
Next-Generation Grade and Survival Expression Biomarkers of Human Gliomas Based on Algorithmically Reconstructed Molecular Pathways

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
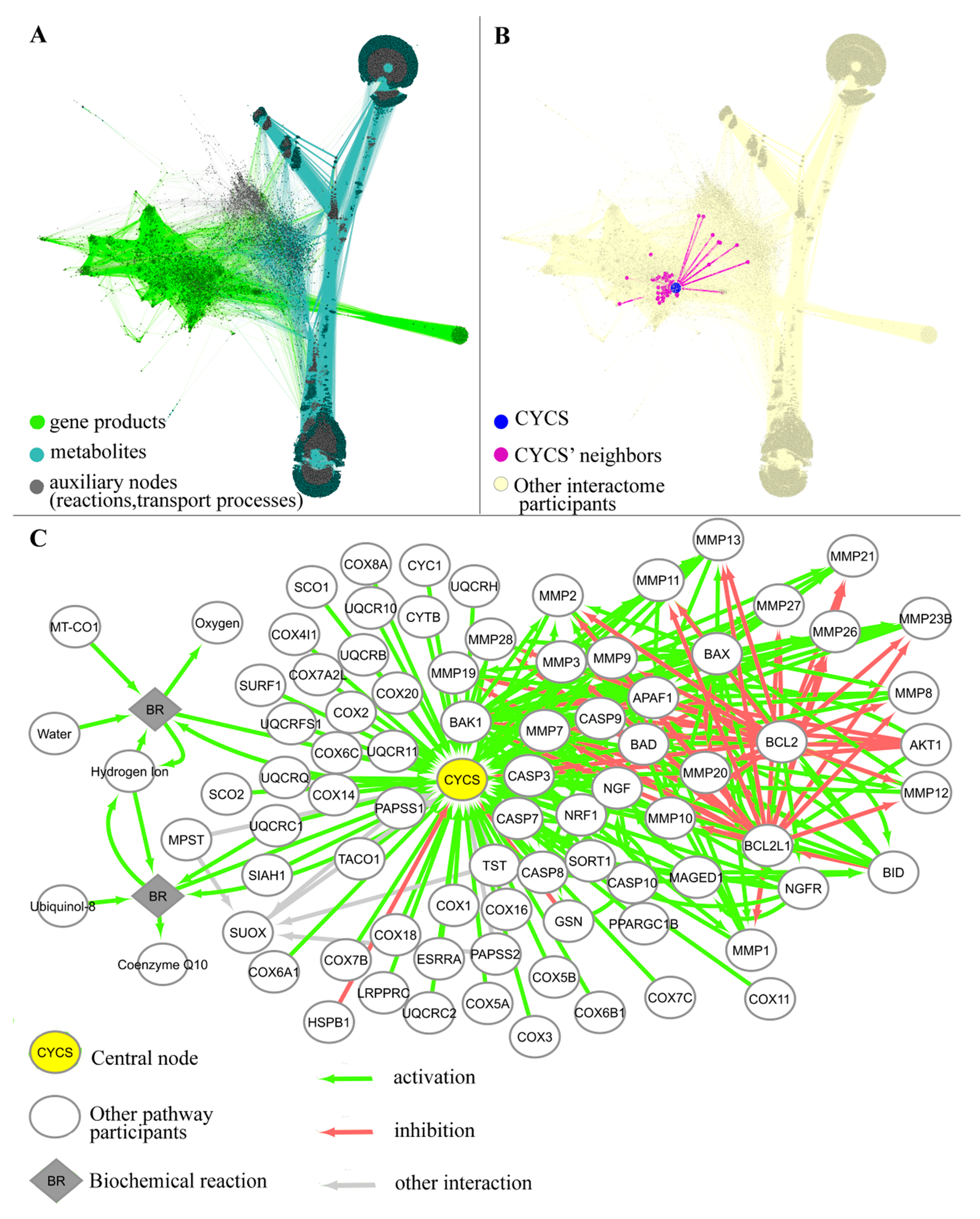
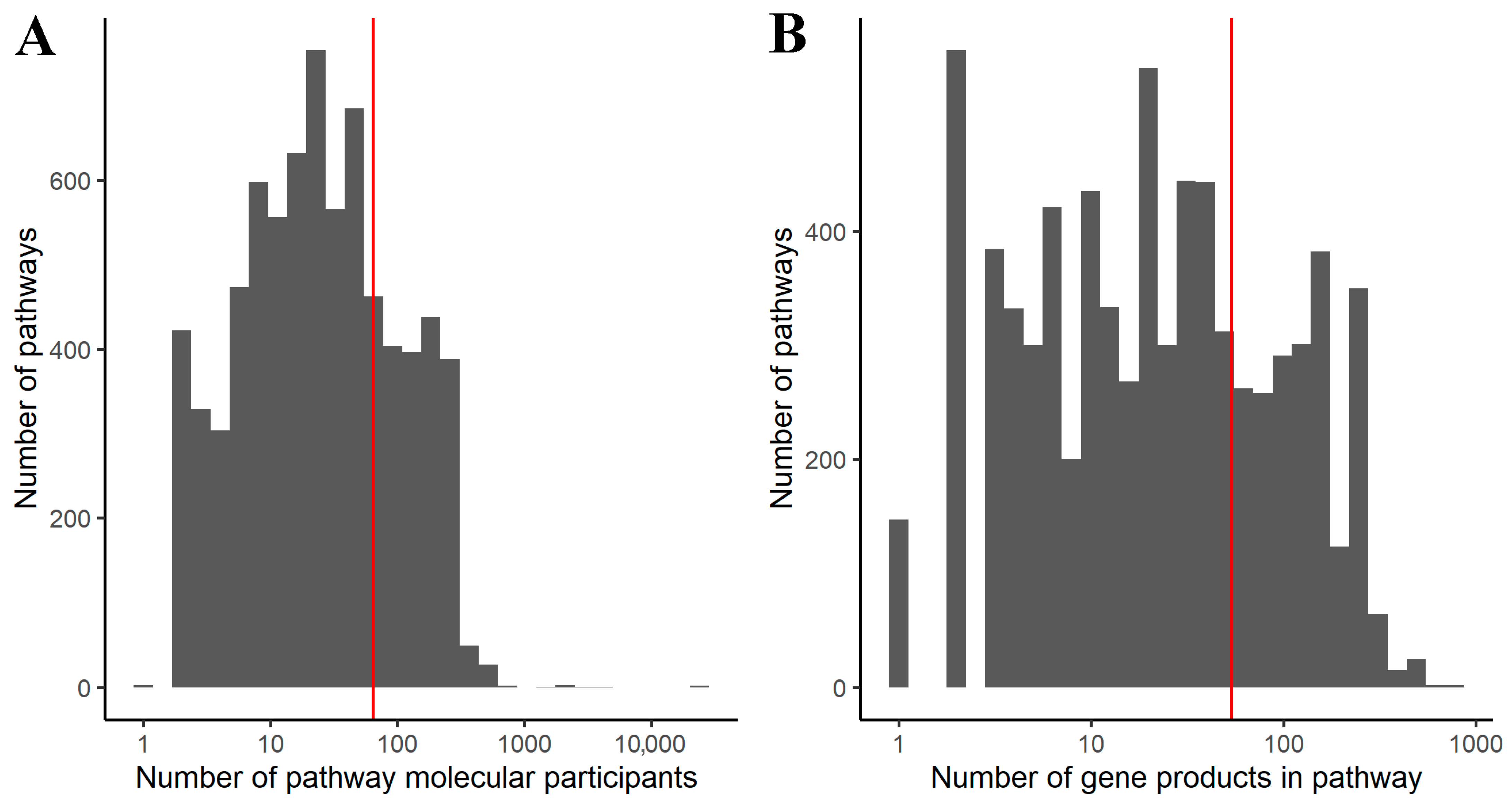
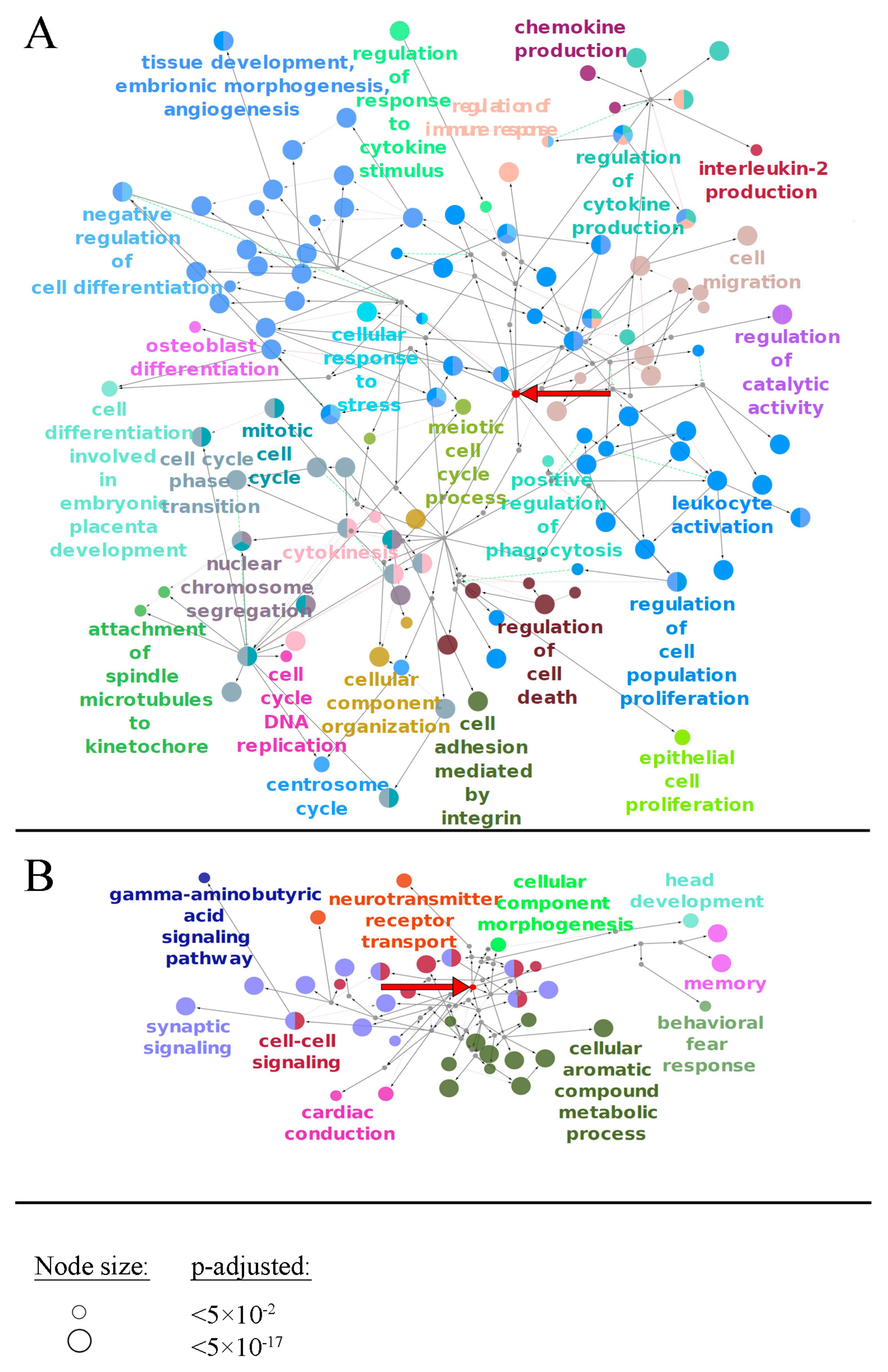
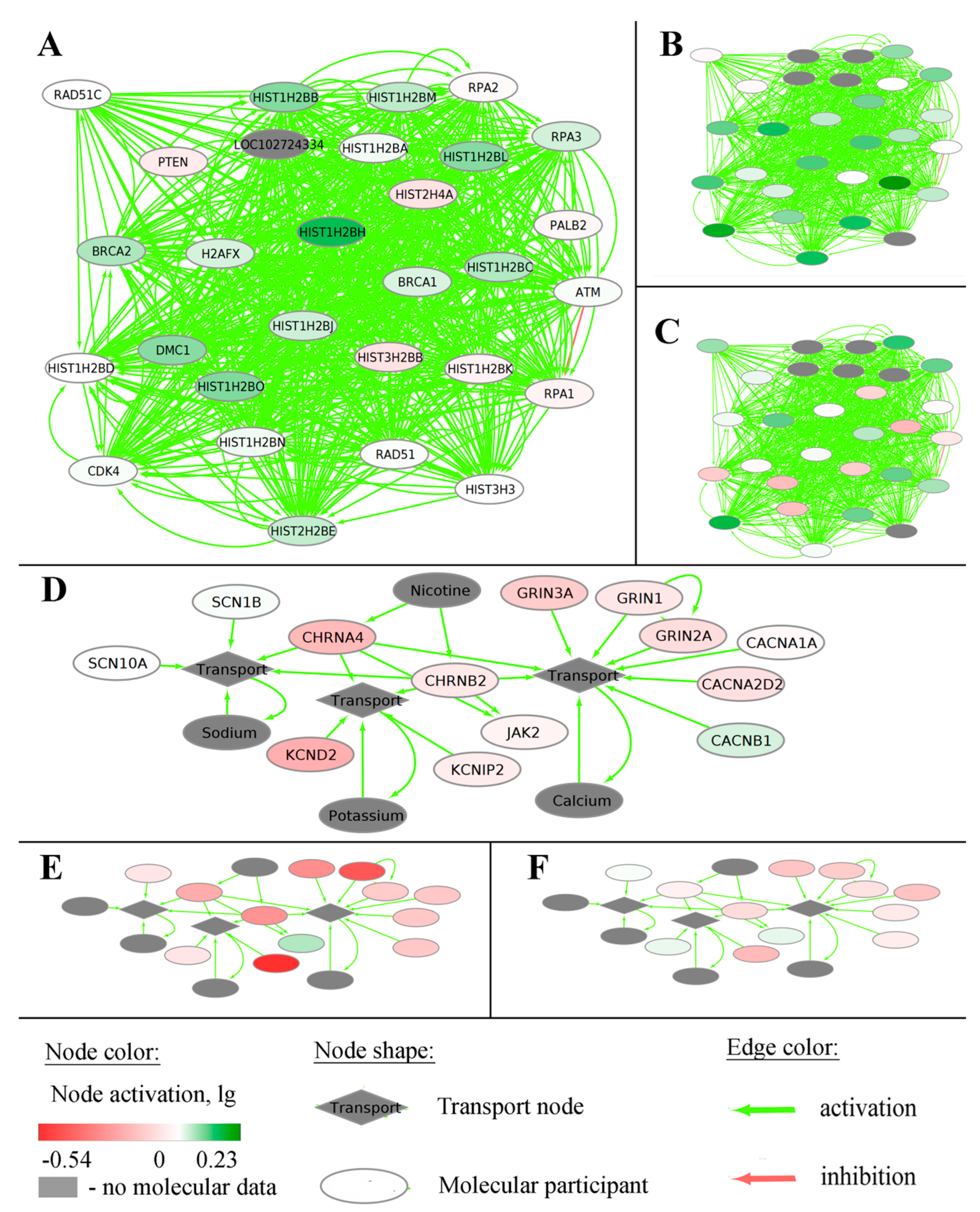
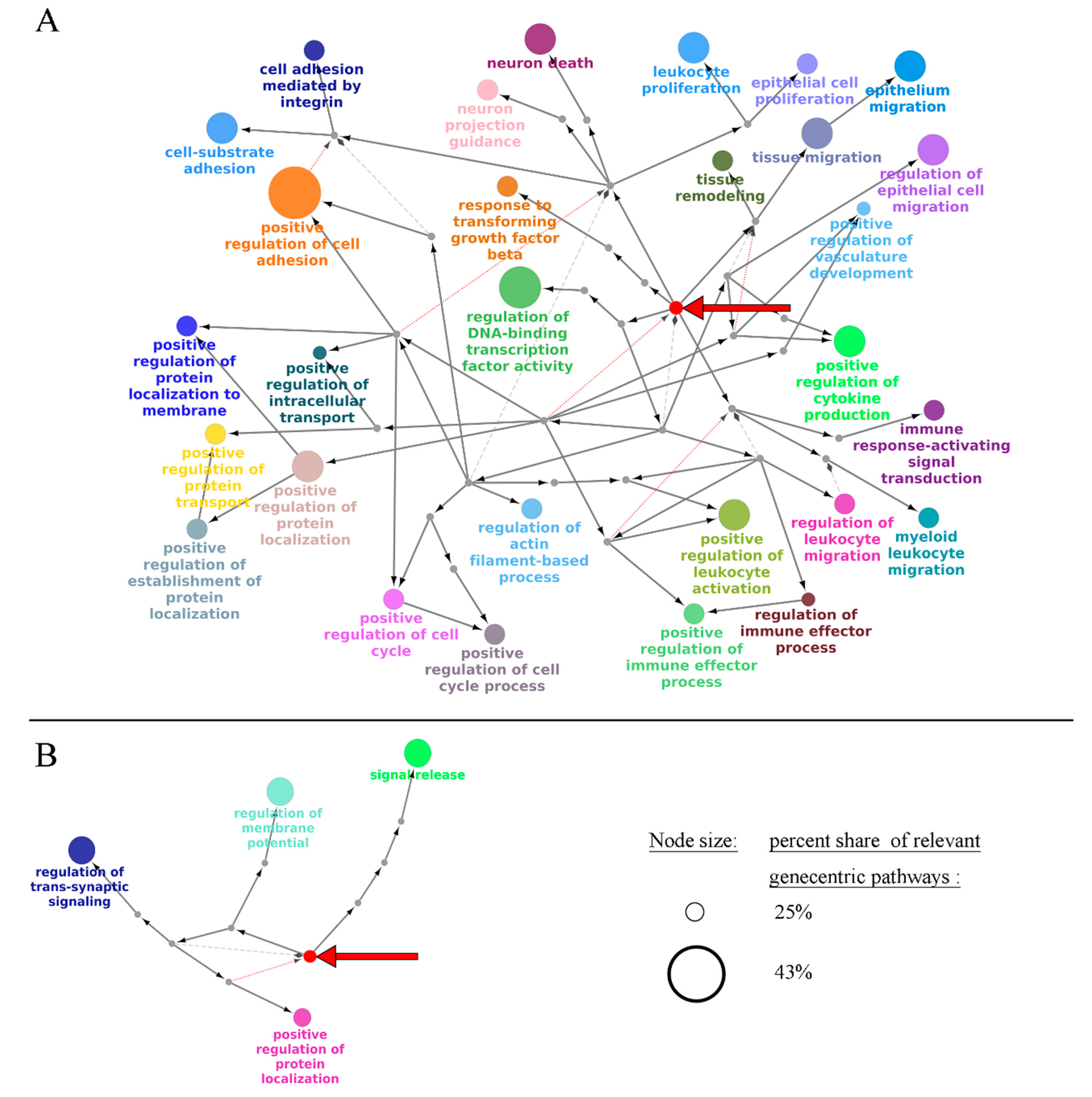
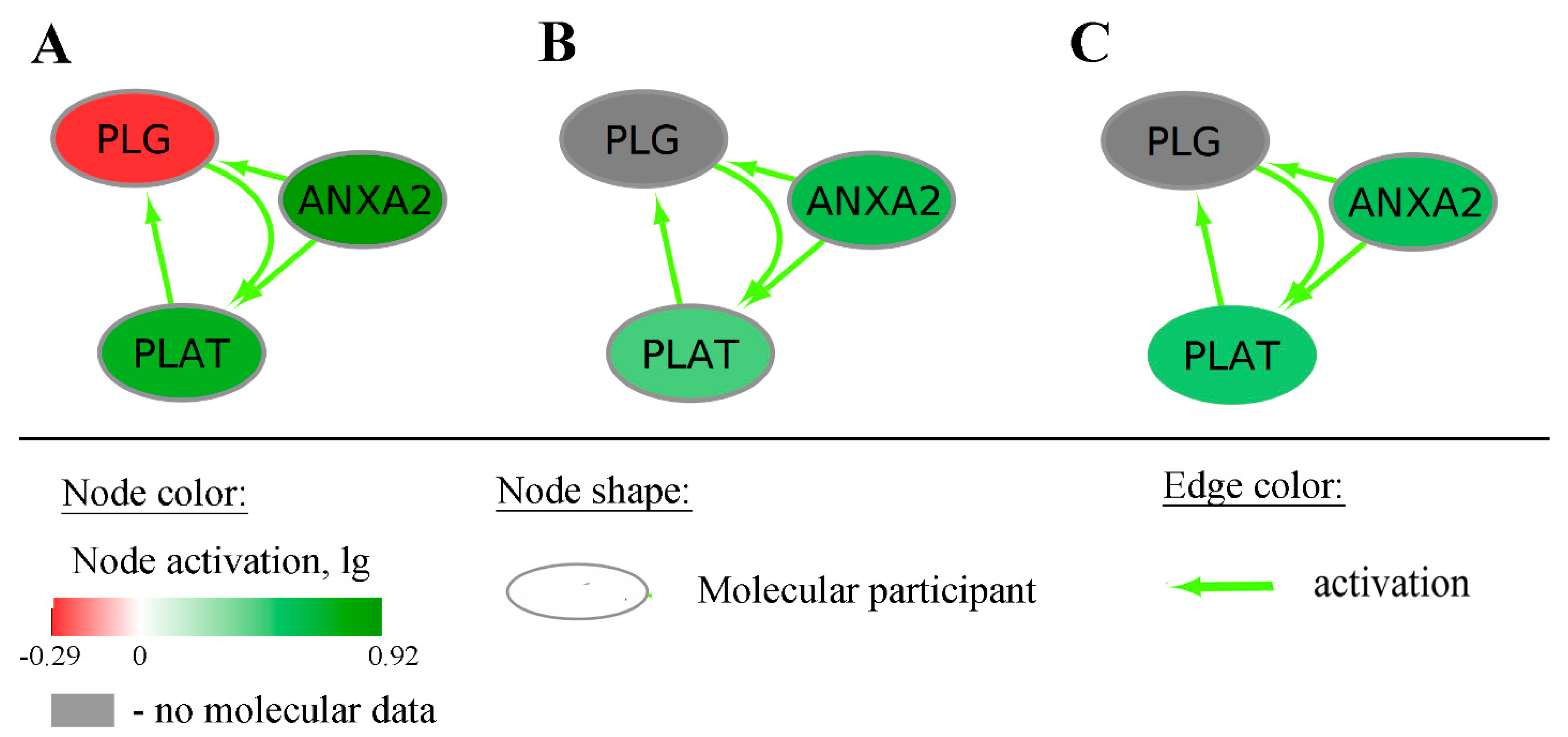
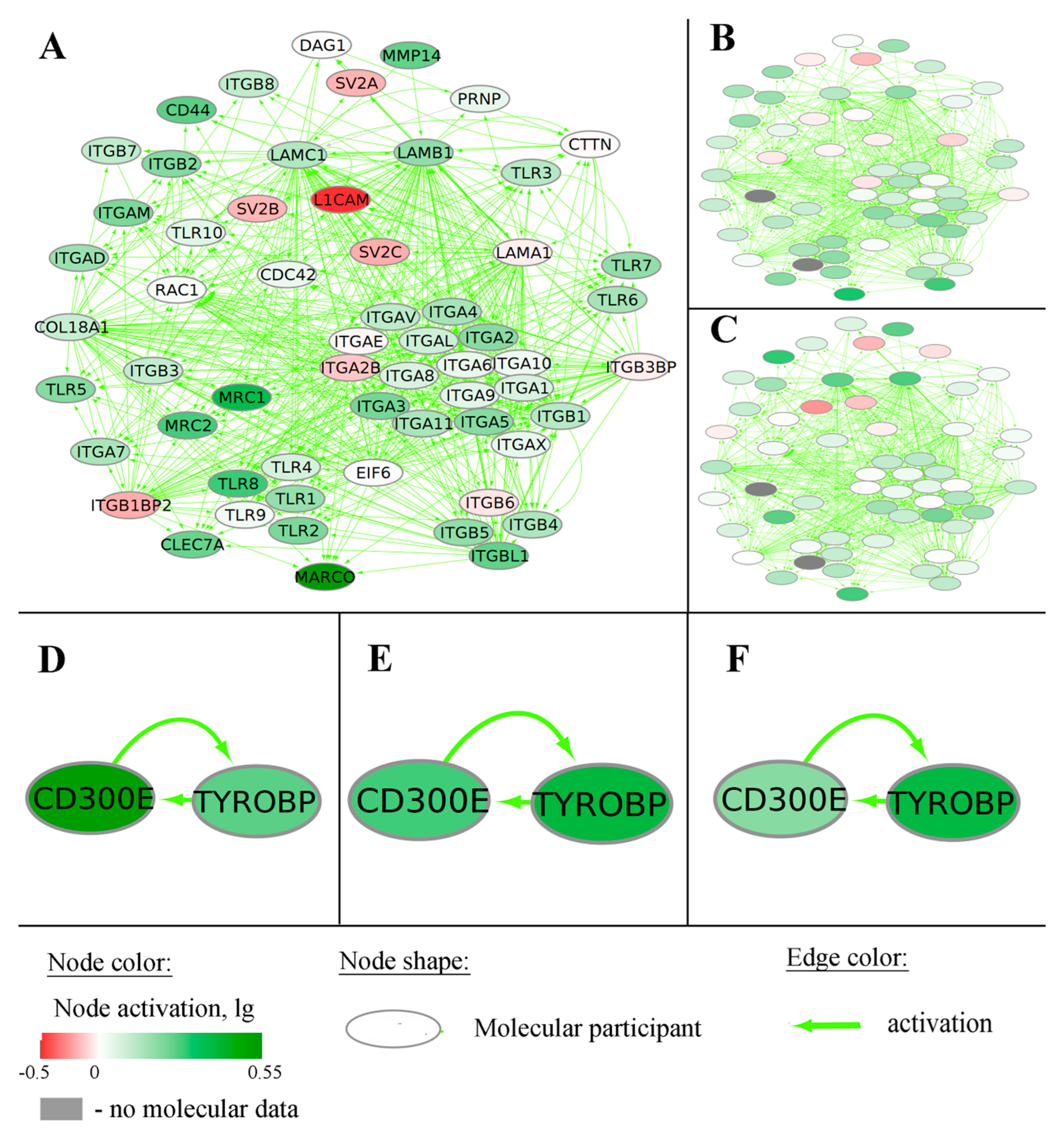
2.1. Generation of Gene-Centric Molecular Pathways
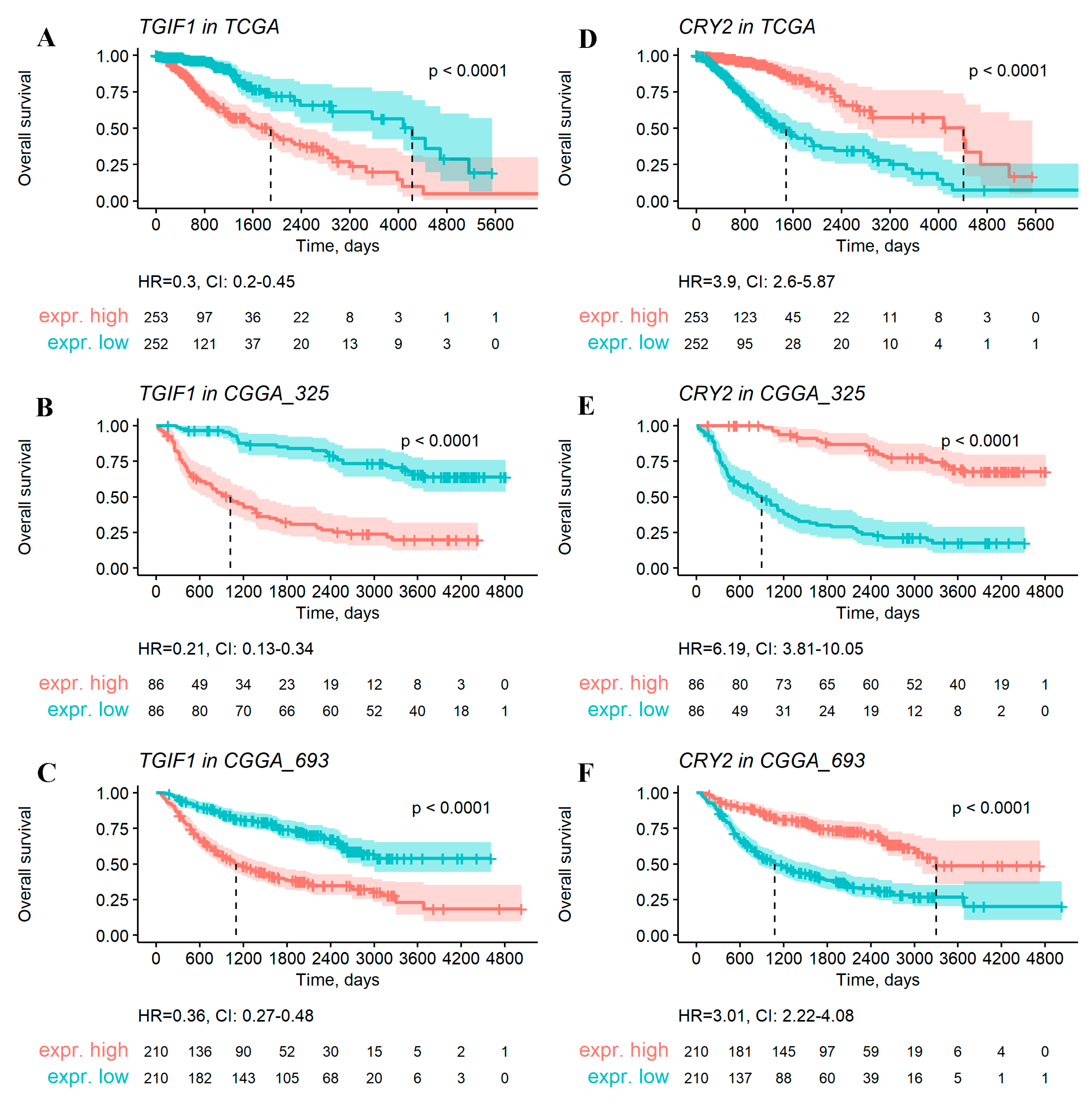
2.2. Single-Gene-Expression Biomarkers as Overall Survival Predictors in Gliomas
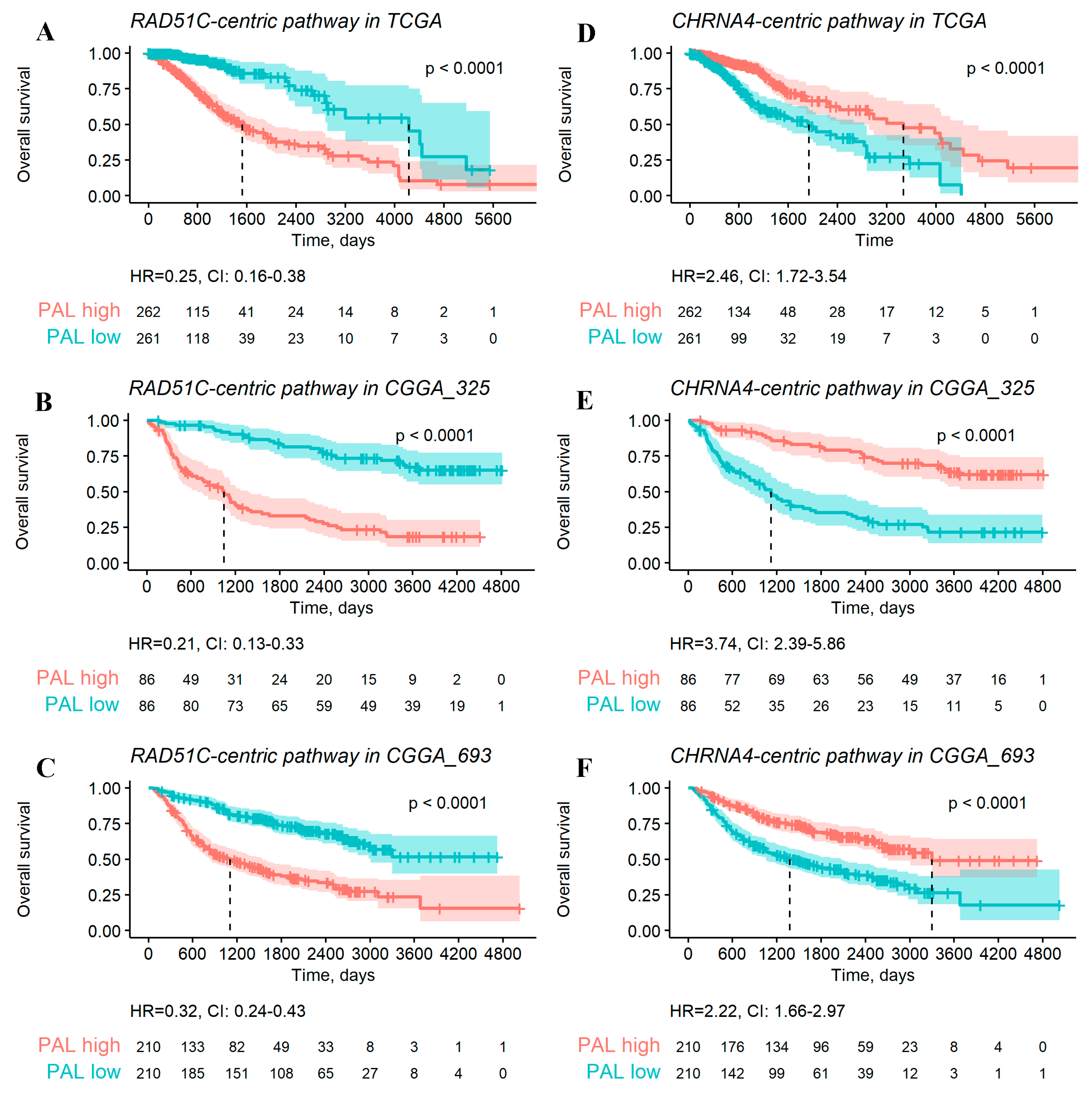
2.3. Gene-Centric Molecular Pathways as Overall Survival Predictors for Gliomas
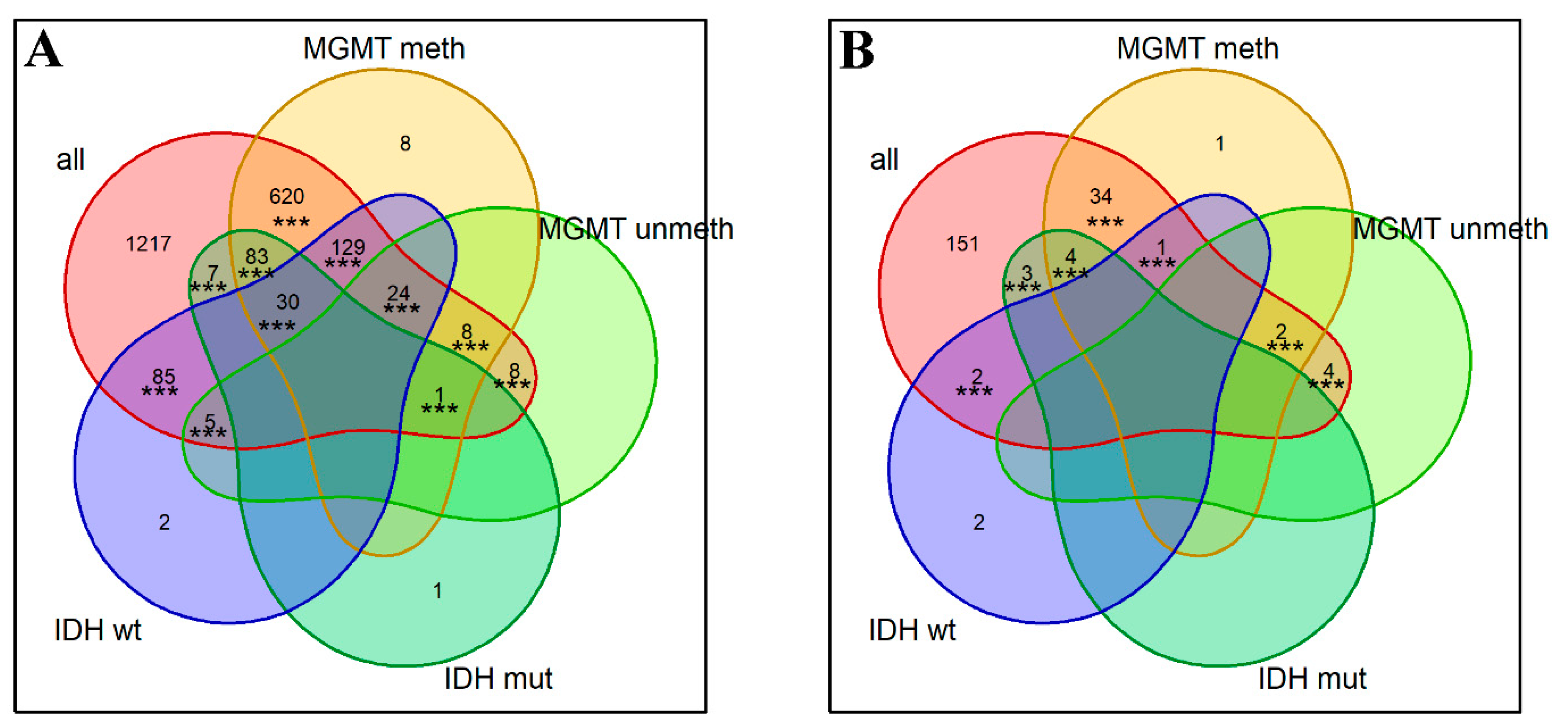
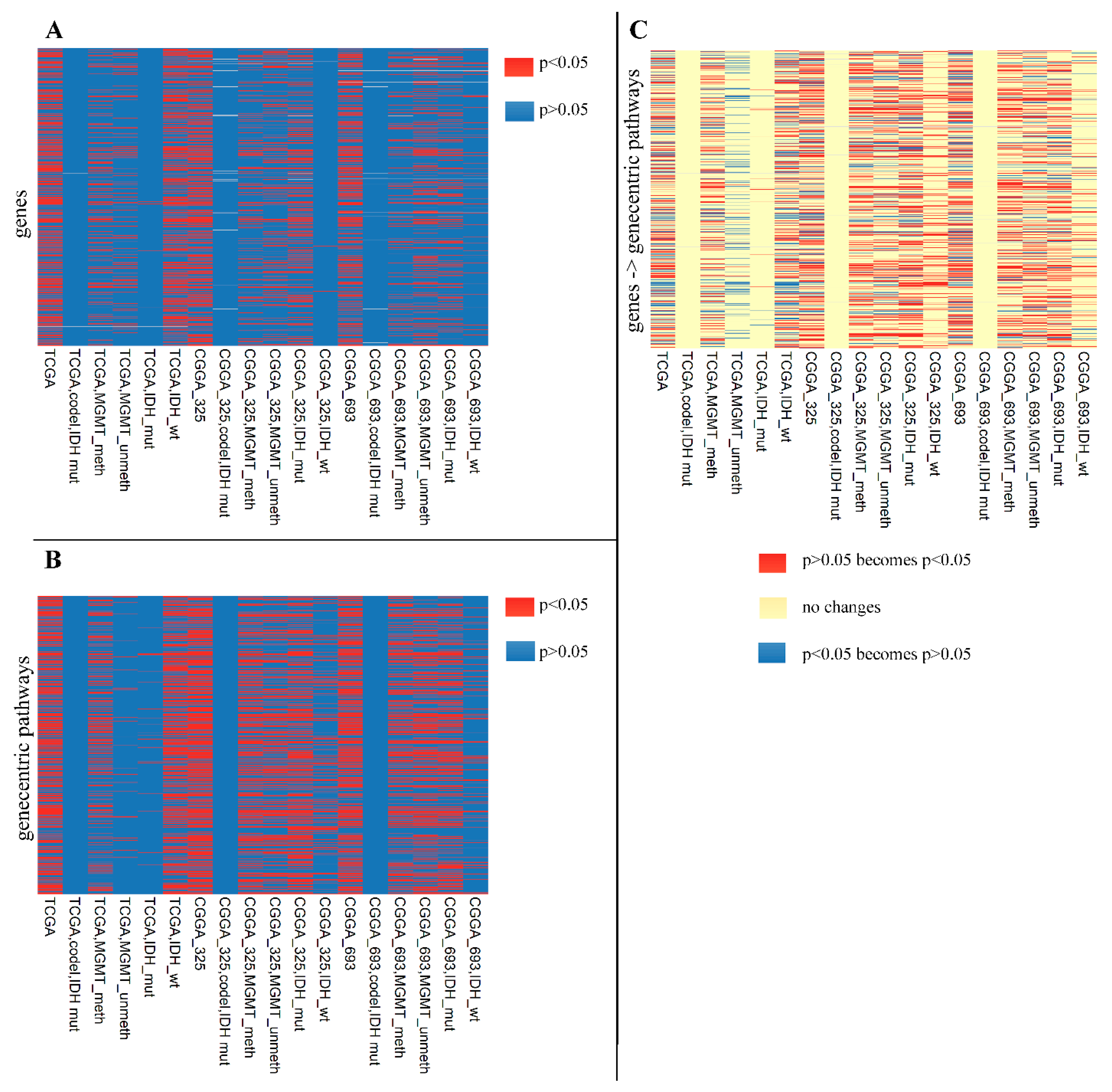
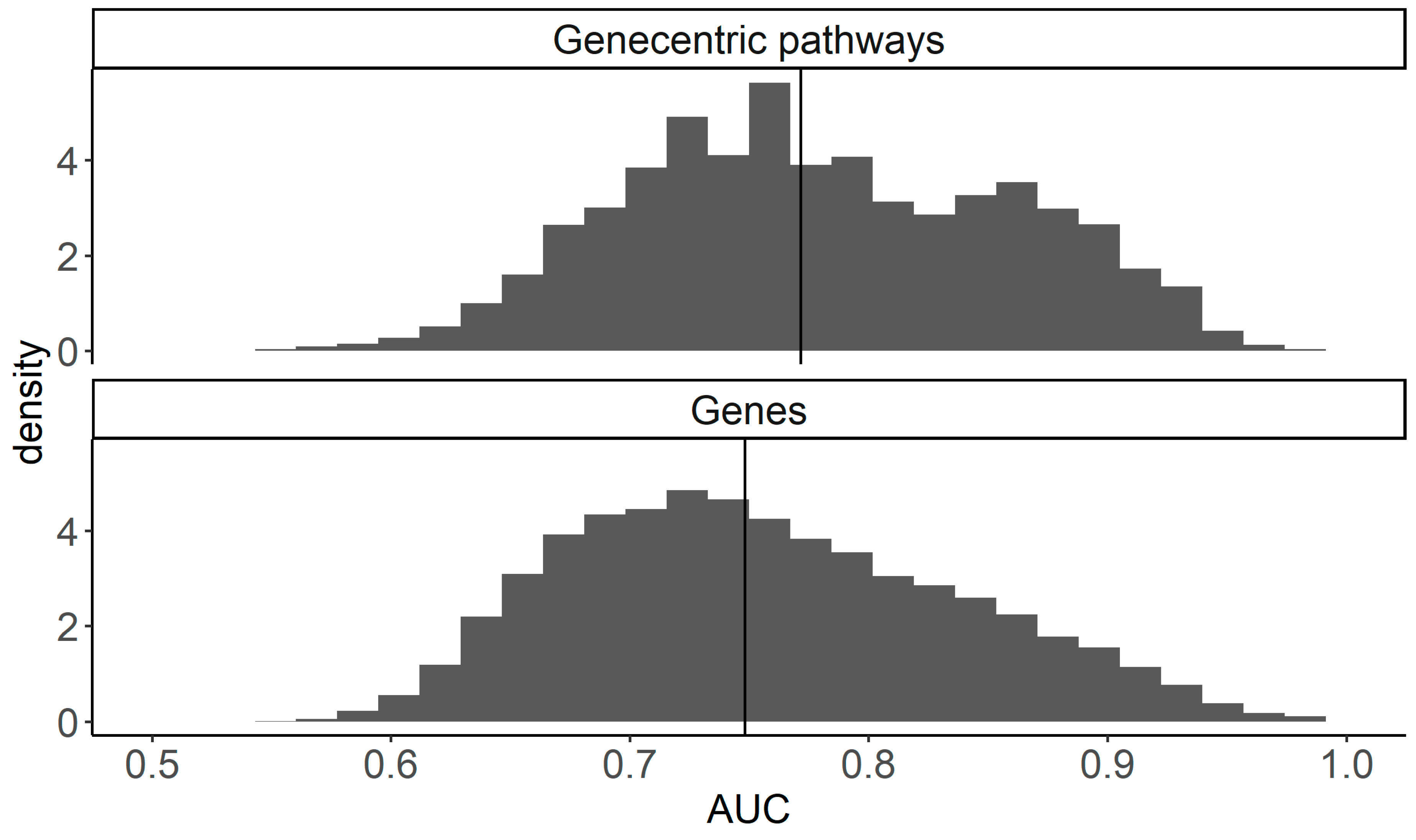
2.4. Performances of Single Gene and Gene-Centric Pathway Biomarkers
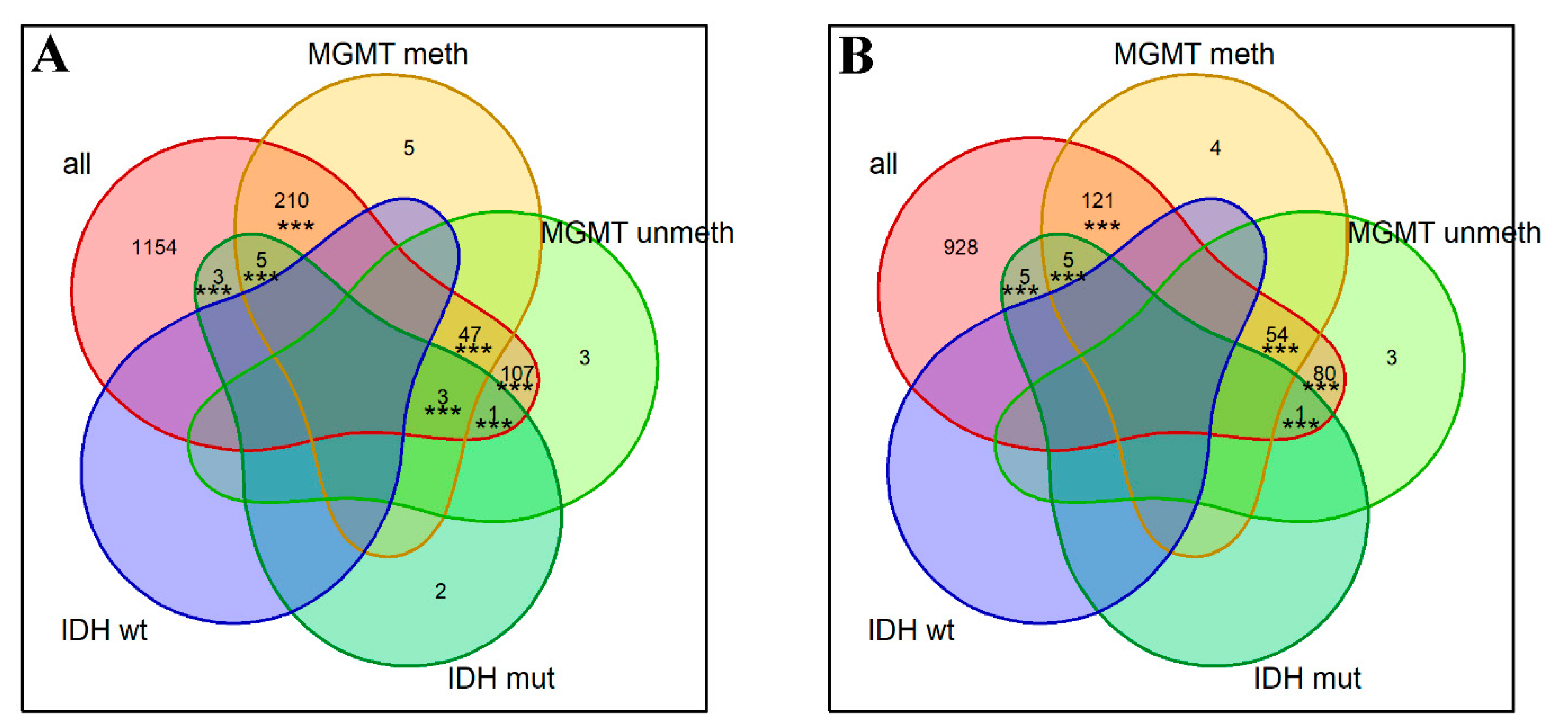
2.5. Individual-Gene and Gene-Centric Pathways as the Histological and Molecular Type Biomarkers
3. Discussion
4. Materials and Methods
4.1. The Cancer Genome Atlas (TCGA) Dataset
4.2. The Chinese Glioma Genome Atlas Dataset CGGA_325
4.3. The Chinese Glioma Genome Atlas Dataset CGGA_693
4.4. Glioma Classification and Diagnosis
4.5. Source Molecular Pathways
4.6. Pathway Activation Level Calculation
4.7. Statistical Analysis
4.8. Gene Ontology Analysis
4.9. Intersection Significance Tests
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nutt, C.L.; Mani, D.R.; Betensky, R.A.; Tamayo, P.; Cairncross, J.G.; Ladd, C.; Pohl, U.; Hartmann, C.; McLaughlin, M.E.; Batchelor, T.T.; et al. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res. 2003, 63, 1602–1607. [Google Scholar]
- Gravendeel, L.A.M.; Kouwenhoven, M.C.M.; Gevaert, O.; De Rooi, J.J.; Stubbs, A.P.; Duijm, J.E.; Daemen, A.; Bleeker, F.E.; Bralten, L.B.C.; Kloosterhof, N.K.; et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009, 69, 9065–9072. [Google Scholar] [CrossRef] [Green Version]
- Freije, W.A.; Castro-Vargas, F.E.; Fang, Z.; Horvath, S.; Cloughesy, T.; Liau, L.M.; Mischel, P.S.; Nelson, S.F. Gene Expression Profiling of Gliomas Strongly Predicts Survival. Cancer Res. 2004, 64, 6503–6510. [Google Scholar] [CrossRef] [Green Version]
- Godard, S.; Getz, G.; Delorenzi, M.; Farmer, P.; Kobayashi, H.; Desbaillets, I.; Nozaki, M.; Diserens, A.-C.; Hamou, M.-F.; Dietrich, P.-Y.; et al. Classification of human astrocytic gliomas on the basis of gene expression: A correlated group of genes with angiogenic activity emerges as a strong predictor of subtypes. Cancer Res. 2003, 63, 6613–6625. [Google Scholar]
- Shai, R.; Shi, T.; Kremen, T.J.; Horvath, S.; Liau, L.M.; Cloughesy, T.F.; Mischel, P.S.; Nelson, S.F. Gene expression profiling identifies molecular subtypes of gliomas. Oncogene 2003, 22, 4918–4923. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, R.; Arao, T.; Yajima, N.; Tsuchiya, N.; Homma, J.; Tanaka, R.; Sano, M.; Oide, A.; Sekijima, M.; Nishio, K. Identification of expressed genes characterizing long-term survival in malignant glioma patients. Oncogene 2006, 25, 5994–6002. [Google Scholar] [CrossRef] [Green Version]
- Shirahata, M.; Oba, S.; Iwao-Koizumi, K.; Saito, S.; Ueno, N.; Oda, M.; Hashimoto, N.; Ishii, S.; Takahashi, J.A.; Kato, K. Using gene expression profiling to identify a prognostic molecular spectrum in gliomas. Cancer Sci. 2009, 100, 165–172. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. An integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 2010, 17, 98. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Zhang, W.; You, G.; Zhang, J.; Han, L.; Bao, Z.; Wang, Y.; Liu, Y.; Jiang, C.; Kang, C.; et al. Molecular classification of gliomas based on whole genome gene expression: A systematic report of 225 samples from the Chinese Glioma Cooperative Group. Neuro. Oncol. 2012, 14, 1432–1440. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Shim, J.K.; Yoon, S.J.; Kim, S.H.; Chang, J.H.; Kang, S.G. Transcriptome profiling-based identification of prognostic subtypes and multi-omics signatures of glioblastoma. Sci. Rep. 2019, 9, 10555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, S.; Pu, J.K.S.; Tsang, A.C.O.; Lee, D.; Man, V.O.Y.; Lui, W.M.; Wong, S.T.S.; Leung, G.K.K. Long non-coding RNA expression profiles predict clinical phenotypes in glioma. Neurobiol. Dis. 2012, 48, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Qian, J.; Wang, Y.Y.; Zhang, J.X.; You, Y.P. Long Noncoding RNA Profiles Reveal Three Molecular Subtypes in Glioma. CNS Neurosci. Ther. 2014, 20, 339. [Google Scholar] [CrossRef] [PubMed]
- Reon, B.J.; Anaya, J.; Zhang, Y.; Mandell, J.; Purow, B.; Abounader, R.; Dutta, A. Expression of lncRNAs in Low-Grade Gliomas and Glioblastoma Multiforme: An In Silico Analysis. PLoS Med. 2016, 13, e1002192. [Google Scholar] [CrossRef] [Green Version]
- Kiran, M.; Chatrath, A.; Tang, X.; Keenan, D.M.; Dutta, A. A prognostic signature for lower-grade gliomas based on expression of long noncoding RNAs. Mol. Neurobiol. 2019, 56, 4786. [Google Scholar] [CrossRef]
- Sorokin, M.; Raevskiy, M.; Zottel, A.; Šamec, N.; Vidmar, M.S.; Matjašič, A.; Zupan, A.; Mlakar, J.; Suntsova, M.; Kuzmin, D.V.; et al. Large-Scale Transcriptomics-Driven Approach Revealed Overexpression of CRNDE as a Poor Survival Prognosis Biomarker in Glioblastoma. Cancers 2021, 13, 3419. [Google Scholar] [CrossRef]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kälin, R.; Van Rooijen, N.; et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Liang, J.; Wu, Z.; Shan, X.; Qiao, H.; Jiang, T. Expression of HLA-DR genes in gliomas: Correlation with clinicopathological features and prognosis. Chin. Neurosurg. J. 2017, 3, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Liu, P.; Li, C.; Luo, Y.; Chen, I.; Liang, W.; Chen, X.; Feng, Y.; Xia, H.; Wang, F. Deregulated expression of the clock genes in gliomas. Technol. Cancer Res. Treat. 2013, 12, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Xiao, W.; Song, T.; Feng, G.; Dai, Z. Expression and Prognostic Significance of p53 in Glioma Patients: A Meta-analysis. Neurochem. Res. 2016, 41, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Ranuncolo, S.M.; Ladeda, V.; Specterman, S.; Varela, M.; Lastiri, J.; Morandi, A.; Matos, E.; De Kier Joffe, E.B.; Puricelli, L.; Pallotta, M.G. CD44 expression in human gliomas. J. Surg. Oncol. 2002, 79, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ning, L.; Zhang, Q.; Ge, Y.; Liu, C.; Bi, S.; Zeng, X.; Nong, W.; Wu, S.; Guo, G.; et al. Expression profile of ACTL8, CTCFL, OIP5 and XAGE3 in glioma and their prognostic significance: A retrospective clinical study. Am. J. Transl. Res. 2020, 12, 7782. [Google Scholar]
- Zhou, K.; Liu, Y.; Zhao, Z.; Wang, Y.; Huang, L.; Chai, R.; Li, G.; Jiang, T. ABCC8 mRNA expression is an independent prognostic factor for glioma and can predict chemosensitivity. Sci. Rep. 2020, 10, 12682. [Google Scholar] [CrossRef] [PubMed]
- Shen, A.; Wang, Y.; Zhao, Y.; Zou, L.; Sun, L.; Cheng, C. Expression of CRM1 in human gliomas and its significance in p27 expression and clinical prognosis. Neurosurgery 2009, 65, 153–159. [Google Scholar] [CrossRef]
- Jovčevska, I.; Zottel, A.; Šamec, N.; Mlakar, J.; Sorokin, M.; Nikitin, D.; Buzdin, A.A.; Komel, R. High FREM2 gene and protein expression are associated with favorable prognosis of IDH-WT glioblastomas. Cancers 2019, 11, 1060. [Google Scholar] [CrossRef] [Green Version]
- Zolotovskaia, M.A.; Tkachev, V.S.; Guryanova, A.A.; Simonov, A.M.; Raevskiy, M.M.; Efimov, V.V.; Wang, Y.; Sekacheva, M.I.; Garazha, A.V.; Borisov, N.M.; et al. OncoboxPD: Human 51 672 molecular pathways database with tools for activity calculating and visualization. Comput. Struct. Biotechnol. J. 2022, 20, 2280–2291. [Google Scholar] [CrossRef]
- Buzdin, A.A.; Sorokin, M.; Borisov, N.M.; Kuzmin, D.; Gudkov, A.; Zolotovskaia, M.A.; Garazha, A. V Algorithmic annotation of functional roles for components of 3044 human molecular pathways. Front. Genet. 2021, 12, 139. [Google Scholar]
- Yang, X.; Hao, J.-Q.; Zhang, Y.; Shi, J.-Y.; Zhu, X.-L.; Xiao, Y.-C.; Bai, H.; Hao, C.-Y. Four Key Genes are Biomarkers Associated with Immunity in Neuroglioma. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Park, Y.; Park, M.; Kim, J.; Ahn, J.; Sim, J.; Bang, J.I.; Heo, J.; Choi, H.; Cho, K.; Lee, M.; et al. NOX2-Induced High Glycolytic Activity Contributes to the Gain of COL5A1-Mediated Mesenchymal Phenotype in GBM. Cancers 2022, 14, 516. [Google Scholar] [CrossRef]
- Tsai, H.F.; Chang, Y.C.; Li, C.H.; Chan, M.H.; Chen, C.L.; Tsai, W.C.; Hsiao, M. Type V collagen alpha 1 chain promotes the malignancy of glioblastoma through PPRC1-ESM1 axis activation and extracellular matrix remodeling. Cell Death Discov. 2021, 7, 313. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Peng, Z.; Wu, Y.; Wang, Y.; Lei, D.; Jiang, X.; Zhao, H.; Fu, P. COL5A1 Serves as a Biomarker of Tumor Progression and Poor Prognosis and May Be a Potential Therapeutic Target in Gliomas. Front. Oncol. 2021, 11, 4749. [Google Scholar] [CrossRef] [PubMed]
- Herting, C.J.; Chen, Z.; Maximov, V.; Duffy, A.; Szulzewsky, F.; Shayakhmetov, D.M.; Hambardzumyan, D. Tumour-associated macrophage-derived interleukin-1 mediates glioblastoma-associated cerebral oedema. Brain 2019, 142, 3834. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Giotti, B.; Kaluzova, M.; Herting, C.J.; Pinero, G.; Vallcorba, M.P.; Cristea, S.; Ross, J.L.; Ackley, J.; Maximov, V.; et al. A paracrine circuit of IL-1β/IL-1R1 between myeloid and tumor cells drives glioblastoma progression. bioRxiv 2022, 2022.04.03.486888. [Google Scholar]
- Lin, H.; Wang, K.; Xiong, Y.; Zhou, L.; Yang, Y.; Chen, S.; Xu, P.; Zhou, Y.; Mao, R.; Lv, G.; et al. Identification of Tumor Antigens and Immune Subtypes of Glioblastoma for mRNA Vaccine Development. Front. Immunol. 2022, 13, 249. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef]
- Zolotovskaia, M.; Tkachev, V.; Sorokin, M.; Garazha, A.; Kim, E.; Kantelhardt, S.R.; Bikar, S.-E.; Zottel, A.; Šamec, N.; Kuzmin, D.; et al. Algorithmically Deduced FREM2 Molecular Pathway Is a Potent Grade and Survival Biomarker of Human Gliomas. Cancers 2021, 13, 4117. [Google Scholar] [CrossRef]
- Szopa, W.; Burley, T.A.; Kramer-Marek, G.; Kaspera, W. Diagnostic and therapeutic biomarkers in glioblastoma: Current status and future perspectives. Biomed Res. Int. 2017, 2017, 8013575. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.L.; Sorokin, M.; Kantelhardt, S.R.; Kalasauskas, D.; Sprang, B.; Fauss, J.; Ringel, F.; Garazha, A.; Albert, E.; Gaifullin, N.; et al. Intratumoral Heterogeneity and Longitudinal Changes in Gene Expression Predict Differential Drug Sensitivity in Newly Diagnosed and Recurrent Glioblastoma. Cancers 2020, 12, 520. [Google Scholar] [CrossRef] [Green Version]
- Kalasauskas, D.; Sorokin, M.; Sprang, B.; Elmasri, A.; Viehweg, S.; Salinas, G.; Opitz, L.; Rave-Fraenk, M.; Schulz-Schaeffer, W.; Kantelhardt, S.R.; et al. Diversity of clinically relevant outcomes resulting from hypofractionated radiation in human glioma stem cells mirrors distinct patterns of transcriptomic changes. Cancers 2020, 12, 570. [Google Scholar] [CrossRef] [Green Version]
- Samii, A.; Sorokin, M.; Kar, S.; Makovskaia, L.; Garazha, A.; Hartmann, C.; Moisseev, A.; Kim, E.; Giese, A.; Buzdin, A. Case of multifocal glioblastoma with four fusion transcripts of ALK, FGFR2, NTRK2, and NTRK3 genes stresses the need for tumor tissue multisampling for transcriptomic analysis. Cold Spring Harb. Mol. Case Stud. 2021, 7, a006100. [Google Scholar] [CrossRef] [PubMed]
- Buzdin, A.A.; Zhavoronkov, A.A.; Korzinkin, M.B.; Roumiantsev, S.A.; Aliper, A.M.; Venkova, L.S.; Smirnov, P.Y.; Borisov, N.M. The OncoFinder algorithm for minimizing the errors introduced by the high-throughput methods of transcriptome analysis. Front. Mol. Biosci. 2014, 1, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisov, N.; Suntsova, M.; Sorokin, M.; Garazha, A.; Kovalchuk, O.; Aliper, A.; Ilnitskaya, E.; Lezhnina, K.; Korzinkin, M.; Tkachev, V.; et al. Data aggregation at the level of molecular pathways improves stability of experimental transcriptomic and proteomic data. Cell Cycle 2017, 16, 1810–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raevskiy, M.; Sorokin, M.; Zakharova, G.; Tkachev, V.; Borisov, N.; Kuzmin, D.; Kremenchutckaya, K.; Gudkov, A.; Kamashev, D.; Buzdin, A. Better Agreement of Human Transcriptomic and Proteomic Cancer Expression Data at the Molecular Pathway Activation Level. Int. J. Mol. Sci. 2022, 23, 2611. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.M.; Ozenberger, B.A.; Ellrott, K.; Sander, C.; Stuart, J.M.; Chang, K.; Creighton, C.J.; et al. The Cancer Genome Atlas Pan-Cancer Analysis Project; Nature Publishing Group: Washington, DC, USA, 2013; Volume 45, pp. 1113–1120. [Google Scholar]
- Bady, P.; Sciuscio, D.; Diserens, A.-C.; Bloch, J.; van den Bent, M.J.; Marosi, C.; Dietrich, P.-Y.; Weller, M.; Mariani, L.; Heppner, F.L.; et al. MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol. 2012, 124, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Meng, F.; Wang, W.; Wang, Z.; Zhang, C.; Jiang, T. Comprehensive RNA-seq transcriptomic profiling in the malignant progression of gliomas. Sci. Data 2017, 4, 170024. [Google Scholar] [CrossRef]
- Bao, Z.S.; Chen, H.M.; Yang, M.Y.; Zhang, C.B.; Yu, K.; Ye, W.L.; Hu, B.Q.; Yan, W.; Zhang, W.; Akers, J.; et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res. 2014, 24, 1765–1773. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Li, Y.; Qian, Z.; Sun, Z.; Xu, K.; Wang, K.; Liu, S.; Fan, X.; Li, S.; Zhang, Z.; et al. A radiomic signature as a non-invasive predictor of progression-free survival in patients with lower-grade gliomas. NeuroImage Clin. 2018, 20, 1070–1077. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, T.; You, G.; Peng, X.; Chen, C.; You, Y.; Yao, K.; Wu, C.; Ma, J.; Sha, Z.; et al. Localizing seizure-susceptible brain regions associated with low-grade gliomas using voxel-based lesion-symptom mapping. Neuro. Oncol. 2015, 17, 282–288. [Google Scholar] [CrossRef] [Green Version]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro. Oncol. 2017, 19, 139–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, D. BioCarta. Biotech Softw. Internet Rep. 2001, 2, 117–120. [Google Scholar] [CrossRef]
- Romero, P.; Wagg, J.; Green, M.L.; Kaiser, D.; Krummenacker, M.; Karp, P.D. Computational prediction of human metabolic pathways from the complete human genome. Genome Biol. 2004, 6, R2. [Google Scholar] [CrossRef] [Green Version]
- QIAGEN—Pathway-Central. Available online: https://www.qiagen.com/us/shop/genes-and-pathways/pathway-central/ (accessed on 19 September 2018).
- Wishart, D.S.; Li, C.; Marcu, A.; Badran, H.; Pon, A.; Budinski, Z.; Patron, J.; Lipton, D.; Cao, X.; Oler, E.; et al. PathBank: A comprehensive pathway database for model organisms. Nucleic Acids Res. 2020, 48, D470–D478. [Google Scholar] [CrossRef]
- Buzdin, A.A.; Prassolov, V.; Zhavoronkov, A.A.; Borisov, N.M. Bioinformatics Meets Biomedicine: OncoFinder, a Quantitative Approach for Interrogating Molecular Pathways Using Gene Expression Data. Methods Mol. Biol. 2017, 1613, 53–83. [Google Scholar]
- Borisov, N.; Sorokin, M.; Garazha, A.; Buzdin, A. Quantitation of Molecular Pathway Activation Using RNA Sequencing Data. Methods Mol. Biol. 2020, 2063, 189–206. [Google Scholar]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov. 2021, 2, 100141. [Google Scholar] [CrossRef]
- Kohl, M.; Wiese, S.; Warscheid, B. Cytoscape: Software for visualization and analysis of biological networks. Methods Mol. Biol. 2011, 696, 291–303. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, M.; Ignatev, K.; Barbara, V.; Vladimirova, U.; Muraveva, A.; Suntsova, M.; Gaifullin, N.; Vorotnikov, I.; Kamashev, D.; Bondarenko, A.; et al. Molecular Pathway Activation Markers Are Associated with Efficacy of Trastuzumab Therapy in Metastatic HER2-Positive Breast Cancer Better than Individual Gene Expression Levels. Biochem. 2020, 85, 758–772. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Biomarkers 1 | Gene-Centric Pathway Biomarkers 1,2 |
|---|---|
| Up-regulated in GBM: ANXA2, CHI3L1, EMP3, ESM1, FN1, GPX8, HSPA6, IGFBP2, ITGA5, LDHA, MMP9, PLAT, PLAUR, PLEK2, SERPINH1, SRPX2, STC1, TAGLN2, TNFRSF12A. Up-regulated in LGG: ALDH5A1, GABBR1, GLUD1, MMD2, NDRG2, SLC25A21-AS1, ZDHHC22. | Up-regulated in GBM: ANXA2-pw, CD44-pw, HRG-pw, ITGB3BP-pw, KLK4-pw, ME2-pw, MFAP2-pw, MFAP5-pw, PAK2-pw, PCOLCE-pw, PLG-pw, PRSS1-pw, PRSS2-pw, SDC4-pw, SERPINB8-pw, SERPINE2-pw, TACC3-pw. Up-regulated in LGG: gdhA-pw, KCND2-pw, KCNIP2-pw, PTPRJ-pw. |
| Mesenchymal vs. Proneural | Classical vs. Proneural | Mesenchymal vs. Classical | |
| Gene biomarkers | 32 | 4 | 440 |
| Gene-centric pathway biomarkers | 283 | 0 | 254 |
| Mesenchymal vs. Proneural | Classical vs. Proneural | Mesenchymal vs. Classical | |
|---|---|---|---|
| Gene biomarkers | - | - | Upregulated in mesenchymal: CD14, EVA1A, FAM20A, MMP19, PLAUR. Upregulated in classical: ACSBG1, C2orf72, ELOVL2, ELOVL2-AS1, FGFR3, HEPACAM, MYO10, NPAS3, PTPRA, SLC24A3 |
| Gene-centric pathway biomarkers 1 | - | - | Upregulated in mesenchymal: CD180-pw, CD300E-pw, MIR100-pw, MIR99A-pw |
| Database/Subtype | Mesenchymal | Proneural | Classical |
|---|---|---|---|
| TCGA | 50 | 46 | 57 |
| CGGA_325 | 34 | 15 | 49 |
| CGGA_693 | 55 | 78 | 57 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zolotovskaia, M.A.; Kovalenko, M.A.; Tkachev, V.S.; Simonov, A.M.; Sorokin, M.I.; Kim, E.; Kuzmin, D.V.; Karademir-Yilmaz, B.; Buzdin, A.A. Next-Generation Grade and Survival Expression Biomarkers of Human Gliomas Based on Algorithmically Reconstructed Molecular Pathways. Int. J. Mol. Sci. 2022, 23, 7330. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137330
Zolotovskaia MA, Kovalenko MA, Tkachev VS, Simonov AM, Sorokin MI, Kim E, Kuzmin DV, Karademir-Yilmaz B, Buzdin AA. Next-Generation Grade and Survival Expression Biomarkers of Human Gliomas Based on Algorithmically Reconstructed Molecular Pathways. International Journal of Molecular Sciences. 2022; 23(13):7330. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137330
Chicago/Turabian StyleZolotovskaia, Marianna A., Max A. Kovalenko, Victor S. Tkachev, Alexander M. Simonov, Maxim I. Sorokin, Ella Kim, Denis V. Kuzmin, Betul Karademir-Yilmaz, and Anton A. Buzdin. 2022. "Next-Generation Grade and Survival Expression Biomarkers of Human Gliomas Based on Algorithmically Reconstructed Molecular Pathways" International Journal of Molecular Sciences 23, no. 13: 7330. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137330