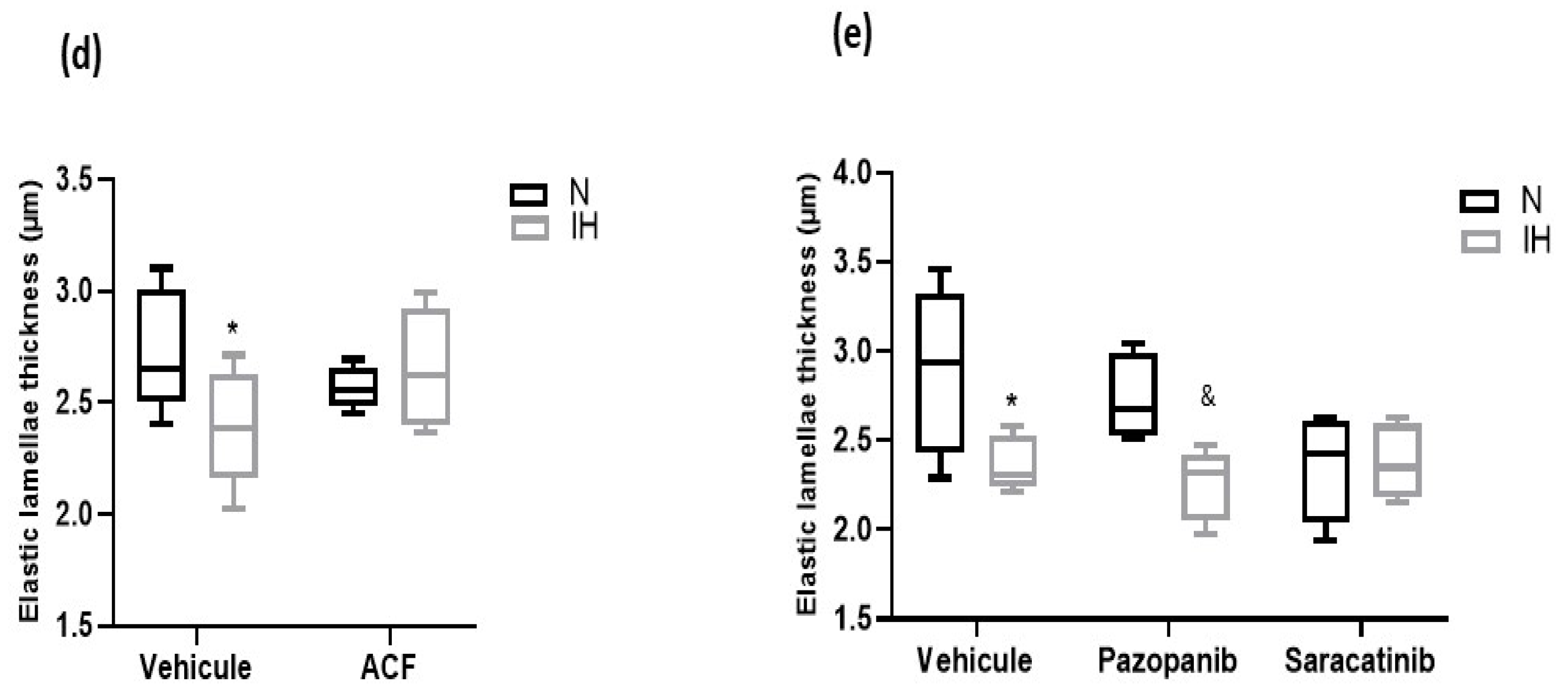
2.3. Inhibiting HIF-1, VEGRF Tyr-Kinases and Src-Kinases Prevents the Effect of Intermittent Hypoxia on Intima-Media Thickness and Elastic Lamella Integrity In Vivo in C57BL/6J Mice
2.4. Inhibiting HIF-1, VEGRF Tyr-Kinases and Src-Kinases Prevents the Favoring Effect of Intermittent Hypoxia on Atherogenesis in ApoE-/- Mice
2.5. Inhibiting VEGRF Tyr-Kinases or Src-Kinases Abolishes the IH-Induced Increase in Arterial Blood Pressures in ApoE-/- Mice
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Intermittent Hypoxia Exposure of Cultured Cells
4.3. Use of Inhibitors in In Vitro Experiments
4.4. In Vitro LDL Transendothelium Passage Assay
4.5. In Vitro Monocyte Transendothelium Migration Assay
4.6. Animals
4.7. Exposure to Intermittent Hypoxia
4.8. Treatment of Mice with VE-Cadherin Cleavage Inhibitors
4.9. Specimen Collection
4.10. sVE Semi-Quantitative Dosage by Western Blot
4.11. Immunofluorescence
4.12. Intima-Media Thickness (IMT) and Elastic Fiber Network Analysis
4.13. Atherosclerotic Lesion Size
4.14. Plasma Total-Cholesterol and LDL-Cholesterol Dosage
4.15. Arterial Blood Pressure Recording
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global burden of cardiovascular diseases and risk factors, 1990–2019. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Benjafield, A.V.; Ayas, N.T.; Eastwood, P.R.; Heinzer, R.; Ip, M.S.M.; Morrell, M.J.; Nunez, C.M.; Patel, S.R.; Penzel, T.; Pépin, J.-L.D.; et al. Estimation of the global prevalence and burden of obstructive sleep apnoea: A literature-based analysis. Lancet Respir. Med. 2019, 7, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Lévy, P.; Kohler, M.; McNicholas, W.T.; Barbé, F.; McEvoy, R.D.; Somers, V.K.; Lavie, L.; Pépin, J.-L. Obstructive sleep apnoea syndrome. Nat. Rev. Dis. Primers 2015, 1, 15015. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, R.D.; Antic, N.A.; Heeley, E.; Luo, Y.; Ou, Q.; Zhang, X.; Mediano, O.; Chen, R.; Drager, L.F.; Liu, Z.; et al. CPAP for prevention of cardiovascular events in obstructive sleep apnea. N. Engl. J. Med. 2016, 375, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Briançon-Marjollet, A.; Henri, M.; Pépin, J.-L.; Lemarié, E.; Lévy, P.; Tamisier, R. Altered in vitro endothelial repair and monocyte migration in obstructive sleep apnea: Implication of VEGF and CRP. Sleep 2014, 37, 1825–1832. [Google Scholar] [CrossRef] [Green Version]
- Briançon-Marjollet, A.; Pépin, J.-L.; Weiss, J.W.; Lévy, P.; Tamisier, R. Intermittent hypoxia upregulates serum VEGF. Sleep Med. 2014, 15, 1425–1426. [Google Scholar] [CrossRef]
- Phillips, S.A.; Olson, E.B.; Lombard, J.H.; Morgan, B.J. Chronic intermittent hypoxia alters NE reactivity and mechanics of skeletal muscle resistance arteries. J. Appl. Physiol. (1985) 2006, 100, 1117–1123. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.; Jacobs, D.R.; Lutsey, P.L.; Brumback, L.; Chirinos, J.A.; Mariani, S.; Redline, S.; Duprez, D.A. Sleep disordered breathing and ECG R-wave to radial artery pulse delay, the multi-ethnic study of atherosclerosis. Sleep Med. 2018, 48, 172–179. [Google Scholar] [CrossRef]
- Dubern, B.; Aggoun, Y.; Boulé, M.; Fauroux, B.; Bonnet, D.; Tounian, P. Arterial alterations in severely obese children with obstructive sleep apnoea. Int. J. Pediatr. Obes. 2010, 5, 230–236. [Google Scholar] [CrossRef]
- Arnaud, C.; Beguin, P.C.; Lantuejoul, S.; Pepin, J.-L.; Guillermet, C.; Pelli, G.; Burger, F.; Buatois, V.; Ribuot, C.; Baguet, J.-P.; et al. The inflammatory preatherosclerotic remodeling induced by intermittent hypoxia is attenuated by RANTES/CCL5 inhibition. Am. J. Respir. Crit. Care Med. 2011, 184, 724–731. [Google Scholar] [CrossRef] [Green Version]
- Utrera, A.; Navarrete, Á.; González-Candia, A.; García-Herrera, C.; Herrera, E.A. Biomechanical and structural responses of the aorta to intermittent hypobaric hypoxia in a rat model. Sci. Rep. 2022, 12, 3790. [Google Scholar] [CrossRef] [PubMed]
- Harki, O.; Boete, Q.; Pépin, J.-L.; Arnaud, C.; Belaidi, E.; Faury, G.; Khouri, C.; Briançon-Marjollet, A. Intermittent hypoxia-related alterations in vascular structure and function: A systematic review and meta-analysis of rodent data. Eur. Respir. J. 2021, 59, 2100866. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S. Mechanisms of cardiovascular disease in obstructive sleep apnoea. J. Thorac. Dis. 2018, 10, S4201–S4211. [Google Scholar] [CrossRef] [PubMed]
- Harki, O.; Tamisier, R.; Pépin, J.-L.; Bailly, S.; Mahmani, A.; Gonthier, B.; Salomon, A.; Vilgrain, I.; Faury, G.; Briançon-Marjollet, A. VE-cadherin cleavage in sleep apnoea: New insights into intermittent hypoxia-related endothelial permeability. Eur. Respir. J. 2021, 58, 2004518. [Google Scholar] [CrossRef] [PubMed]
- Polena, H.; Creuzet, J.; Dufies, M.; Sidibé, A.; Khalil-Mgharbel, A.; Salomon, A.; Deroux, A.; Quesada, J.-L.; Roelants, C.; Filhol, O.; et al. The tyrosine-kinase inhibitor sunitinib targets vascular endothelial (VE)-cadherin: A marker of response to antitumoural treatment in metastatic renal cell carcinoma. Br. J. Cancer 2018, 118, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Sidibé, A.; Mannic, T.; Arboleas, M.; Subileau, M.; Gulino-Debrac, D.; Bouillet, L.; Jan, M.; Vandhuick, T.; Le Loët, X.; Vittecoq, O.; et al. Soluble VE-cadherin in rheumatoid arthritis patients correlates with disease activity: Evidence for tumor necrosis factor α-induced VE-cadherin cleavage. Arthritis Rheum. 2012, 64, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, LDL deposition, and cardiovascular risk factors—A review. Cardiovasc. Res. 2018, 114, 35–52. [Google Scholar] [CrossRef]
- Arnaud, C.; Bouyon, S.; Recoquillon, S.; Brasseur, S.; Lemarié, E.; Briançon-Marjollet, A.; Gonthier, B.; Toral, M.; Faury, G.; Martinez, M.C.; et al. Nonmuscle Myosin Light Chain Kinase: A Key Player in Intermittent Hypoxia-Induced Vascular Alterations. J. Am. Heart Assoc. 2018, 7, e007893. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Yang, Y.; Zhang, H.; Du, Y.; Jiao, X.; Yu, H.; Wang, Y.; Lv, Q.; Li, F.; Sun, Q.; et al. Salidroside ameliorated intermittent hypoxia-aggravated endothelial barrier disruption and atherosclerosis via the CAMP/PKA/RhoA signaling pathway. Front. Pharmacol. 2021, 12, 723922. [Google Scholar] [CrossRef]
- Makarenko, V.V.; Usatyuk, P.V.; Yuan, G.; Lee, M.M.; Nanduri, J.; Natarajan, V.; Kumar, G.K.; Prabhakar, N.R. Intermittent hypoxia-induced endothelial barrier dysfunction requires ROS-dependent MAP kinase activation. Am. J. Physiol. Cell Physiol. 2014, 306, C745–C752. [Google Scholar] [CrossRef] [Green Version]
- Zychowski, K.E.; Sanchez, B.; Pedrosa, R.P.; Lorenzi-Filho, G.; Drager, L.F.; Polotsky, V.Y.; Campen, M.J. Serum from obstructive sleep apnea patients induces inflammatory responses in coronary artery endothelial cells. Atherosclerosis 2016, 254, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manuneedhi Cholan, P.; Cartland, S.P.; Dang, L.; Rayner, B.S.; Patel, S.; Thomas, S.R.; Kavurma, M.M. TRAIL Protects against endothelial dysfunction in vivo and inhibits angiotensin-II-induced oxidative stress in vascular endothelial cells in vitro. Free Radic. Biol. Med. 2018, 126, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Wessel, F.; Winderlich, M.; Holm, M.; Frye, M.; Rivera-Galdos, R.; Vockel, M.; Linnepe, R.; Ipe, U.; Stadtmann, A.; Zarbock, A.; et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-Cadherin. Nat. Immunol. 2014, 15, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Fraemohs, L.; Dejana, E. The role of junctional adhesion molecules in vascular inflammation. Nat. Rev. Immunol. 2007, 7, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Ba, J.; Peng, H.; Chen, Y.; Gao, Y. Effects and mechanism analysis of vascular endothelial growth factor and salvianolic acid B on 125I-low density lipoprotein permeability of the rabbit aortary endothelial cells. Cell Biochem. Biophys. 2014, 70, 1533–1538. [Google Scholar] [CrossRef]
- Hermant, B.; Bibert, S.; Concord, E.; Dublet, B.; Weidenhaupt, M.; Vernet, T.; Gulino-Debrac, D. Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J. Biol. Chem. 2003, 278, 14002–14012. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, F.; Fan, L.; Zhang, W.; Wang, T.; Du, Y.; Bai, X. Baicalin alleviates atherosclerosis by relieving oxidative stress and inflammatory responses via inactivating the NF-ΚB and P38 MAPK signaling pathways. Biomed. Pharmacother. 2018, 97, 1673–1679. [Google Scholar] [CrossRef]
- Miyazaki, T.; Taketomi, Y.; Takimoto, M.; Lei, X.-F.; Arita, S.; Kim-Kaneyama, J.; Arata, S.; Ohata, H.; Ota, H.; Murakami, M.; et al. M-calpain induction in vascular endothelial cells on human and mouse atheromas and its roles in VE-Cadherin disorganization and atherosclerosis. Circulation 2011, 124, 2522–2532. [Google Scholar] [CrossRef] [Green Version]
- Soeki, T.; Tamura, Y.; Shinohara, H.; Sakabe, K.; Onose, Y.; Fukuda, N. Elevated concentration of soluble vascular endothelial cadherin is associated with coronary atherosclerosis. Circ. J. 2004, 68, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Jang, E.; Robert, J.; Rohrer, L.; von Eckardstein, A.; Lee, W.L. Transendothelial transport of lipoproteins. Atherosclerosis 2020, 315, 111–125. [Google Scholar] [CrossRef]
- Millán, J.; Hewlett, L.; Glyn, M.; Toomre, D.; Clark, P.; Ridley, A.J. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to Caveola- and F-Actin-Rich domains. Nat. Cell Biol. 2006, 8, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Keiper, T.; Al-Fakhri, N.; Chavakis, E.; Athanasopoulos, A.N.; Isermann, B.; Herzog, S.; Saffrich, R.; Hersemeyer, K.; Bohle, R.M.; Haendeler, J.; et al. The role of junctional adhesion molecule-C (JAM-C) in oxidized LDL-mediated leukocyte recruitment. FASEB J. 2005, 19, 2078–2080. [Google Scholar] [CrossRef] [PubMed]
- Koenen, R.R.; Pruessmeyer, J.; Soehnlein, O.; Fraemohs, L.; Zernecke, A.; Schwarz, N.; Reiss, K.; Sarabi, A.; Lindbom, L.; Hackeng, T.M.; et al. Regulated release and functional modulation of junctional adhesion molecule A by Disintegrin metalloproteinases. Blood 2009, 113, 4799–4809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, T.S.; Jiang, L.; Lynch, R.M.; Zohar, Y. Permeability of epithelial/endothelial barriers in transwells and microfluidic bilayer devices. Micromachines 2019, 10, 533. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.; Wang, S.; Sohrabi, S.; Orr, C.; He, R.; Shi, W.; Liu, Y. Characterization of vascular permeability using a biomimetic microfluidic blood vessel model. Biomicrofluidics 2017, 11, 024102. [Google Scholar] [CrossRef]
- Suarez-Giron, M.C.; Castro-Grattoni, A.; Torres, M.; Farré, R.; Barbé, F.; Sánchez-de-la-Torre, M.; Gozal, D.; Picado, C.; Montserrat, J.M.; Almendros, I. Acetylsalicylic acid prevents intermittent hypoxia-induced vascular remodeling in a murine model of sleep apnea. Front. Physiol. 2018, 9, 600. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Huang, S.; Li, X.; Li, X.; Huang, S.; Zhang, Y.; Chen, Z.-Y. Endothelial tyrosine kinase receptor B prevents VE-Cadherin cleavage and protects against atherosclerotic lesion development in ApoE−/− Mice. Oncotarget 2015, 6, 30640–30649. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, L.; You, J.; Cao, J.; Fu, X. Effect of 7-difluoromethyl-5, 4’-dimethoxygenistein on aorta atherosclerosis in hyperlipidemia ApoE(-/-) mice induced by a cholesterol-rich diet. Drug Des. Devel. Ther. 2013, 7, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Bian, F.; Yang, X.; Zhou, F.; Wu, P.-H.; Xing, S.; Xu, G.; Li, W.; Chi, J.; Ouyang, C.; Zhang, Y.; et al. C-reactive protein promotes atherosclerosis by increasing LDL transcytosis across endothelial cells. Br. J. Pharmacol. 2014, 171, 2671–2684. [Google Scholar] [CrossRef]
- Liu, J.; Guo, Z.; Zhang, Y.; Wu, T.; Ma, Y.; Lai, W.; Guo, Z. LCK inhibitor attenuates atherosclerosis in ApoE-/- Mice via regulating T cell differentiation and reverse cholesterol transport. J. Mol. Cell Cardiol. 2020, 139, 87–97. [Google Scholar] [CrossRef]
- Bourghardt, J.; Bergström, G.; Krettek, A.; Sjöberg, S.; Borén, J.; Tivesten, A. The endogenous estradiol metabolite 2-methoxyestradiol reduces atherosclerotic lesion formation in female apolipoprotein E-deficient mice. Endocrinology 2007, 148, 4128–4132. [Google Scholar] [CrossRef] [Green Version]
- Christoph, M.; Ibrahim, K.; Hesse, K.; Augstein, A.; Schmeisser, A.; Braun-Dullaeus, R.C.; Simonis, G.; Wunderlich, C.; Quick, S.; Strasser, R.H.; et al. Local inhibition of hypoxia-inducible factor reduces neointima formation after arterial injury in ApoE-/- mice. Atherosclerosis 2014, 233, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, C.; Poulain, L.; Lévy, P.; Dematteis, M. Inflammation contributes to the atherogenic role of intermittent hypoxia in apolipoprotein-E knock out mice. Atherosclerosis 2011, 219, 425–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier-Veyret, E.; Bäck, M.; Arnaud, C.; Belaïdi, E.; Tamisier, R.; Lévy, P.; Arnol, N.; Perrin, M.; Pépin, J.-L.; Stanke-Labesque, F. Cysteinyl-leukotriene pathway as a new therapeutic target for the treatment of atherosclerosis related to obstructive sleep apnea syndrome. Pharmacol. Res. 2018, 134, 311–319. [Google Scholar] [CrossRef]
- Gautier-Veyret, E.; Arnaud, C.; Bäck, M.; Pépin, J.-L.; Petri, M.H.; Baguet, J.-P.; Tamisier, R.; Lévy, P.; Stanke-Labesque, F. Intermittent hypoxia-activated cyclooxygenase pathway: Role in atherosclerosis. Eur. Respir. J. 2013, 42, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennequin, L.F.; Allen, J.; Breed, J.; Curwen, J.; Fennell, M.; Green, T.P.; Lambert-van der Brempt, C.; Morgentin, R.; Norman, R.A.; Olivier, A.; et al. N-(5-Chloro-1,3-Benzodioxol-4-Yl)-7-[2-(4-Methylpiperazin-1-Yl)Ethoxy]-5- (Tetrahydro-2H-Pyran-4-Yloxy)Quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J. Med. Chem. 2006, 49, 6465–6488. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Liu, J.; Huang, X.; Jiang, Q. Adverse effects of Dasatinib on glucose-lipid metabolism in patients with chronic myeloid leukaemia in the chronic phase. Sci. Rep. 2019, 9, 17601. [Google Scholar] [CrossRef]
- Rea, D.; Mirault, T.; Cluzeau, T.; Gautier, J.-F.; Guilhot, F.; Dombret, H.; Messas, E. Early onset hypercholesterolemia induced by the 2nd-generation tyrosine kinase inhibitor nilotinib in patients with chronic phase-chronic myeloid leukemia. Haematologica 2014, 99, 1197–1203. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.; Liu, C.; Li, P.; Mai, Z.; Tan, X.; Chen, S.; Zhou, Z.; Tang, Z.; Miao, J.; Liu, L.; et al. Risk of dyslipidemia associated with VEGF/VEGFR inhibitors: A meta-analysis. Transl. Oncol. 2020, 13, 100779. [Google Scholar] [CrossRef]
- Pinkhas, D.; Ho, T.; Smith, S. Assessment of pazopanib-related hypertension, cardiac dysfunction and identification of clinical risk factors for their development. Cardiooncology 2017, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Študentová, H.; Indráková, J.; Petrová, P.; Kamínek, M.; Kalábová, H.; Šrámek, V.; Adam, T.; Melichar, B. Risk factors of atherosclerosis during systemic therapy targeting vascular endothelial growth factor. Oncol. Lett. 2016, 11, 939–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidibé, A.; Imhof, B.A. VE-Cadherin phosphorylation decides: Vascular permeability or diapedesis. Nat. Immunol. 2014, 15, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Minoves, M.; Morand, J.; Perriot, F.; Chatard, M.; Gonthier, B.; Lemarié, E.; Menut, J.-B.; Polak, J.; Pépin, J.-L.; Godin-Ribuot, D.; et al. An innovative intermittent hypoxia model for cell cultures allowing fast Po2 oscillations with minimal gas consumption. Am. J. Physiol. Cell Physiol. 2017, 313, C460–C468. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harki, O.; Bouyon, S.; Sallé, M.; Arco-Hierves, A.; Lemarié, E.; Demory, A.; Chirica, C.; Vilgrain, I.; Pépin, J.-L.; Faury, G.; et al. Inhibition of Vascular Endothelial Cadherin Cleavage Prevents Elastic Fiber Alterations and Atherosclerosis Induced by Intermittent Hypoxia in the Mouse Aorta. Int. J. Mol. Sci. 2022, 23, 7012. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137012
Harki O, Bouyon S, Sallé M, Arco-Hierves A, Lemarié E, Demory A, Chirica C, Vilgrain I, Pépin J-L, Faury G, et al. Inhibition of Vascular Endothelial Cadherin Cleavage Prevents Elastic Fiber Alterations and Atherosclerosis Induced by Intermittent Hypoxia in the Mouse Aorta. International Journal of Molecular Sciences. 2022; 23(13):7012. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137012
Chicago/Turabian StyleHarki, Olfa, Sophie Bouyon, Marine Sallé, Alejandro Arco-Hierves, Emeline Lemarié, Alexandra Demory, Carole Chirica, Isabelle Vilgrain, Jean-Louis Pépin, Gilles Faury, and et al. 2022. "Inhibition of Vascular Endothelial Cadherin Cleavage Prevents Elastic Fiber Alterations and Atherosclerosis Induced by Intermittent Hypoxia in the Mouse Aorta" International Journal of Molecular Sciences 23, no. 13: 7012. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137012