
Synthesis of Shape-Memory Polyurethanes: Combined Experimental and Simulation Studies

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Syntheses
2.2.1. Bis(methylcarbonate)decamethylene
Bis(methylcarbonate)decamethylene Based on 1,10-Decanediol with an 8 Molar Excess of DMC (E_BMC_8)
BMC Based on 1,10-Decanediol with a 10 Molar Excess of DMC (E_BMC_10)
BMC Based on 1,10-Decanediol with an 18 Molar Excess of DMC (E_BMC_18)
2.2.2. Oligo(decamethylene carbonate) Diols
Oligo(decamethylene carbonate) Diol with an Average Molar Mass of 3000 g × mol−1 (E_OCD_3000)
Oligo(decamethylene carbonate) Diol with a Number Average Molar Mass of 5000 g × mol−1 (E_OCD_5000) after the Described Synthesis Step
2.2.3. Synthesis of Urethane Prepolymers PCUUs
2.2.4. Chain Extending of the PCURs Prepolymers with Water Vapor–Formation of PCUUs
2.3. Characterization Techniques
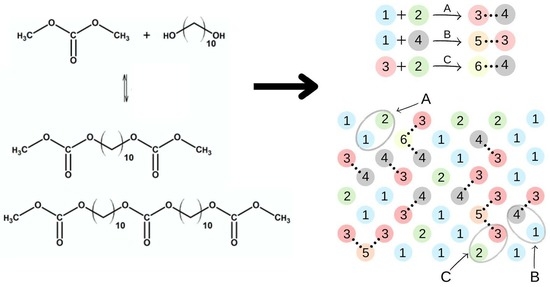
2.4. Computer Simulations: The Model and the Method
3. Results and Discussion
3.1. Characterization of PCUUs
3.2. Mechanical Tests
3.3. Thermal Analyses
3.4. Shape-Memory Properties
3.5. Results of Monte Carlo Simulations
3.5.1. Synthesis of Bis(methylcarbonate)decamethylene
3.5.2. Synthesis of Oligo(decamethylene carbonate) Diols
3.5.3. Urethane Prepolymers
3.5.4. Curing of Urethane Prepolymers with Water Vapor
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Polyurethane Elastomer Market by End-User and Geography—Forecast and Analysis 2022–2026. Available online: www.technavio.com (accessed on 2 June 2022).
- Akindoyo, J.O.; Beg, M.D.H.; Ghazali, S.; Islam, M.R.; Jeyaratnam, N.; Yuvaraj, A.R. Polyurethane Types, Synthesis and Applications—A Review. RSC Adv. 2016, 6, 114453–114482. [Google Scholar] [CrossRef] [Green Version]
- Ge, C.; Lian, D.; Cui, S.; Gao, J.; Lu, J. Highly Selective CO2 Capture on Waste Polyurethane Foam-Based Activated Carbon. Processes 2019, 7, 592. [Google Scholar] [CrossRef] [Green Version]
- Gavgani, J.N.; Adelnia, H.; Gudarzi, M.M. Intumescent Flame Retardant Polyurethane/Reduced Graphene Oxide Composites with Improved Mechanical, Thermal, and Barrier Properties. J. Mater. Sci. 2014, 49, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Kucinska-Lipka, J.; Gubanska, I.; Janik, H.; Sienkiewicz, M. Fabrication of Polyurethane and Polyurethane Based Composite Fibres by the Electrospinning Technique for Soft Tissue Engineering of Cardiovascular System. Mater. Sci. Eng. C 2015, 46, 166–176. [Google Scholar] [CrossRef]
- Bil, M.; Kijeńska-Gawrońska, E.; Głodkowska-Mrówka, E.; Manda-Handzlik, A.; Mrówka, P. Design and in Vitro Evaluation of Electrospun Shape Memory Polyurethanes for Self-Fitting Tissue Engineering Grafts and Drug Delivery Systems. Mater. Sci. Eng. C 2020, 110, 110675. [Google Scholar] [CrossRef]
- Janik, H.; Marzec, M. A Review: Fabrication of Porous Polyurethane Scaffolds. Mater. Sci. Eng. C 2015, 48, 586–591. [Google Scholar] [CrossRef]
- Davis, F.J.; Mitchell, G.R. Polyurethane Based Materials with Applications in Medical Devices. In Bio-Materials and Prototyping Applications in Medicine; Springer: Boston, MA, USA, 2008; pp. 27–48. [Google Scholar] [CrossRef]
- George Drobny, J. 9—Thermoplastic Polyurethane Elastomers. In Handbook of Thermoplastic Elastomers; Plastics Design Library: Oxford, UK, 2014; pp. 233–253. [Google Scholar] [CrossRef]
- Mazurek-Budzyńska, M.; Behl, M.; Razzaq, M.Y.; Nöchel, U.; Rokicki, G.; Lendlein, A. Hydrolytic Stability of Aliphatic Poly(Carbonate-Urea-Urethane)s: Influence of Hydrocarbon Chain Length in Soft Segment. Polym. Degrad. Stab. 2019, 161, 283–297. [Google Scholar] [CrossRef]
- Christenson, E.M.; Anderson, J.M.; Hiltner, A. Oxidative Mechanisms of Poly(Carbonate Urethane) and Poly(Ether Urethane) Biodegradation: In Vivo and In Vitro Correlations. J. Biomed. Mater. Res. Part A 2004, 70, 245–255. [Google Scholar] [CrossRef]
- Srivastava, R.; Srinivas, D.; Ratnasamy, P. Syntheses of Polycarbonate and Polyurethane Precursors Utilizing CO2 over Highly Efficient, Solid as-Synthesized MCM-41 Catalyst. Tetrahedron Lett. 2006, 47, 4213–4217. [Google Scholar] [CrossRef]
- Ubaghs, L.; Fricke, N.; Keul, H.; Höcker, H. Polyurethanes with Pendant Hydroxyl Groups: Synthesis and Characterization. Macromol. Rapid Commun. 2004, 25, 517–521. [Google Scholar] [CrossRef]
- Tomczyk, K.M.; Parzuchowski, P.G.; Rokicki, G. Oligocarbonate Diols from Ethylene Carbonate—Optimization of the Synthesis Process. J. Appl. Polym. Sci. 2010, 120, 683–691. [Google Scholar] [CrossRef]
- Tomczyk, K.M.; Parzuchowski, P.G.; Kozakiewicz, J.; Przybylski, J.; Rokicki, G. Synthesis of Oligocarbonate Diols from a “Green Monomer”—Dimethyl Carbonate—As Soft Segments for Poly(Urethane-Urea) Elastomers. Polimery 2010, 55, 366–372. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Smith, N.; Jones, E.; Finch, D.S.; Cameron, R.E. Analysis and Evaluation of a Biomedical Polycarbonate Urethane Tested in an in Vitro Study and an Ovine Arthroplasty Model. Part I: Materials Selection and Evaluation. Biomaterials 2005, 26, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Meng, F.; Cheng, R.; Deng, C.; Feijen, J.; Zhong, Z. Advanced Drug and Gene Delivery Systems Based on Functional Biodegradable Polycarbonates and Copolymers. J. Control. Release 2014, 190, 398–414. [Google Scholar] [CrossRef] [PubMed]
- Elsner, J.J.; Mezape, Y.; Hakshur, K.; Shemesh, M.; Linder-Ganz, E.; Shterling, A.; Eliaz, N. Wear Rate Evaluation of a Novel Polycarbonate-Urethane Cushion Form Bearing for Artificial Hip Joints. Acta Biomater. 2010, 6, 4698–4707. [Google Scholar] [CrossRef] [PubMed]
- Mazurek-Budzyńska, M.; Razzaq, M.Y.; Rokicki, G.; Behl, M.; Lendlein, A. High-Strain Shape-Memory Properties of Poly(Carbonate-Urea-Urethane)s Based on Aliphatic Oligocarbonates and L-Lysine Diisocyanate. Soft Mater. Biomater. 2017, 2, 2529–2536. [Google Scholar] [CrossRef]
- Mazurek-Budzyńska, M.; Yasar, M.; Tomczyk, K.; Rokicki, G.; Behl, M.; Lendlein, A. Poly(Carbonate-Urea-Urethane) Networks Exhibiting High-Strain Shape-Memory Effect †. Polym. Adv. Technol. 2016, 28, 1285–1293. [Google Scholar] [CrossRef]
- Mazurek-Budzyńska, M.; Behl, M.; Neumann, R.; Lendlein, A. 4D-Actuators by 3D-Printing Combined with Water-Based Curing. Mater. Today Commun. 2022, 30, 102966. [Google Scholar] [CrossRef]
- Balko, J.; Fernández-D’Arlas, B.; Pöselt, E.; Dabbous, R.; Müller, A.J.; Thurn-Albrecht, T. Clarifying the Origin of Multiple Melting of Segmented Thermoplastic Polyurethanes by Fast Scanning Calorimetry. Macromolecules 2017, 50, 7672–7680. [Google Scholar] [CrossRef]
- Fernández-D’Arlas, B.; Balko, J.; Baumann, R.P.; Pöselt, E.; Dabbous, R.; Eling, B.; Thurn-Albrecht, T.; Müller, A.J. Tailoring the Morphology and Melting Points of Segmented Thermoplastic Polyurethanes by Self-Nucleation. Macromolecules 2016, 49, 7952–7964. [Google Scholar] [CrossRef]
- Fernández-D’Arlas, B.; Baumann, R.P.; Pöselt, E.; Müller, A.J. Influence of Composition on the Isothermal Crystallisation of Segmented Thermoplastic Polyurethanes. CrystEngComm 2017, 19, 4720–4733. [Google Scholar] [CrossRef]
- Asensio, M.; Costa, V.; Nohales, A.; Bianchi, O.; Gómez, C.M. Tunable Structure and Properties of Segmented Thermoplastic Polyurethanes as a Function Offlexible Segment. Polymers 2019, 11, 1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilgor, I.; Yilgor, E.; Guler, I.G.; Ward, T.C.; Wilkes, G.L. FTIR Investigation of the Influence of Diisocyanate Symmetry on the Morphology Development in Model Segmented Polyurethanes. Polymer 2006, 47, 4105–4114. [Google Scholar] [CrossRef]
- Castro, J.M.; López-Serrano, F.; Camargo, R.E.; Macosko, C.W.; Tirrell, M. Onset of Phase Separation in Segmented Urethane Polymerization. J. Appl. Polym. Sci. 1981, 26, 2067–2076. [Google Scholar] [CrossRef]
- Yilgör, I.; Yilgör, E.; Wilkes, G.L. Critical Parameters in Designing Segmented Polyurethanes and Their Effect on Morphology and Properties: A Comprehensive Review. Polymer 2015, 58, A1–A36. [Google Scholar] [CrossRef]
- Sun, X.D.; Sung, C.S.P. Cure Characterization in Polyurethane and Model Urethane Reactions by an Intrinsic Fluorescence Technique. Macromolecules 1996, 29, 3198–3202. [Google Scholar] [CrossRef]
- Lee, K.J.; Eichinger, B.E. Computer Simulation of the Structure and Elasticity of Polyurethane Networks: 1. Polyoxypropylene Tetrols and Hexamethylene Diisocyanate. Polymer 1990, 31, 406–413. [Google Scholar] [CrossRef]
- Lee, K.J.; Eichinger, B.E. Computer Simulation of the Structure and Elasticity of Polyurethane Networks: 2. Polyoxypropylene Triols and 4,4′-Diphenylmethane Diisocyanate. Polymer 1990, 31, 414–422. [Google Scholar] [CrossRef]
- Lempesis, N.; In’T Veld, P.J.; Rutledge, G.C. Atomistic Simulation of a Thermoplastic Polyurethane and Micromechanical Modeling. Macromolecules 2017, 50, 7399–7409. [Google Scholar] [CrossRef]
- Yildirim, E.; Yurtsever, M. The Role of Diisocyanate and Soft Segment on the Intersegmental Interactions in Urethane and Urea Based Segmented Copolymers: A DFT Study. Comput. Theor. Chem. 2014, 1035, 28–38. [Google Scholar] [CrossRef]
- Tobushi, H.; Okumura, K.; Hayashi, S.; Ito, N. Thermomechanical Constitutive Model of Shape Memory Polymers. Mech. Mater. 2001, 33, 545–554. [Google Scholar] [CrossRef]
- Park, S.; Moon, J.; Kim, B.; Cho, M. Multi-Scale Coarse-Grained Molecular Dynamics Simulation to Investigate the Thermo-Mechanical Behavior of Shape-Memory Polyurethane Copolymers. Polymer 2021, 213, 123228. [Google Scholar] [CrossRef]
- van Duin, A.C.T.; Dasgupta, S.; Lorant, F. ReaxFF: A Reactive Force Field for Hydrocarbons. J. Phys. Chem. 2001, 105, 9396–9409. [Google Scholar] [CrossRef] [Green Version]
- Yong, X.; Kuksenok, O.; Balazs, A.C. Modeling Free Radical Polymerization Using Dissipative Particle Dynamics. Polymer 2015, 72, 217–225. [Google Scholar] [CrossRef]
- Gavrilov, A.A.; Chertovich, A.V. Copolymerization of Partly Incompatible Monomers: An Insight from Computer Simulations. Macromolecules 2017, 50, 4677–4685. [Google Scholar] [CrossRef] [Green Version]
- D’Hooge, D.R.; Van Steenberge, P.H.M.; Reyniers, M.F.; Marin, G.B. The Strength of Multi-Scale Modeling to Unveil the Complexity of Radical Polymerization. Prog. Polym. Sci. 2016, 58, 59–89. [Google Scholar] [CrossRef]
- Wang, L.; Broadbelt, L.J. Tracking Explicit Chain Sequence in Kinetic Monte Carlo Simulations. Macromol. Theory Simul. 2011, 20, 54–64. [Google Scholar] [CrossRef]
- Turgman-Cohen, S.; Genzer, J. Simultaneous Bulk- and Surface-Initiated Controlled Radical Polymerization from Planar Substrates. J. Am. Chem. Soc. 2011, 133, 17567–17569. [Google Scholar] [CrossRef]
- Genzer, J. In Silico Polymerization: Computer Simulation of Controlled Radical Polymerization in Bulk and on Flat Surfaces. Macromolecules 2006, 39, 7157–7169. [Google Scholar] [CrossRef]
- Marien, Y.W.; Van Steenberge, P.H.M.; D’Hooge, D.R.; Marin, G.B. Particle by Particle Kinetic Monte Carlo Tracking of Reaction and Mass Transfer Events in Miniemulsion Free Radical Polymerization. Macromolecules 2019, 52, 1408–1423. [Google Scholar] [CrossRef]
- Drache, M.; Drache, G. Simulating Controlled Radical Polymerizations with McPolymer—A Monte Carlo Approach. Polymers 2012, 4, 1416–1442. [Google Scholar] [CrossRef] [Green Version]
- Polanowski, P.; Pakula, T. Studies of Polymer Conformation and Dynamics in Two Dimensions Using Simulations Based on the Dynamic Lattice Liquid (DLL) Model. J. Chem. Phys. 2002, 117, 4022–4029. [Google Scholar] [CrossRef]
- Polanowski, P.; Jeszka, J.K.; Sikorski, A. Dynamic Properties of Linear and Cyclic Chains in Two Dimensions. Computer Simulation Studies. Macromolecules 2014, 47, 4830–4839. [Google Scholar] [CrossRef]
- Polanowski, P.; Jeszka, J.K.; Matyjaszewski, K. Synthesis of Star Polymers by “Core-First” One-Pot Method via ATRP: Monte Carlo Simulations. Polymer 2014, 55, 2552–2561. [Google Scholar] [CrossRef]
- Polanowski, P.; Jeszka, J.K.; Matyjaszewski, K. Star Polymer Synthesis and Gelation in ATRP Copolymerization: Monte Carlo Simulations. Polymer 2013, 54, 1979–1986. [Google Scholar] [CrossRef]
- Polanowski, P.; Hałagan, K.; Pietrasik, J.; Jeszka, J.K.; Matyjaszewski, K. Growth of Polymer Brushes by “Grafting from” via ATRP—Monte Carlo Simulations. Polymer 2017, 130, 267–279. [Google Scholar] [CrossRef]
- Hałagan, K.; Banaszak, M.; Jung, J.; Polanowski, P.; Sikorski, A. Dynamics of Opposing Polymer Brushes: A Computer Simulation Study. Polymers 2021, 13, 2758. [Google Scholar] [CrossRef]
- Halagan, K.; Banaszak, M.; Jung, J.; Polanowski, P.; Sikorski, A. Polymerization and Structure of Opposing Polymer Brushes Studied by Computer Simulations. Polymers 2021, 13, 4294. [Google Scholar] [CrossRef]
- Bain, E.D.; Turgman-Cohen, S.; Genzer, J. Progress in Computer Simulation of Bulk, Confined, and Surface-Initiated Polymerizations. Macromol. Theory Simul. 2013, 22, 8–30. [Google Scholar] [CrossRef]
- Tobita, H.; Yanase, F. Monte Carlo Simulation of Controlled/Living Radical Polymerization in Emulsified Systems. Macromol. Theory Simul. 2007, 16, 476–488. [Google Scholar] [CrossRef]
- Kratz, K.; Madbouly, S.A.; Wagermaier, W.; Lendlein, A. Temperature-Memory Polymer Networks with Crystallizable Controlling Units. Adv. Mater. 2011, 23, 4058–4062. [Google Scholar] [CrossRef] [PubMed]
- Çaykara, T.; Turan, E. Effect of the Amount and Type of the Crosslinker on the Swelling Behavior of Temperature-Sensitive Poly(N-Tert-Butylacrylamide-Co-Acrylamide) Hydrogels. Colloid Polym. Sci. 2006, 284, 1038–1048. [Google Scholar] [CrossRef]
- Shieh, Y.-T.; Liu, G.-L. Effects of Carbon Nanotubes on Crystallization and MeltingBehavior of Poly(L-Lactide) via DSC and TMDSC Studies. J. Polym. Sci. Part B Polym. Phys. 2007, 45, 1870–1881. [Google Scholar] [CrossRef]
- Yan, B.; Gu, S.; Zhang, Y. Polylactide-Based Thermoplastic Shape Memory Polymer Nanocomposites. Eur. Polym. J. 2013, 49, 366–378. [Google Scholar] [CrossRef]
- Kozakiewicz, J.; Rokicki, G.; Przybylski, J.; Sylwestrzak, K.; Parzuchowski, P.G.; Tomczyk, K.M. Studies on the Effect of Curing Conditions on the Curing Rate and Mechanical Properties of Moisture-Cured Poly(Urethane-Urea) Elastomers Containing Oligocarbonate Segments. Polimery 2011, 56, 564–570. [Google Scholar] [CrossRef]
- Arun, A.; Baack, K.K.J.; Gaymans, R. Polyurethane Tri-Block Copolymers—Synthesis, Mechanical, Elastic, and Rheological Properties. Polym. Eng. Sci. 2010, 50, 747–755. [Google Scholar] [CrossRef]
- Gaymans, R.J. Segmented Copolymers with Monodisperse Crystallizable Hard Segments: Novel Semi-Crystalline Materials. Prog. Polym. Sci. 2011, 36, 713–748. [Google Scholar] [CrossRef]
- Husken, D.; Gaymans, A.R.J. The Tensile Properties of Poly(Ethylene Oxide)-Based Segmented Block Copolymers in the Dry and Wet State. J. Mater. Sci. 2009, 44, 2656–2664. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Mn | Mw | Đ |
|---|---|---|---|
| g × mol−1 | g × mol−1 | - | |
| E_PCUU3000_3 | 23,550 a | 2.008 × 106 a | 85.3 a |
| E_PCUU3000_2.5 | 56,290 a | 1.674 × 106 a | 29.7 a |
| E_PCUU3000_2 | 43,750 a | 2.665 × 106 a | 60.9 a |
| E_PCUU3000_1.5 | 64,590 a | 1.169 × 106 a | 18.1 a |
| E_OCD_5000 | 7610 b | 19,220 b | 2.5 b |
| E_OCD_3000 | 5950 b 2910 c | 13,850 b 3970 c | 2.3 b 1.4 c |
| Sample | Gf | Qm | HS a | σ | ε | EYoung |
|---|---|---|---|---|---|---|
| % | % | wt.% | MPa | % | MPa | |
| E_PCUU3000_3.5 | 45 ± 4 | 8430 ± 700 | 21 ± 1 | 28 ± 3 | 840 ± 40 | 67 ± 15 |
| E_PCUU3000_3 | 0 | - | 18 ± 1 | 24 ± 3 | 990 ± 35 | 91 ± 7 |
| E_PCUU3000_2.5 | 0 | - | 15 ± 1 | 23 ± 3 | 950 ± 20 | 108 ± 11 |
| E_PCUU3000_2 | 0 | - | 13 ± 1 | 27 ± 2 | 960 ± 40 | 112 ± 9 |
| E_PCUU3000_1.5 | 0 | - | 10 ± 1 | 34 ± 5 | 1070 ± 40 | 164 ± 58 |
| E_PCUU5000_3 | 61 ± 18 | 4480 ± 670 | 12 ± 1 | 23 ± 4 | 1100 ± 70 | 172 ± 30 |
| E_PCUU5000_2 | 51 ± 12 | 5840 ± 290 | 8 ± 1 | 22 ± 3 | 1020 ± 45 | 101 ± 22 |
| Sample | Tc | ΔHc | Tm | ΔHm |
|---|---|---|---|---|
| °C | J × g−1 | °C | J × g−1 | |
| E_PCUU3000_3.5 | −12 ± 1 | 13 ± 1 | 32 ± 1 | 12 ± 1 |
| E_PCUU3000_3 | −7 ± 1 | 19 ± 1 | 38 ± 1 | 18 ± 1 |
| E_PCUU3000_2.5 | −9 ± 1 | 16 ± 1 | 41 ± 1 | 21 ± 1 |
| E_PCUU3000_2 | −6 ± 1 | 20 ± 1 | 43 ± 1 | 22 ± 1 |
| E_PCUU3000_1.5 | 0 ± 1 | 24 ± 1 | 47 ± 1 | 25 ± 1 |
| E_PCUU5000_3 | 11 ± 1 | 30 ± 1 | 50 ± 1 | 30 ± 1 |
| E_PCUU5000_2 | 19 ± 1 | 35 ± 1 | 53 ± 1 | 62 ± 1 |
| E_OCD_3000 | 40 ± 1 | 77 ± 1 | 57 ± 1 | 76 ± 1 |
| E_OCD_5000 | 39 ± 1 | 79 ± 1 | 58 ± 1 | 78 ± 1 |
| E_BMC _8 | 42 ± 1 | 123 ± 1 | 63 ± 1 | 104 ± 1 |
| Sample | Tg tanδ | Tg E′ | Tg E″ | Tm |
|---|---|---|---|---|
| °C | °C | °C | °C | |
| E_PCUU3000_3.5 | −2 ± 1 | −29 ± 1 | −18 ± 1 | 51 ± 1 |
| E_PCUU3000_3 | −7 ± 1 | −33 ± 1 | −22 ± 1 | 51 ± 1 |
| E_PCUU3000_2.5 | −11 ± 1 | −31 ± 1 | −21 ± 1 | 49 ± 1 |
| E_PCUU3000_2 | −15 ± 1 | −33 ± 1 | −23 ± 1 | 51 ± 1 |
| E_PCUU3000_1.5 | −11 ± 1 | −32 ± 1 | −22 ± 1 | 57 ± 1 |
| E_PCUU5000_3 | −16 ± 1 | −31 ± 1 | −23 ± 1 | 51 ± 1 |
| E_PCUU5000_2 | −17 ± 1 | −29 ± 1 | −25 ± 1 | 50 ± 1 |
| Sample | Tg | Tm | ΔHm | |||||
|---|---|---|---|---|---|---|---|---|
| 1st Run | 1st Run * | 1st Run | 1st Run * | 1st Run | 2nd Run | 1st Run * | 2nd Run * | |
| °C | °C | J × g−1 | ||||||
| E_PCUU3000_3.5 | −27 ± 2 | −31 ± 2 | 42 ± 2 | 42 ± 2 | 17 ± 1 | 23 ± 1 | 25 ± 1 | 23 ± 1 |
| E_PCUU3000_3 | −30 ± 2 | −34 ± 2 | 43 ± 2 | 43 ± 2 | 25 ± 1 | 27 ± 1 | 40 ± 1 | 20 ± 1 |
| E_PCUU3000_2.5 | −30 ± 2 | −34 ± 2 | 46 ± 2 | 44 ± 2 | 44 ± 1 | 40 ± 1 | 45 ± 1 | 34 ± 1 |
| E_PCUU3000_2 | −37 ± 2 | −35 ± 2 | 44 ± 2 | 43 ± 2 | 40 ± 1 | 35 ± 1 | 48 ± 1 | 27 ± 1 |
| E_PCUU3000_1.5 | −32 ± 2 | −34 ± 2 | 47 ± 2 | 47 ± 2 | 46 ± 1 | 36 ± 1 | 37 ± 1 | 31 ± 1 |
| E_PCUU5000_3 | −33 ± 2 | −36 ± 2 | 55 ± 2 | 53 ± 2 | 70 ± 1 | 41 ± 1 | 60 ± 1 | 28 ± 1 |
| Sample | Rr | Rf |
|---|---|---|
| % | % | |
| E_PCUU3000_3.5 | 99.6 ± 0.2 | 94.5 ± 0.1 |
| E_PCUU3000_3 | 99.6 ± 0.1 | 95.8 ± 0.1 |
| E_PCUU3000_2.5 | 99.4 ± 0.4 | 98.8 ± 0.1 |
| E_PCUU3000_2 | 99.0 ± 0.6 | 98.2 ± 0.5 |
| E_PCUU3000_1.5 | nd | nd |
| E_PCUU5000_3 | 99.3 ± 0.0 * | 99.3 ± 0.6 * |
| E_PCUU5000_2 | 98.5 ± 0.7 * | 98.6 ± 0.3 * |
| Sample | Molar Ratio DMC:Diol | Mass Contribution Dimers: Longer Homologues |
|---|---|---|
| E_BMC_18 | 18:1 | 98:2 |
| E_BMC_10 | 10:1 | 95:5 |
| E_BMC_8 | 8:1 | 90:10 |
| S_BMC_18 | 18:1 | 93:7 |
| S_BMC_10 | 10:1 | 88:12 |
| S_BMC_8 | 8:1 | 84:16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rolińska, K.; Mazurek-Budzyńska, M.; Parzuchowski, P.G.; Wołosz, D.; Balk, M.; Gorący, K.; El Fray, M.; Polanowski, P.; Sikorski, A. Synthesis of Shape-Memory Polyurethanes: Combined Experimental and Simulation Studies. Int. J. Mol. Sci. 2022, 23, 7064. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137064
Rolińska K, Mazurek-Budzyńska M, Parzuchowski PG, Wołosz D, Balk M, Gorący K, El Fray M, Polanowski P, Sikorski A. Synthesis of Shape-Memory Polyurethanes: Combined Experimental and Simulation Studies. International Journal of Molecular Sciences. 2022; 23(13):7064. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137064
Chicago/Turabian StyleRolińska, Karolina, Magdalena Mazurek-Budzyńska, Paweł G. Parzuchowski, Dominik Wołosz, Maria Balk, Krzysztof Gorący, Miroslawa El Fray, Piotr Polanowski, and Andrzej Sikorski. 2022. "Synthesis of Shape-Memory Polyurethanes: Combined Experimental and Simulation Studies" International Journal of Molecular Sciences 23, no. 13: 7064. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137064