
Virus-Mediated Inhibition of Apoptosis in the Context of EBV-Associated Diseases: Molecular Mechanisms and Therapeutic Perspectives

 ,
, 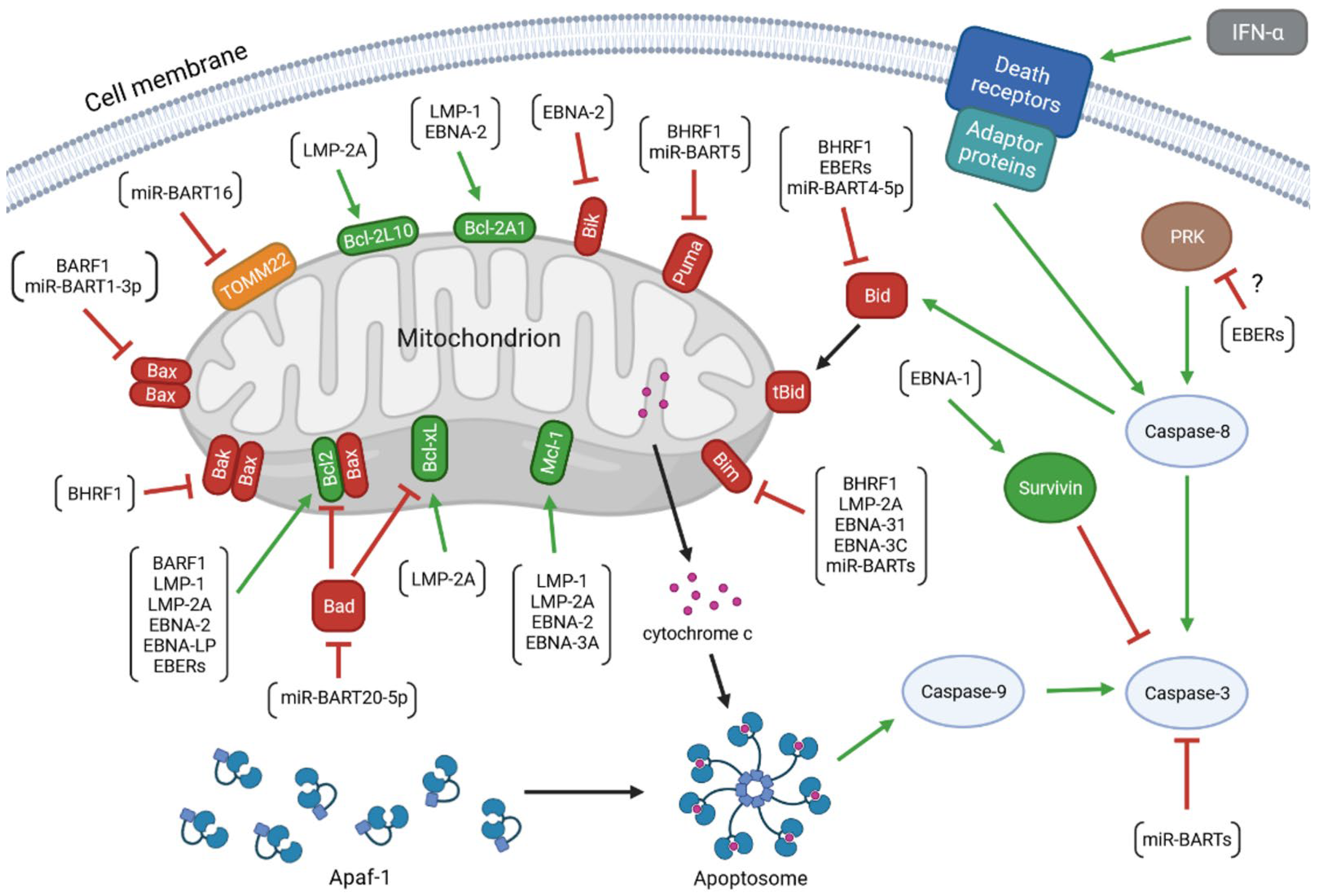{kind=link}
{kind=link}
Abstract
:1. Introduction
2. EBV and Its Pathogenicity
3. The Anti-Apoptotic Activity of EBV Latent Proteins in the Development of Virus-Driven Diseases
3.1. BHRF1
3.2. BARF1
3.3. LMP-2A
3.4. EBNA-3C
3.5. Other EBV Latent Proteins: LMP-1, EBNA-1, -2, -3A and LP
4. The Anti-Apoptotic Activity of EBV RNA Molecules in the Development of Virus-Driven Diseases
4.1. EBERs
4.2. miRNAs
4.2.1. miR-BARTs
4.2.2. miR-BHRF1s
5. EBV-Encoded Anti-Apoptotic Molecules as the Potential Targets for Anti-Viral Drugs: Therapeutic Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175. [Google Scholar] [CrossRef] [PubMed]
- Ibtisham, F.; Zhao, Y.; Nawab, A.; Liguang, H.; Wu, J.; Xiao, M.; Zhao, Z.; An, L. The Effect of High Temperature on Viability, Proliferation, Apoptosis and Anti-oxidant Status of Chicken Embryonic Fibroblast Cells. Braz. J. Poult. Sci. 2018, 20, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Kim, G.; Kim, D.; Yoo, J.; Kim, D.-K.; Kim, H. Numerical Study on Effective Conditions for the Induction of Apoptotic Temperatures for Various Tumor Aspect Ratios Using a Single Continuous-Wave Laser in Photothermal Therapy Using Gold Nanorods. Cancers 2019, 11, 764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanojevic-Pirkovic, M.; Andelkovic, M.; Lukovic, J.; Zelen, I.; Milosavljevic, Z.; Mitrovic, M.; Jurisic, V.; Nikolic, I. UV irradiation induces apoptosis in the human endometrial stromal cell line (ThESC). J. BUON 2020, 25, 1541–1546. [Google Scholar]
- Sample, A.; He, Y.Y. Autophagy in UV Damage Response. Photochem. Photobiol. 2017, 93, 943–955. [Google Scholar] [CrossRef]
- Ritchhart, C.; Joy, A. Reversal of drug-induced gingival overgrowth by UV-mediated apoptosis of gingival fibroblasts—An in vitro study. Ann. Anat. 2018, 217, 7–11. [Google Scholar] [CrossRef]
- Lim, J.R.; Lee, H.J.; Jung, Y.H.; Kim, J.S.; Chae, C.W.; Kim, S.Y.; Han, H.J. Ethanol-activated CaMKII signaling induces neuronal apoptosis through Drp1-mediated excessive mitochondrial fission and JNK1-dependent NLRP3 inflammasome activation. Cell Commun. Signal. 2020, 18, 123. [Google Scholar] [CrossRef]
- Li, C.; Li, J.; Xu, G.; Sun, H. Influence of Chronic Ethanol Consumption on Apoptosis and Autophagy Following Transient Focal Cerebral Ischemia in Male Mice. Sci. Rep. 2020, 10, 6164. [Google Scholar] [CrossRef] [Green Version]
- Bauer, C.; Stotter, C.; Jeyakumar, V.; Niculescu-Morzsa, E.; Simlinger, B.; Rodríguez Ripoll, M.; Klestil, T.; Franek, F.; Nehrer, S. Concentration-Dependent Effects of Cobalt and Chromium Ions on Osteoarthritic Chondrocytes. Cartilage 2019, 13, 908S–919S. [Google Scholar] [CrossRef] [Green Version]
- Genchi, G.; Sinicropi, M.S.; Lauria, G.; Carocci, A.; Catalano, A. The Effects of Cadmium Toxicity. Int. J. Environ. Res. Public Health 2020, 17, 3782. [Google Scholar] [CrossRef]
- Brokatzky, D.; Kretz, O.; Häcker, G. Apoptosis Functions in Defense against Infection of Mammalian Cells with Environmental Chlamydiae. Infect. Immun. 2020, 88, e00851-19. [Google Scholar] [CrossRef] [PubMed]
- Carloni, G.; Fioretti, D.; Rinaldi, M.; Ponzetto, A. Heterogeneity and coexistence of oncogenic mechanisms involved in HCV-associated B-cell lymphomas. Crit. Rev. Oncol. Hematol. 2019, 138, 156–171. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimmons, L.; Cartlidge, R.; Chang, C.; Sejic, N.; Galbraith, L.C.A.; Suraweera, C.D.; Croom-Carter, D.; Dewson, G.; Tierney, R.J.; Bell, A.I.; et al. EBV BCL-2 homologue BHRF1 drives chemoresistance and lymphomagenesis by inhibiting multiple cellular pro-apoptotic proteins. Cell Death Differ. 2020, 27, 1554–1568. [Google Scholar] [CrossRef] [PubMed]
- Jahan, S.; Khaliq, S.; Siddiqi, M.H.; Ijaz, B.; Ahmad, W.; Ashfaq, U.A.; Hassan, S. Anti-apoptotic effect of HCV core gene of genotype 3a in Huh-7 cell line. Virol. J. 2011, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Hösel, M.; Quasdorff, M.; Ringelhan, M.; Kashkar, H.; Debey-Pascher, S.; Sprinzl, M.F.; Bockmann, J.-H.; Arzberger, S.; Webb, D.; von Olshausen, G.; et al. Hepatitis B Virus Activates Signal Transducer and Activator of Transcription 3 Supporting Hepatocyte Survival and Virus Replication. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 339. [Google Scholar] [CrossRef] [PubMed]
- Ryerson, M.R.; Richards, M.M.; Kvansakul, M.; Hawkins, C.J.; Shisler, J.L. Vaccinia Virus Encodes a Novel Inhibitor of Apoptosis That Associates with the Apoptosome. J. Virol. 2017, 91, e01385-17. [Google Scholar] [CrossRef] [Green Version]
- Veyer, D.L.; Carrara, G.; Maluquer de Motes, C.; Smith, G.L. Vaccinia virus evasion of regulated cell death. Immunol. Lett. 2017, 186, 68–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Zhang, Y.J. Interference of apoptosis by hepatitis B virus. Viruses 2017, 9, 230. [Google Scholar] [CrossRef] [Green Version]
- Manners, O.; Murphy, J.C.; Coleman, A.; Hughes, D.J.; Whitehouse, A. Contribution of the KSHV and EBV lytic cycles to tumourigenesis. Curr. Opin. Virol. 2018, 32, 60–70. [Google Scholar] [CrossRef]
- Hawkins, J.B.; Delgado-Eckert, E.; Thorley-Lawson, D.A.; Shapiro, M. The Cycle of EBV Infection Explains Persistence, the Sizes of the Infected Cell Populations and Which Come under CTL Regulation. PLoS Pathog. 2013, 9, e1003685. [Google Scholar] [CrossRef]
- He, H.-P.; Luo, M.; Cao, Y.-L.; Lin, Y.-X.; Zhang, H.; Zhang, X.; Ou, J.-Y.; Yu, B.; Chen, X.; Xu, M.; et al. Structure of Epstein-Barr virus tegument protein complex BBRF2-BSRF1 reveals its potential role in viral envelopment. Nat. Commun. 2020, 11, 5405. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.P.; Chen, M.R. Conquering the nuclear envelope barriers by ebv lytic replication. Viruses 2021, 13, 702. [Google Scholar] [CrossRef] [PubMed]
- Zanella, L.; Riquelme, I.; Buchegger, K.; Abanto, M.; Ili, C.; Brebi, P. A reliable Epstein-Barr Virus classification based on phylogenomic and population analyses. Sci. Rep. 2019, 9, 9829. [Google Scholar] [CrossRef] [Green Version]
- Cai, Q.; Chen, K.; Young, K.H. Epstein–Barr virus-positive T/NK-cell lymphoproliferative disorders. Exp. Mol. Med. 2015, 47, e133. [Google Scholar] [CrossRef] [PubMed]
- Dugan, J.P.; Coleman, C.B.; Haverkos, B. Opportunities to Target the Life Cycle of Epstein-Barr Virus (EBV) in EBV-Associated Lymphoproliferative Disorders. Front. Oncol. 2019, 9, 127. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.; Robertson, E.S. Impact of EBV essential nuclear protein EBNA-3C on B-cell proliferation and apoptosis. Future Microbiol. 2013, 8, 323. [Google Scholar] [CrossRef] [Green Version]
- Caetano, B.F.R.; Jorge, B.A.S.; Müller-Coan, B.G.; Elgui de Oliveira, D. Epstein-Barr virus microRNAs in the pathogenesis of human cancers. Cancer Lett. 2021, 499, 14–23. [Google Scholar] [CrossRef]
- Graham, B.S.; Lynch, D.T. Cancer, Burkitt Lymphoma; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Hutcheson, R.L.; Chakravorty, A.; Sugden, B. Burkitt Lymphomas Evolve to Escape Dependencies on Epstein-Barr Virus. Front. Cell. Infect. Microbiol. 2021, 10, 606412. [Google Scholar] [CrossRef]
- Sinclair, A.J.; Moalwi, M.H.; Amoaten, T. Is ebv associated with breast cancer in specific geographic locations? Cancers 2021, 13, 819. [Google Scholar] [CrossRef]
- Stanland, L.J.; Luftig, M.A. The role of ebv-induced hypermethylation in gastric cancer tumorigenesis. Viruses 2020, 12, 1222. [Google Scholar] [CrossRef]
- Grewal, R.; Irimie, A.; Naidoo, N.; Mohamed, N.; Petrushev, B.; Chetty, M.; Tomuleasa, C.; Abayomi, E.A. Hodgkin’s lymphoma and its association with EBV and HIV infection. Crit. Rev. Clin. Lab. Sci. 2018, 55, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Knecht, H.; Mai, S. LMP1 and dynamic progressive telomere dysfunction: A major culprit in EBV-associated Hodgkin’s lymphoma. Viruses 2017, 9, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crombie, J.L.; LaCasce, A.S. Epstein Barr virus associated B-cell lymphomas and iatrogenic lymphoproliferative disorders. Front. Oncol. 2019, 9, 109. [Google Scholar] [CrossRef] [Green Version]
- Murthy, S.L.; Hitchcock, M.A.; Endicott-Yazdani, T.R.; Watson, J.T.; Krause, J.R. Epstein-Barr virus–positive diffuse large B-cell lymphoma. Proc. (Bayl. Univ. Med. Cent.) 2017, 30, 443. [Google Scholar] [CrossRef] [Green Version]
- Shannon-Lowe, C.; Rickinson, A.B.; Bell, A.I. Epstein-barr virus-associated lymphomas. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160271. [Google Scholar] [CrossRef] [PubMed]
- Becnel, D.; Abdelghani, R.; Nanbo, A.; Avilala, J.; Kahn, J.; Li, L.; Lin, Z. Pathogenic role of epstein–barr virus in lung cancers. Viruses 2021, 13, 877. [Google Scholar] [CrossRef]
- Hong, S.; Liu, D.; Luo, S.; Fang, W.; Zhan, J.; Fu, S.; Zhang, Y.; Wu, X.; Zhou, H.; Chen, X.; et al. The genomic landscape of Epstein-Barr virus-associated pulmonary lymphoepithelioma-like carcinoma. Nat. Commun. 2019, 10, 3108. [Google Scholar] [CrossRef] [Green Version]
- Kheir, F.; Zhao, M.; Strong, M.J.; Yu, Y.; Nanbo, A.; Flemington, E.K.; Morris, G.F.; Reiss, K.; Li, L.; Lin, Z. Detection of Epstein-Barr Virus Infection in Non-Small Cell Lung Cancer. Cancers 2019, 11, 759. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Jia, K.; Lv, H.; Wang, S.Q.; Wu, Y.; Lei, H.; Chen, X. EBV-Positive Gastric Cancer: Current Knowledge and Future Perspectives. Front. Oncol. 2020, 10, 2096. [Google Scholar] [CrossRef]
- Tavakoli, A.; Monavari, S.H.; Solaymani Mohammadi, F.; Kiani, S.J.; Armat, S.; Farahmand, M. Association between Epstein-Barr virus infection and gastric cancer: A systematic review and meta-analysis. BMC Cancer 2020, 20, 493. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Z.; Zeng, B.; Hu, G.; Gan, R. Epstein–Barr virus-associated gastric cancer: A distinct subtype. Cancer Lett. 2020, 495, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Tang, Y.; Wang, J.; Guo, C.; Wang, Y.; Xiang, B.; Zhou, M.; Li, X.; Wu, X.; Li, Y.; et al. The emerging role of Epstein-Barr virus encoded microRNAs in nasopharyngeal carcinoma. J. Cancer 2018, 9, 2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hau, P.M.; Lung, H.L.; Wu, M.; Tsang, C.M.; Wong, K.L.; Mak, N.K.; Lo, K.W. Targeting Epstein-Barr Virus in Nasopharyngeal Carcinoma. Front. Oncol. 2020, 10, 600. [Google Scholar] [CrossRef] [PubMed]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein–Barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef]
- Chen, Z.; Gao, L.; Wang, M.; Tang, Y.; Zhao, S.; Liu, W. Epstein-Barr virus positive peripheral T cell lymphoma with novel variants in STAT5B of a pediatric patient: A case report. BMC Cancer 2018, 18, 373. [Google Scholar] [CrossRef]
- Allen, P.B.; Lechowicz, M.J. Management of NK/T-Cell Lymphoma, Nasal Type. J. Oncol. Pract. 2019, 15, 513–520. [Google Scholar] [CrossRef]
- Tse, E.; Kwong, Y.L. The diagnosis and management of NK/T-cell lymphomas. J. Hematol. Oncol. 2017, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Camilleri-Broët, S.; Davi, F.; Feuillard, J.; Bourgeois, C.; Seilhean, D.; Hauw, J.J.; Raphaël, M. High Expression of Latent Membrane Protein 1 of Epstein-Barr Virus and BCL-2 Oncoprotein in Acquired Immunodeficiency Syndrome-Related Primary Brain Lymphomas. Blood 1995, 86, 432–435. [Google Scholar] [CrossRef] [Green Version]
- Buonavoglia, A.; Leone, P.; Prete, M.; Solimando, A.G.; Guastadisegno, C.; Lanave, G.; Camero, M.; Martella, V.; Muzio, L.L.; Racanelli, V. Epstein–Barr Virus in Salivary Samples from Systemic Lupus Erythematosus Patients with Oral Lesions. J. Clin. Med. 2021, 10, 4995. [Google Scholar] [CrossRef]
- Janz, S. Myc translocations in B cell and plasma cell neoplasms. DNA Repair 2006, 5, 1213–1224. [Google Scholar] [CrossRef]
- Hui, K.F.; Yeung, P.L.; Tam, K.P.; Chiang, A.K.S. Counteracting survival functions of EBNA3C in Epstein-Barr virus (EBV)-driven lymphoproliferative diseases by combination of SAHA and bortezomib. Oncotarget 2018, 9, 25101–25114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Barlow, E.A.; Ma, S.; Hagemeier, S.R.; Duellman, S.J.; Burgess, R.R.; Tellam, J.; Khanna, R.; Kenney, S.C. Hsp90 inhibitors block outgrowth of EBV-infected malignant cells in vitro and in vivo through an EBNA1-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 3146–3151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Nie, K.; Redmond, D.; Liu, Y.; Elemento, O.; Knowles, D.M.; Tam, W. EBV-miR-BHRF1-2 targets PRDM1/Blimp1: Potential role in EBV lymphomagenesis. Leukemia 2016, 30, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Fitzsimmons, L.; Boyce, A.J.; Wei, W.; Chang, C.; Croom-Carter, D.; Tierney, R.J.; Herold, M.J.; Bell, A.I.; Strasser, A.; Kelly, G.L.; et al. Coordinated repression of BIM and PUMA by Epstein-Barr virus latent genes maintains the survival of Burkitt lymphoma cells. Cell Death Differ. 2018, 25, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Gu, B.; Chen, X.; Wang, Y.; Li, P.; Wang, K. The Function and Therapeutic Potential of Epstein-Barr Virus-Encoded MicroRNAs in Cancer. Mol. Ther.-Nucleic Acids 2019, 17, 657–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poling, B.C.; Price, A.M.; Luftig, M.A.; Cullen, B.R. The Epstein-Barr Virus miR-BHRF1 microRNAs Regulate Viral Gene Expression in cis. Virology 2017, 512, 113. [Google Scholar] [CrossRef]
- De Re, V.; Caggiari, L.; De Zorzi, M.; Fanotto, V.; Miolo, G.; Puglisi, F.; Cannizzaro, R.; Canzonieri, V.; Steffan, A.; Farruggia, P.; et al. Epstein-Barr virus BART microRNAs in EBV- associated Hodgkin lymphoma and gastric cancer. Infect. Agents Cancer 2020, 15, 42. [Google Scholar] [CrossRef]
- Torres, K.; Landeros, N.; Wichmann, I.A.; Polakovicova, I.; Aguayo, F.; Corvalan, A.H. EBV miR-BARTs and human lncRNAs: Shifting the balance in competing endogenous RNA networks in EBV-associated gastric cancer. Biochim. Biophys. Acta-Mol. Basis Dis. 2021, 1867, 166049. [Google Scholar] [CrossRef]
- Lo, A.K.F.; Kwok, W.L.; Sai, W.T.; Hing, L.W.; Hui, J.W.Y.; Ka, F.T.; Hayward, S.D.; Yiu, L.C.; Yu, L.L.; Takada, K.; et al. Epstein-Barr Virus Infection Alters Cellular Signal Cascades in Human Nasopharyngeal Epithelial Cells. Neoplasia 2006, 8, 173. [Google Scholar] [CrossRef] [Green Version]
- Mckenzie, J.; El-Guindy, A. Epstein-Barr Virus Lytic Cycle Reactivation. Curr. Top. Microbiol. Immunol. 2015, 391, 237–261. [Google Scholar] [CrossRef]
- Murata, T.; Sugimoto, A.; Inagaki, T.; Yanagi, Y.; Watanabe, T.; Sato, Y.; Kimura, H. Molecular Basis of Epstein–Barr Virus Latency Establishment and Lytic Reactivation. Viruses 2021, 13, 2344. [Google Scholar] [CrossRef]
- Rosemarie, Q.; Sugden, B. Epstein-Barr Virus: How Its Lytic Phase Contributes to Oncogenesis. Microorganisms 2020, 8, 1824. [Google Scholar] [CrossRef]
- Song, H.; Lim, Y.; Im, H.; Bae, J.M.; Kang, G.H.; Ahn, J.; Baek, D.; Kim, T.Y.; Yoon, S.S.; Koh, Y. Interpretation of EBV infection in pan-cancer genome considering viral life cycle: LiEB (Life cycle of Epstein-Barr virus). Sci. Rep. 2019, 9, 3465. [Google Scholar] [CrossRef] [Green Version]
- Hoebe, E.; Wille, C.; Hagemeier, S.; Kenney, S.; Greijer, A.; Middeldorp, J. Epstein–Barr Virus Gene BARF1 Expression is Regulated by the Epithelial Differentiation Factor ΔNp63α in Undifferentiated Nasopharyngeal Carcinoma. Cancers 2018, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Novalic, Z.; van Rossen, T.M. Agents and Approaches for Lytic Induction Therapy of Epstein-Barr Virus Associated Malignancies. Med. Chem. 2016, 6, 449–466. [Google Scholar] [CrossRef]
- Chen, L.W.; Hung, C.H.; Wang, S.S.; Yen, J.B.; Liu, A.C.; Hung, Y.H.; Chang, P.J. Expression and regulation of the BKRF2, BKRF3 and BKRF4 genes of Epstein-Barr virus. Virus Res. 2018, 256, 76–89. [Google Scholar] [CrossRef]
- Oudejans, J.J.; Van Den Brule, A.J.C.; Jiwa, N.M.; De Bruin, P.C.; Ossenkoppele, G.J.; Van Der Valk, P.; Walboomers, J.M.M.; Meijer, C.J.L.M. BHRF1, the Epstein-Barr Virus (EBV) Homologue of the BCL-2 Proto-oncogene, Is Transcribed in EBV-Associated B-Cell Lymphomas and in Reactive Lymphocytes. Blood 1995, 86, 1893–1902. [Google Scholar] [CrossRef] [Green Version]
- Lo, A.K.F.; Dawson, C.W.; Lung, H.L.; Wong, K.L.; Young, L.S. The Therapeutic Potential of Targeting BARF1 in EBV-Associated Malignancies. Cancers 2020, 12, 1940. [Google Scholar] [CrossRef]
- Germini, D.; Sall, F.B.; Shmakova, A.; Wiels, J.; Dokudovskaya, S.; Drouet, E.; Vassetzky, Y. Oncogenic Properties of the EBV ZEBRA Protein. Cancers 2020, 12, 1479. [Google Scholar] [CrossRef]
- Vilmen, G.; Glon, D.; Siracusano, G.; Lussignol, M.; Shao, Z.; Hernandez, E.; Perdiz, D.; Quignon, F.; Mouna, L.; Poüs, C.; et al. BHRF1, a BCL2 viral homolog, disturbs mitochondrial dynamics and stimulates mitophagy to dampen type I IFN induction. Autophagy 2021, 17, 1296. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2017, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Opferman, J.T.; Kothari, A. Anti-apoptotic BCL-2 family members in development. Cell Death Differ. 2018, 25, 37. [Google Scholar] [CrossRef]
- Andreu-Fernández, V.; Sancho, M.; Genovés, A.; Lucendo, E.; Todt, F.; Lauterwasser, J.; Funk, K.; Jahreis, G.; Pérez-Payá, E.; Mingarro, I.; et al. Bax transmembrane domain interacts with prosurvival Bcl-2 proteins in biological membranes. Proc. Natl. Acad. Sci. USA 2017, 114, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, J.M.; Riemer, J. Apoptosis inducing factor and mitochondrial NADH dehydrogenases: Redox-controlled gear boxes to switch between mitochondrial biogenesis and cell death. Biol. Chem. 2021, 402, 289–297. [Google Scholar] [CrossRef]
- Benítez-Guzmán, A.; Arriaga-Pizano, L.; Morán, J.; Gutiérrez-Pabello, J.A. Endonuclease G takes part in AIF-mediated caspase-independent apoptosis in Mycobacterium bovis-infected bovine macrophages. Vet. Res. 2018, 49, 69. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.Y.; Wang, X.Y.; Wei, Q.Y.; Xu, Y.M.; Lau, A.T.Y. Potency and Selectivity of SMAC/DIABLO Mimetics in Solid Tumor Therapy. Cells 2020, 9, 1012. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Che, K.; Zhao, Z.; Liu, S.; Xing, X.; Luo, B. Sequence analysis of Epstein-Barr virus (EBV) early genes BARF1 and BHRF1 in NK/T cell lymphoma from Northern China. Virol. J. 2015, 12, 135. [Google Scholar] [CrossRef] [Green Version]
- Desbien, A.L.; Kappler, J.W.; Marrack, P. The Epstein-Barr virus Bcl-2 homolog, BHRF1, blocks apoptosis by binding to a limited amount of Bim. Proc. Natl. Acad. Sci. USA 2009, 106, 5663–5668. [Google Scholar] [CrossRef] [Green Version]
- Kvansakul, M.; Wei, A.H.; Fletcher, J.I.; Willis, S.N.; Chen, L.; Roberts, A.W.; Huang, D.C.S.; Colman, P.M. Structural basis for apoptosis inhibition by Epstein-Barr virus bhrf1. PLoS Pathog. 2010, 6, e1001236. [Google Scholar] [CrossRef] [Green Version]
- Fitzsimmons, L.; Kelly, G.L. EBV and apoptosis: The viral master regulator of cell fate? Viruses 2017, 9, 339. [Google Scholar] [CrossRef] [Green Version]
- Chiang, A.K.; Tao, Q.; Srivastava, G.; Ho, F.C. Nasal NK- and T-cell lymphomas share the same type of Epstein-Barr virus latency as nasopharyngeal carcinoma and Hodgkin’s disease. Int. J. Cancer 1996, 68, 285–290. [Google Scholar] [CrossRef]
- Hayes, D.P.; Brink, A.A.T.P.; Vervoort, M.B.H.J.; Middeldorp, J.M.; Meijer, C.J.L.M.; Van Den Brule, A.J.C. Expression of Epstein-Barr virus (EBV) transcripts encoding homologues to important human proteins in diverse EBV associated diseases. J. Clin. Pathol.-Mol. Pathol. 1999, 52, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernhardt, K.; Haar, J.; Tsai, M.H.; Poirey, R.; Feederle, R.; Delecluse, H.J. A Viral microRNA Cluster Regulates the Expression of PTEN, p27 and of a bcl-2 Homolog. PLoS Pathog. 2016, 12, e1005405. [Google Scholar] [CrossRef]
- Kawanishi, M.; Tada-Oikawa, S.; Kawanishi, S. Epstein-Barr virus BHRF1 functions downstream of Bid cleavage and upstream of mitochondrial dysfunction to inhibit TRAIL-induced apoptosis in BJAB cells. Biochem. Biophys. Res. Commun. 2002, 297, 682–687. [Google Scholar] [CrossRef]
- Kelly, G.L.; Long, H.M.; Stylianou, J.; Thomas, W.A.; Leese, A.; Bell, A.I.; Bornkamm, G.W.; Mautner, J.; Rickinson, A.B.; Rowe, M. An Epstein-Barr Virus Anti-Apoptotic Protein Constitutively Expressed in Transformed Cells and Implicated in Burkitt Lymphomagenesis: The Wp/BHRF1 Link. PLoS Pathog. 2009, 5, e1000341. [Google Scholar] [CrossRef]
- McCarthy, N.J.; Hazlewood, S.A.; Huen, D.S.; Rickinson, A.B.; Williams, G.T. The Epstein-Barr virus gene BHRF1, a homologue of the cellular oncogene bcl-2, inhibits apoptosis induced by gamma radiation and chemotherapeutic drugs. Adv. Exp. Med. Biol. 1996, 406, 83–97. [Google Scholar] [CrossRef]
- Song, S.; Jiang, Z.; Spezia-Lindner, D.E.; Liang, T.; Xu, C.; Wang, H.; Tian, Y.; Bai, Y. BHRF1 Enhances EBV Mediated Nasopharyngeal Carcinoma Tumorigenesis through Modulating Mitophagy Associated with Mitochondrial Membrane Permeabilization Transition. Cells 2020, 9, 1158. [Google Scholar] [CrossRef]
- Sheng, W.; Decaussin, G.; Sumner, S.; Ooka, T. N-terminal domain of BARF1 gene encoded by Epstein-Barr virus is essential for malignant transformation of rodent fibroblasts and activation of BCL-2. Oncogene 2001, 20, 1176–1185. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; Decaussin, G.; Ligout, A.; Takada, K.; Ooka, T. Malignant transformation of Epstein-Barr virus-negative Akata cells by introduction of the BARF1 gene carried by Epstein-Barr virus. J. Virol. 2003, 77, 3859–3865. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Tsao, S.W.; Ooka, T.; Nicholls, J.M.; Cheung, H.W.; Fu, S.; Wong, Y.C.; Wang, X. Anti-apoptotic role of BARF1 in gastric cancer cells. Cancer Lett. 2006, 238, 90–103. [Google Scholar] [CrossRef]
- Casao, A.; Mata-Campuzano, M.; Ordás, L.; Cebrián-Pérez, J.A.; Muiño-Blanco, T.; Martínez-Pastor, F. Cleaved PARP-1, an Apoptotic Marker, can be Detected in Ram Spermatozoa. Reprod. Domest. Anim. 2015, 50, 688–691. [Google Scholar] [CrossRef]
- Mashimo, M.; Onishi, M.; Uno, A.; Tanimichi, A.; Nobeyama, A.; Mori, M.; Yamada, S.; Negi, S.; Bu, X.; Kato, J.; et al. The 89-kDa PARP1 cleavage fragment serves as a cytoplasmic PAR carrier to induce AIF-mediated apoptosis. J. Biol. Chem. 2021, 296, 100046. [Google Scholar] [CrossRef]
- Cen, O.; Longnecker, R. Latent Membrane Protein 2 (LMP2). Curr. Top. Microbiol. Immunol. 2015, 391, 151–180. [Google Scholar] [CrossRef]
- Ma, S.-D.; Tsai, M.-H.; Romero-Masters, J.C.; Ranheim, E.A.; Huebner, S.M.; Bristol, J.A.; Delecluse, H.-J.; Kenney, S.C. Latent Membrane Protein 1 (LMP1) and LMP2A Collaborate To Promote Epstein-Barr Virus-Induced B Cell Lymphomas in a Cord Blood-Humanized Mouse Model but Are Not Essential. J. Virol. 2017, 91, 1928–1944. [Google Scholar] [CrossRef] [Green Version]
- Vrzalikova, K.; Ibrahim, M.; Nagy, E.; Vockerodt, M.; Perry, T.; Wei, W.; Woodman, C.; Murray, P. Co-Expression of the Epstein-Barr Virus-Encoded Latent Membrane Proteins and the Pathogenesis of Classic Hodgkin Lymphoma. Cancers 2018, 10, 285. [Google Scholar] [CrossRef] [Green Version]
- Casola, S.; Otipoby, K.L.; Alimzhanov, M.; Humme, S.; Uyttersprot, N.; Kutok, J.L.; Carroll, M.C.; Rajewsky, K. B cell receptor signal strength determines B cell fate. Nat. Immunol. 2004, 5, 317–327. [Google Scholar] [CrossRef]
- Mancao, C.; Altmann, M.; Jungnickel, B.; Hammerschmidt, W. Rescue of “crippled” germinal center B cells from apoptosis by Epstein-Barr virus. Blood 2005, 106, 4339. [Google Scholar] [CrossRef]
- Mancao, C.; Hammerschmidt, W. Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival. Blood 2007, 110, 3715–3721. [Google Scholar] [CrossRef] [Green Version]
- Portis, T.; Longnecker, R. Epstein-Barr virus (EBV) LMP2A mediates B-lymphocyte survival through constitutive activation of the Ras/PI3K/Akt pathway. Oncogene 2004, 23, 8619–8628. [Google Scholar] [CrossRef] [Green Version]
- Minamitani, T.; Yasui, T.; Ma, Y.; Zhou, H.; Okuzaki, D.; Tsai, C.Y.; Sakakibara, S.; Gewurz, B.E.; Kieff, E.; Kikutani, H. Evasion of affinity-based selection in germinal centers by Epstein-Barr virus LMP2A. Proc. Natl. Acad. Sci. USA 2015, 112, 11612–11617. [Google Scholar] [CrossRef] [Green Version]
- Fish, K.; Comoglio, F.; Shaffer, A.L.; Ji, Y.; Pan, K.T.; Scheich, S.; Oellerich, A.; Doebele, C.; Ikeda, M.; Schaller, S.J.; et al. Rewiring of B cell receptor signaling by Epstein-Barr virus LMP2A. Proc. Natl. Acad. Sci. USA 2020, 117, 26318–26327. [Google Scholar] [CrossRef]
- El-Sharkawy, A.; Al Zaidan, L.; Malki, A. Epstein–Barr Virus-Associated Malignancies: Roles of Viral Oncoproteins in Carcinogenesis. Front. Oncol. 2018, 8, 265. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Post, C.B. Insights into the allosteric regulation of Syk association with receptor ITAM, a multi-state equilibrium. Phys. Chem. Chem. Phys. 2016, 18, 5807–5818. [Google Scholar] [CrossRef] [Green Version]
- Engels, N.; Yigit, G.; Emmerich, C.H.; Czesnik, D.; Schild, D.; Wienands, J. Epstein-Barr virus LMP2A signaling in statu nascendi mimics a B cell antigen receptor-like activation signal. Cell Commun. Signal. 2012, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Lin, W.H.; Chen, S.Y.; Longnecker, R.; Tsai, S.C.; Chen, C.L.; Tsai, C.H. Syk Tyrosine Kinase Mediates Epstein-Barr Virus Latent Membrane Protein 2A-induced Cell Migration in Epithelial Cells. J. Biol. Chem. 2006, 281, 8806–8814. [Google Scholar] [CrossRef] [Green Version]
- Mielcarska, M.B.; Bossowska-Nowicka, M.; Gregorczyk-Zboroch, K.P.; Wyżewski, Z.; Szulc-Dąbrowska, L.; Gieryńska, M.; Toka, F.N. Syk and Hrs Regulate TLR3-Mediated Antiviral Response in Murine Astrocytes. Oxidative Med. Cell. Longev. 2019, 2019, 6927380. [Google Scholar] [CrossRef] [Green Version]
- Tomaszewski-Flick, M.J.; Rowe, D.T. Minimal protein domain requirements for the intracellular localization and self-aggregation of Epstein-Barr Virus Latent Membrane Protein 2. Virus Genes 2007, 35, 225–234. [Google Scholar] [CrossRef]
- Gobessi, S.; Laurenti, L.; Longo, P.G.; Carsetti, L.; Berno, V.; Sica, S.; Leone, G.; Efremov, D.G. Inhibition of constitutive and BCR-induced Syk activation downregulates Mcl-1 and induces apoptosis in chronic lymphocytic leukemia B cells. Leukemia 2009, 23, 686–697. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Ma, J.; Sun, Y.; Tang, M.; Kong, L. Effect and mechanism of PI3K/AKT/mTOR signaling pathway in the apoptosis of GC-1 cells induced by nickel nanoparticles. Chemosphere 2020, 255, 126913. [Google Scholar] [CrossRef]
- Wang, C.; Jin, A.; Huang, W.; Ling Tsang, L.; Cai, Z.; Zhou, X.; Chen, H.; Chan, H.C. Up-regulation of Bcl-2 by CD147 Through ERK Activation Results in Abnormal Cell Survival in Human Endometriosis. J. Clin. Endocrinol. Metab. 2015, 100, E955–E963. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Qi, Z.; Zhao, J.; Liao, M.; Wen, S.; Manyi, Y. Phosphorylation of Bcl-2 plays an important role in glycochenodeoxycholate-induced survival and chemoresistance in HCC. Oncol. Rep. 2017, 38, 1742–1750. [Google Scholar] [CrossRef] [Green Version]
- Ahn, I.E.; Brown, J.R. Targeting Bruton’s Tyrosine Kinase in CLL. Front. Immunol. 2021, 12, 2137. [Google Scholar] [CrossRef]
- Capello, D.; Gaidano, G. Post-Transplant Lymphoproliferative Disorders: Role of Viral Infection, Genetic Lesions And Antigen Stimulation In The Pathogenesis Of The Disease. Mediterr. J. Hematol. Infect. Dis. 2009, 1, e2009018. [Google Scholar] [CrossRef]
- Merchant, M.; Longnecker, R. LMP2A Survival and Developmental Signals Are Transmitted through Btk-Dependent and Btk-Independent Pathways. Virology 2001, 291, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.S.; Teutsch, M.; Dong, Z.; Wortis, H.H. An essential role for Bruton’s [corrected] tyrosine kinase in the regulation of B-cell apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93, 10966–10971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef]
- Pfeiffer, A.; Schneider, J.; Bueno, D.; Dolga, A.; Voss, T.D.; Lewerenz, J.; Wüllner, V.; Methner, A. Bcl-x L knockout attenuates mitochondrial respiration and causes oxidative stress that is compensated by pentose phosphate pathway activity. Free Radic. Biol. Med. 2017, 112, 350–359. [Google Scholar] [CrossRef]
- Solvason, N.; Wu, W.W.; Kabra, N.; Lund-Johansen, F.; Roncarolo, M.G.; Behrens, T.W.; Grillot, D.A.M.; Nunez, G.; Lees, E.; Howard, M. Transgene Expression of bcl-xL Permits Anti-immunoglobulin (Ig)–induced Proliferation in xid B Cells. J. Exp. Med. 1998, 187, 1081. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.E.; Vigorito, E.; McAdam, S.; Reynolds, H.M.; Caraux, A.; Colucci, F.; Turner, M. PLCgamma2 regulates Bcl-2 levels and is required for survival rather than differentiation of marginal zone and follicular B cells. Eur. J. Immunol. 2004, 34, 2237–2247. [Google Scholar] [CrossRef]
- He, Z.; O’Neal, J.; Wilson, W.C.; Mahajan, N.; Luo, J.; Wang, Y.; Su, M.Y.; Lu, L.; Skeath, J.B.; Bhattacharya, D.; et al. Deletion of Rb1 induces both hyperproliferation and cell death in murine germinal center B cells. Exp. Hematol. 2016, 44, 161–165.e4. [Google Scholar] [CrossRef] [Green Version]
- Grande, B.M.; Gerhard, D.S.; Jiang, A.; Griner, N.B.; Abramson, J.S.; Alexander, T.B.; Allen, H.; Ayers, L.W.; Bethony, J.M.; Bhatia, K.; et al. Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood 2019, 133, 1313–1324. [Google Scholar] [CrossRef]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef]
- Murer, A.; McHugh, D.; Caduff, N.; Kalchschmidt, J.; Barros, M.; Zbinden, A.; Capaul, R.; Niedobitek, G.; Allday, M.; Chijioke, O.; et al. EBV persistence without its EBNA3A and 3C oncogenes in vivo. PLoS Pathog. 2018, 14, e1007039. [Google Scholar] [CrossRef]
- Jha, H.C.; Shukla, S.K.; Lu, J.; Aj, M.P.; Banerjee, S.; Robertson, E.S. Dissecting the contribution of EBNA3C domains important for EBV-induced B-cell growth and proliferation. Oncotarget 2015, 6, 30115–30129. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Uppal, T.; Strahan, R.; Dabral, P.; Verma, S.C. The modulation of apoptotic pathways by gammaherpesviruses. Front. Microbiol. 2016, 7, 585. [Google Scholar] [CrossRef]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Feroz, W.; Sheikh, A.M.A. Exploring the multiple roles of guardian of the genome: P53. Egypt. J. Med. Hum. Genet. 2020, 21, 49. [Google Scholar] [CrossRef]
- Gordon, E.M.; Ravicz, J.R.; Liu, S.; Chawla, S.P.; Hall, F.L. Cell cycle checkpoint control: The cyclin G1/Mdm2/p53 axis emerges as a strategic target for broad-spectrum cancer gene therapy-A review of molecular mechanisms for oncologists. Mol. Clin. Oncol. 2018, 9, 115–134. [Google Scholar] [CrossRef] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Lee, S.; Park, J.Y.; Lee, S.Y.; Kim, H.S. Induction of p53-Dependent Apoptosis by Prostaglandin A2. Biomolecules 2020, 10, 492. [Google Scholar] [CrossRef] [Green Version]
- Ménard, M.; Costechareyre, C.; Ichim, G.; Blachier, J.; Neves, D.; Jarrosson-Wuilleme, L.; Depping, R.; Koster, J.; Saintigny, P.; Mehlen, P.; et al. Hey1- and p53-dependent TrkC proapoptotic activity controls neuroblastoma growth. PLoS Biol. 2018, 16, e2002912. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Shang, Q.; Zhang, D.; Guo, C.; Lin, Q.; Guo, Z.; Deng, C. Potential synergism of Bim with p53 in mice with Myc-induced lymphoma in a mouse lymphoma model. Mol. Med. Rep. 2012, 5, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Chen, G.; Yun, J.-P.; Lai, P. Association of p53 with Bid Induces Cell Death in Response to Etoposide Treatment in Hepatocellular Carcinoma. Curr. Cancer Drug Targets 2009, 9, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct Activation of Bax by p53 Mediates Mitochondrial Membrane Permeabilization and Apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2017, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Leu, J.I.J.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a Bak–Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef]
- Wang, J.; Guo, W.; Zhou, H.; Luo, N.; Nie, C.; Zhao, X.; Yuan, Z.; Liu, X.; Wei, Y.; Wang, J.; et al. Mitochondrial p53 phosphorylation induces Bak-mediated and caspase-independent cell death. Oncotarget 2015, 6, 17192–17205. [Google Scholar] [CrossRef] [Green Version]
- Yi, F.; Saha, A.; Murakami, M.; Kumar, P.; Knight, J.S.; Cai, Q.; Choudhuri, T.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C targets p53 and modulates its transcriptional and apoptotic activities. Virology 2009, 388, 236. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, B.G.; Murakami, M.; Cai, Q.; Verma, S.C.; Lan, K.; Robertson, E.S. Epstein-Barr Virus Nuclear Antigen 3C Interacts with and Enhances the Stability of the c-Myc Oncoprotein. J. Virol. 2008, 82, 4082. [Google Scholar] [CrossRef] [Green Version]
- Polager, S.; Ginsberg, D. p53 and E2f: Partners in life and death. Nat. Rev. Cancer 2009, 9, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zheng, S.; Yu, Q. The E2F family and the role of E2F1 in apoptosis. Int. J. Biochem. Cell Biol. 2009, 41, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Lu, J.; Morizur, L.; Upadhyay, S.K.; AJ, M.P.; Robertson, E.S. E2F1 Mediated Apoptosis Induced by the DNA Damage Response Is Blocked by EBV Nuclear Antigen 3C in Lymphoblastoid Cells. PLoS Pathog. 2012, 8, e1002573. [Google Scholar] [CrossRef] [Green Version]
- Bell, L.A.; Ryan, K.M. Life and death decisions by E2F-1. Cell Death Differ. 2004, 11, 137–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, J.K.; Fredersdorf, S.; Kouzarides, T.; Martin, K.; Lu, X. E2F1-induced apoptosis requires DNA binding but not transactivation and is inhibited by the retinoblastoma protein through direct interaction. Genes Dev. 1997, 11, 1840–1852. [Google Scholar] [CrossRef] [Green Version]
- Nikitin, P.A.; Yan, C.M.; Forte, E.; Bocedi, A.; Tourigny, J.P.; White, R.E.; Allday, M.J.; Patel, A.; Dave, S.S.; Kim, W.; et al. An ATM/Chk2-mediated DNA damage responsive signaling pathwaysuppresses Epstein-Barr virus transformation of primary human Bcells. Cell Host Microbe 2010, 8, 510. [Google Scholar] [CrossRef] [Green Version]
- Molina-Privado, I.; Rodríguez-Martínez, M.; Rebollo, P.; Martín-Pérez, D.; Artiga, M.J.; Menárguez, J.; Flemington, E.K.; Piris, M.A.; Campanero, M.R. E2F1 expression is deregulated and plays an oncogenic role in sporadic Burkitt’s lymphoma. Cancer Res. 2009, 69, 4052. [Google Scholar] [CrossRef] [Green Version]
- Price, A.M.; Dai, J.; Bazot, Q.; Patel, L.; Nikitin, P.A.; Djavadian, R.; Winter, P.S.; Salinas, C.A.; Barry, A.P.; Wood, K.C.; et al. Epstein-Barr virus ensures B cell survival by uniquely modulating apoptosis at early and late times after infection. eLife 2017, 6, e22509. [Google Scholar] [CrossRef]
- Romero-Masters, J.C.; Ohashi, M.; Djavadian, R.; Eichelberg, M.R.; Hayes, M.; Bristol, J.A.; Ma, S.; Ranheim, E.A.; Gumperz, J.; Johannsen, E.C.; et al. An EBNA3C-deleted Epstein-Barr virus (EBV) mutant causes B-cell lymphomas with delayed onset in a cord blood-humanized mouse model. PLoS Pathog. 2018, 14, e1007221. [Google Scholar] [CrossRef] [Green Version]
- Egle, A.; Harris, A.W.; Bouillet, P.; Cory, S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 6164–6169. [Google Scholar] [CrossRef] [Green Version]
- Richter-Larrea, J.A.; Robles, E.F.; Fresquet, V.; Beltran, E.; Rullan, A.J.; Agirre, X.; Calasanz, M.J.; Panizo, C.; Richter, J.A.; Hernandez, J.M.; et al. Reversion of epigenetically mediated BIM silencing overcomes chemoresistance in Burkitt lymphoma. Blood 2010, 116, 2531–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderton, E.; Yee, J.; Smith, P.; Crook, T.; White, R.E.; Allday, M.J. Two Epstein–Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: Clues to the pathogenesis of Burkitt’s lymphoma. Oncogene 2007, 27, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschos, K.; Smith, P.; Anderton, E.; Middeldorp, J.M.; White, R.E. Epstein-Barr Virus Latency in B Cells Leads to Epigenetic Repression and CpG Methylation of the Tumour Suppressor Gene Bim. PLoS Pathog 2009, 5, 1000492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, H.C.; Lu, J.; Saha, A.; Cai, Q.; Banerjee, S.; Prasad, M.A.J.; Robertson, E.S. EBNA3C-Mediated Regulation of Aurora Kinase B Contributes to Epstein-Barr Virus-Induced B-Cell Proliferation through Modulation of the Activities of the Retinoblastoma Protein and Apoptotic Caspases. J. Virol. 2013, 87, 12121–12138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldenson, B.; Crispino, J.D. The aurora kinases in cell cycle and leukemia. Oncogene 2014, 34, 537–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Lu, J.; Cai, Q.; Sun, Z.; Jha, H.C.; Robertson, E.S. EBNA3C Augments Pim-1 Mediated Phosphorylation and Degradation of p21 to Promote B-Cell Proliferation. PLoS Pathog. 2014, 10, e1004304. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S.; Bose, P.; Patel, K.; Roy, S.G.; Gain, C.; Gowda, H.; Robertson, E.S.; Saha, A. Transcriptional and epigenetic modulation of autophagy promotes EBV oncoprotein EBNA3C induced B-cell survival. Cell Death Dis. 2018, 9, 605. [Google Scholar] [CrossRef]
- Zhang, R.; Varela, M.; Forn-Cuní, G.; Torraca, V.; van der Vaart, M.; Meijer, A.H. Deficiency in the autophagy modulator Dram1 exacerbates pyroptotic cell death of Mycobacteria-infected macrophages. Cell Death Dis. 2020, 11, 277. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Zhu, Z.; Wu, J.; She, H.; Han, R.; Xu, H.; Qin, Z.H. DRAM1 regulates autophagy and cell proliferation via inhibition of the phosphoinositide 3-kinase-Akt-mTOR-ribosomal protein S6 pathway. Cell Commun. Signal. 2019, 17, 28. [Google Scholar] [CrossRef] [Green Version]
- Harrison, B.; Kraus, M.; Burch, L.; Stevens, C.; Craig, A.; Gordon-Weeks, P.; Hupp, T.R. DAPK-1 binding to a linear peptide motif in MAP1B stimulates autophagy and membrane blebbing. J. Biol. Chem. 2008, 283, 9999–10014. [Google Scholar] [CrossRef] [Green Version]
- Nowosad, A.; Besson, A. CDKN1B/p27 regulates autophagy via the control of Ragulator and MTOR activity in amino acid-deprived cells. Autophagy 2020, 16, 2297–2298. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, B.; Rowe, M.; Walls, D. The bfl-1 gene is transcriptionally upregulated by the Epstein-Barr virus LMP1, and its expression promotes the survival of a Burkitt’s lymphoma cell line. J. Virol. 2000, 74, 6652–6658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Q.; He, C.; Mao, Z.R. Epstein-Barr virus interactions with the Bcl-2 protein family and apoptosis in human tumor cells. J. Zhejiang Univ. Sci. B 2013, 14, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes-González, A.M.; Contreras-Paredes, A.; Manzo-Merino, J.; Lizano, M. The modulation of apoptosis by oncogenic viruses. Virol. J. 2013, 10, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, Y.; Wong, J.H.Y.; Robertson, E.S. Targeted Therapies for Epstein-Barr Virus-Associated Lymphomas. Cancers 2020, 12, 2565. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.-C.; Hsieh, S.-S.; Hsu, W.-L.; Chen, Y.-F.; Ho, H.-H.; Sheu, L.-F. Expression of Epstein-Barr nuclear antigen 1 in gastric carcinoma cells is associated with enhanced tumorigenicity and reduced cisplatin sensitivity. Int. J. Oncol. 2010, 36, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Holowaty, M.N.; Zeghouf, M.; Wu, H.; Tellam, J.; Athanasopoulos, V.; Greenblatt, J.; Frappier, L. Protein profiling with Epstein-Barr nuclear antigen-1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7. J. Biol. Chem. 2003, 278, 29987–29994. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Murakami, M.; Verma, S.C.; Cai, Q.; Haldar, S.; Kaul, R.; Wasik, M.A.; Middeldorp, J.; Robertson, E.S. Epstein-Barr Virus nuclear antigen 1 (EBNA1) confers resistance to apoptosis in EBV-positive B-lymphoma cells through up-regulation of survivin. Virology 2011, 410, 64–75. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhou, Y.; Wang, J.; Wang, J.; Yang, J.; Zhai, Y.; Li, B. Dual silencing of Bcl-2 and Survivin by HSV-1 vector shows better antitumor efficacy in higher PKR phosphorylation tumor cells in vitro and in vivo. Cancer Gene Ther. 2015, 22, 380–386. [Google Scholar] [CrossRef]
- Frappier, L. Contributions of Epstein–Barr Nuclear Antigen 1 (EBNA1) to Cell Immortalization and Survival. Viruses 2012, 4, 1537. [Google Scholar] [CrossRef] [Green Version]
- Pegman, P.M.; Smith, S.M.; D’Souza, B.N.; Loughran, S.T.; Maier, S.; Kempkes, B.; Cahill, P.A.; Simmons, M.J.; Gélinas, C.; Walls, D. Epstein-Barr Virus Nuclear Antigen 2 trans -Activates the Cellular Antiapoptotic bfl-1 Gene by a CBF1/RBPJκ-Dependent Pathway. J. Virol. 2006, 80, 8133–8144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohlhof, H.; Hampel, F.; Hoffmann, R.; Burtscher, H.; Weidle, U.H.; Hölzel, M.; Eick, D.; Zimber-Strobl, U.; Strobl, L.J. Notch1, Notch2, and Epstein-Barr virus–encoded nuclear antigen 2 signaling differentially affects proliferation and survival of Epstein-Barr virus–infected B cells. Blood 2009, 113, 5506–5515. [Google Scholar] [CrossRef] [PubMed]
- Campion, E.M.; Hakimjavadi, R.; Loughran, S.T.; Phelan, S.; Smith, S.M.; D’Souza, B.N.; Tierney, R.J.; Bell, A.I.; Cahill, P.A.; Walls, D. Repression of the Proapoptotic Cellular BIK/NBK Gene by Epstein-Barr Virus Antagonizes Transforming Growth Factor β1-Induced B-Cell Apoptosis. J. Virol. 2014, 88, 5001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Lee, K.H.; Weidner, M.; Osborne, B.A.; Hayward, S.D. Epstein-Barr virus EBNA2 blocks Nur77-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2002, 99, 11878–11883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Lee, K.-H.; Farrell, C.J.; Ling, P.D.; Kempkes, B.; Park, J.H.; Hayward, S.D. EBNA2 Is Required for Protection of Latently Epstein-Barr Virus-Infected B Cells against Specific Apoptotic Stimuli. J. Virol. 2004, 78, 12694–12697. [Google Scholar] [CrossRef] [Green Version]
- Foo, J.; Bellot, G.; Pervaiz, S.; Alonso, S. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 2022, 30, 679–692. [Google Scholar] [CrossRef]
- Szymula, A.; Palermo, R.D.; Bayoumy, A.; Groves, I.J.; Ba Abdullah, M.; Holder, B.; White, R.E. Epstein-Barr virus nuclear antigen EBNA-LP is essential for transforming naïve B cells, and facilitates recruitment of transcription factors to the viral genome. PLoS Pathog. 2018, 14, e1006890. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, G.; Nakajima, K.; Kawaguchi, Y.; Yamanashi, Y.; Hirai, K. Epstein-Barr virus (EBV) nuclear antigen leader protein (EBNA-LP) forms complexes with a cellular anti-apoptosis protein Bcl-2 or its EBV counterpart BHRF1 through HS1-associated protein X-1. Microbiol. Immunol. 2003, 47, 91–99. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Nakajima, K.; Igarashi, M.; Morita, T.; Tanaka, M.; Suzuki, M.; Yokoyama, A.; Matsuda, G.; Kato, K.; Kanamori, M.; et al. Interaction of Epstein-Barr Virus Nuclear Antigen Leader Protein (EBNA-LP) with HS1-Associated Protein X-1: Implication of Cytoplasmic Function of EBNA-LP. J. Virol. 2000, 74, 10104. [Google Scholar] [CrossRef] [Green Version]
- Ruf, I.K.; Lackey, K.A.; Warudkar, S.; Sample, J.T. Protection from Interferon-Induced Apoptosis by Epstein-Barr Virus Small RNAs Is Not Mediated by Inhibition of PKR. J. Virol. 2005, 79, 14562–14569. [Google Scholar] [CrossRef] [Green Version]
- Iwakiri, D.; Takada, K. Role of EBERs in the Pathogenesis of EBV Infection. Adv. Cancer Res. 2010, 107, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Vrzalikova, K.; Pugh, M.; Mundo, L.; Murray, P. The contribution of ebv to the pathogenesis of classical hodgkin lymphoma. Ann. Lymphoma 2021, 5, 30. [Google Scholar] [CrossRef]
- Ahmed, W.; Tariq, S.; Khan, G. Tracking EBV-encoded RNAs (EBERs) from the nucleus to the excreted exosomes of B-lymphocytes. Sci. Rep. 2018, 8, 15438. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, D. Epstein-Barr Virus-Encoded RNAs: Key Molecules in Viral Pathogenesis. Cancers 2014, 6, 1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komano, J.; Maruo, S.; Kurozumi, K.; Oda, T.; Takada, K. Oncogenic role of Epstein-Barr virus-encoded RNAs in Burkitt’s lymphoma cell line Akata. J. Virol. 1999, 73, 9827–9831. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.L.; Wang, X.; Chang, R.C.C.; Jin, D.Y.; Feng, H.; Wang, Q.; Lo, K.W.; Huang, D.P.; Yuen, P.W.; Takada, K.; et al. Stable expression of EBERs in immortalized nasopharyngeal epithelial cells confers resistance to apoptotic stress. Mol. Carcinog. 2005, 44, 92–101. [Google Scholar] [CrossRef]
- Cesaro, T.; Michiels, T. Inhibition of PKR by Viruses. Front. Microbiol. 2021, 12, 757238. [Google Scholar] [CrossRef]
- Greifenegger, N.; Jäger, M.; Kunz-Schughart, L.A.; Wolf, H.; Schwarzmann, F. Epstein-Barr virus small RNA (EBER) genes: Differential regulation during lytic viral replication. J. Virol. 1998, 72, 9323–9328. [Google Scholar] [CrossRef] [Green Version]
- Nanbo, A.; Inoue, K.; Adachi-Takasawa, K.; Takada, K. Epstein-Barr virus RNA confers resistance to interferon-alpha-induced apoptosis in Burkitt’s lymphoma. EMBO J. 2002, 21, 954–965. [Google Scholar] [CrossRef]
- Vuyisich, M.; Spanggord, R.J.; Beal, P.A. The binding site of the RNA-dependent protein kinase (PKR) on EBER1 RNA from Epstein–Barr virus. EMBO Rep. 2002, 3, 622. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Gamil, A.A.A.; Munang’andu, H.M.; Evensen, Ø. Apoptosis Induction by dsRNA-Dependent Protein Kinase R (PKR) in EPC Cells via Caspase 8 and 9 Pathways. Viruses 2018, 10, 526. [Google Scholar] [CrossRef] [Green Version]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A kinase to remember. Front. Mol. Neurosci. 2019, 11, 480. [Google Scholar] [CrossRef] [Green Version]
- Eichhorn, S.W.; Guo, H.; McGeary, S.E.; Rodriguez-Mias, R.A.; Shin, C.; Baek, D.; Hsu, S.H.; Ghoshal, K.; Villén, J.; Bartel, D.P. MRNA Destabilization Is the dominant effect of mammalian microRNAs by the time substantial repression ensues. Mol. Cell 2014, 56, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Lima, J.F.; Cerqueira, L.; Figueiredo, C.; Oliveira, C.; Azevedo, N.F.; Frias, R.B.; Biomode, P.; Mestre Jos, E.; Veiga, I.-A.; Braga, P. Anti-miRNA oligonucleotides: A comprehensive guide for design. RNA Biol. 2018, 15, 338–352. [Google Scholar] [CrossRef] [Green Version]
- Choy, E.Y.W.; Siu, K.L.; Kok, K.H.; Lung, R.W.M.; Tsang, C.M.; To, K.F.; Kwong, D.L.W.; Tsao, S.W.; Jin, D.Y. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J. Exp. Med. 2008, 205, 2551–2560. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Kowdley, K.V. MicroRNAs in Common Human Diseases. Genom. Proteom. Bioinform. 2012, 10, 246. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Lowey, B.; Sodroski, C.; Krishnamurthy, S.; Alao, H.; Cha, H.; Chiu, S.; El-Diwany, R.; Ghany, M.G.; Liang, T.J. Cellular microRNA networks regulate host dependency of hepatitis C virus infection. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Min, K.; Kim, J.Y.; Lee, S.K. Epstein-Barr virus miR-BART1-3p suppresses apoptosis and promotes migration of gastric carcinoma cells by targeting DAB2. Int. J. Biol. Sci. 2020, 16, 694. [Google Scholar] [CrossRef] [Green Version]
- Hassani, A.; Khan, G. Epstein-Barr Virus and miRNAs: Partners in Crime in the Pathogenesis of Multiple Sclerosis? Front. Immunol. 2019, 10, 695. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.M.; Yu, Y.; Zhao, H.P. EBV-BART-6-3p and cellular microRNA-197 compromise the immune defense of host cells in EBV-positive Burkitt lymphoma. Mol. Med. Rep. 2017, 15, 1877–1883. [Google Scholar] [CrossRef] [Green Version]
- Marquitz, A.R.; Mathur, A.; Nam, C.S.; Raab-Traub, N. The Epstein-Barr Virus BART microRNAs target the pro-apoptotic protein Bim. Virology 2011, 412, 392. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki-Ushiku, A.; Kunita, A.; Isogai, M.; Hibiya, T.; Ushiku, T.; Takada, K.; Fukayama, M. Profiling of Virus-Encoded MicroRNAs in Epstein-Barr Virus-Associated Gastric Carcinoma and Their Roles in Gastric Carcinogenesis. J. Virol. 2015, 89, 5581–5591. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Choi, H.; Lee, S.K. Epstein–Barr virus miR-BART20-5p regulates cell proliferation and apoptosis by targeting BAD. Cancer Lett. 2015, 356, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Cartron, P.; Er, E.; Oliver, L.; Juin, P.; Armstrong, L.; Bornstein, P.; Mihara, K.; Manon, S.; Vallette, F. TOM22, a core component of the mitochondria outer membrane protein translocation pore, is a mitochondrial receptor for the proapoptotic protein Bax translocase of the outer membrane of mitochondria. Cell Death Differ. 2007, 14, 785–794. [Google Scholar] [CrossRef] [Green Version]
- Dölken, L.; Malterer, G.; Erhard, F.; Kothe, S.; Friedel, C.C.; Suffert, G.; Marcinowski, L.; Motsch, N.; Barth, S.; Beitzinger, M.; et al. Systematic analysis of viral and cellular microRNA targets in cells latently infected with human γ-herpesviruses by RISC immunoprecipitation assay. Cell Host Microbe 2010, 7, 324–334. [Google Scholar] [CrossRef]
- Harold, C.; Cox, D.; Riley, K.J. Epstein-Barr viral microRNAs target caspase 3. Virol. J. 2016, 13, 145. [Google Scholar] [CrossRef] [Green Version]
- Caenepeel, S.; Brown, S.P.; Belmontes, B.; Moody, G.; Keegan, K.S.; Chui, D.; Whittington, D.A.; Huang, X.; Poppe, L.; Cheng, A.C.; et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discov. 2018, 8, 1582–1597. [Google Scholar] [CrossRef] [Green Version]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef] [Green Version]
- Pullarkat, V.A.; Lacayo, N.J.; Jabbour, E.; Rubnitz, J.E.; Bajel, A.; Laetsch, T.W.; Leonard, J.; Colace, S.I.; Khaw, S.L.; Fleming, S.A.; et al. Venetoclax and Navitoclax in Combination with Chemotherapy in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia and Lymphoblastic Lymphoma. Cancer Discov. 2021, 11, 1440–1453. [Google Scholar] [CrossRef]
- Ramsey, H.E.; Fischer, M.A.; Lee, T.; Gorska, A.E.; Arrate, M.P.; Fuller, L.; Boyd, K.L.; Strickland, S.A.; Sensintaffar, J.; Hogdal, L.J.; et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 2018, 8, 1566–1581. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Xie, C.; Lung, H.L.; Lo, K.W.; Law, G.L.; Mak, N.K.; Wong, K.L. EBNA1-targeted inhibitors: Novel approaches for the treatment of Epstein-Barr virus-associated cancers. Theranostics 2018, 8, 5307. [Google Scholar] [CrossRef]
- Zhu, J.; Kamara, S.; Cen, D.; Tang, W.; Gu, M.; Ci, X.; Chen, J.; Wang, L.; Zhu, S.; Jiang, P.; et al. Generation of novel affibody molecules targeting the EBV LMP2A N-terminal domain with inhibiting effects on the proliferation of nasopharyngeal carcinoma cells. Cell Death Dis. 2020, 11, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yu, F.; Wu, W.; Wang, Y.; Ding, H.; Qian, L. Epstein-Barr virus-encoded microRNAs as regulators in host immune responses. Int. J. Biol. Sci. 2018, 14, 565. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Claret, F.X.; Wu, W. MicroRNAs as Therapeutic Targets in Nasopharyngeal Carcinoma. Front. Oncol. 2019, 9, 756. [Google Scholar] [CrossRef] [Green Version]
- Skalsky, R.L.; Corcoran, D.L.; Gottwein, E.; Frank, C.L.; Kang, D.; Hafner, M.; Nusbaum, J.D.; Feederle, R.; Delecluse, H.J.; Luftig, M.A.; et al. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 2012, 8, e1002484. [Google Scholar] [CrossRef]
- Neudecker, V.; Brodsky, K.S.; Kreth, S.; Ginde, A.A.; Eltzschig, H.K. Emerging Roles for MicroRNAs in Perioperative Medicine. Anesthesiology 2016, 124, 489. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wyżewski, Z.; Mielcarska, M.B.; Gregorczyk-Zboroch, K.P.; Myszka, A. Virus-Mediated Inhibition of Apoptosis in the Context of EBV-Associated Diseases: Molecular Mechanisms and Therapeutic Perspectives. Int. J. Mol. Sci. 2022, 23, 7265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137265
Wyżewski Z, Mielcarska MB, Gregorczyk-Zboroch KP, Myszka A. Virus-Mediated Inhibition of Apoptosis in the Context of EBV-Associated Diseases: Molecular Mechanisms and Therapeutic Perspectives. International Journal of Molecular Sciences. 2022; 23(13):7265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137265
Chicago/Turabian StyleWyżewski, Zbigniew, Matylda Barbara Mielcarska, Karolina Paulina Gregorczyk-Zboroch, and Anna Myszka. 2022. "Virus-Mediated Inhibition of Apoptosis in the Context of EBV-Associated Diseases: Molecular Mechanisms and Therapeutic Perspectives" International Journal of Molecular Sciences 23, no. 13: 7265. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137265