
Ferroptosis: A Promising Therapeutic Target for Neonatal Hypoxic-Ischemic Brain Injury


1
Department of Pediatrics, University of Nebraska Medical Center, Omaha, NE 68198, USA
2
Children’s Hospital & Medical Center, Omaha, NE 68114, USA
3
Child Health Research Institute, Omaha, NE 68198, USA
4
Munroe-Meyer Institute for Genetics and Rehabilitation, University of Nebraska Medical Center, Omaha, NE 68198, USA
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(13), 7420; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137420
Submission received: 9 June 2022
/
Revised: 30 June 2022
/
Accepted: 1 July 2022
/
Published: 4 July 2022
(This article belongs to the Special Issue Molecular Mechanisms of Adaptation to Hypoxia)
Abstract
:Ferroptosis is a type of programmed cell death caused by phospholipid peroxidation that has been implicated as a mechanism in several diseases resulting from ischemic-reperfusion injury. Most recently, ferroptosis has been identified as a possible key injury mechanism in neonatal hypoxic-ischemic brain injury (HIBI). This review summarizes the current literature regarding the different ferroptotic pathways, how they may be activated after neonatal HIBI, and which current or investigative interventions may attenuate ferroptotic cell death associated with neonatal HIBI.
1. Introduction
Neonatal hypoxic-ischemic encephalopathy—the clinical phenotype resulting from neonatal hypoxic-ischemic brain injury (HIBI)—is a devastating neurological injury following decreased oxygen and blood flow to the brain around the time of birth. Every year, nearly 1.2 million newborns worldwide suffer from encephalopathy, and despite treatment with therapeutic hypothermia, almost half of infants with moderate to severe HIBI die or suffer from significant developmental disability [1,2]. Since high morbidity and mortality occur after neonatal HIBI despite available therapy, novel supplemental therapies are needed.
Ferroptosis is a relatively recently described mechanism of cell death that is distinct from other types of cell death such as apoptosis, necrosis, necroptosis, etc. [3]. Ferroptosis was first described in 2012 by Dixon, et al. [4]. In their investigation of non-apoptotic mechanisms of cell death, they documented that cell death induced by erastin or RAS-selective lethal 3 (RSL3) lacked the characteristic features of apoptosis such as mitochondrial cytochrome c release and caspase activation. Instead, they demonstrated that the cell death mechanism that they termed ferroptosis was primarily characterized morphologically by iron-dependent accumulation of lipid reactive oxygen species and alterations in several ferroptosis-related genes such as iron response element binding protein 2 (IREB2), citrate synthase (CS), and acyl-CoA synthetase family member 2 (ACSF2).
The modulation of ferroptosis has been recognized as a potential target for novel therapies against numerous diseases [5]. Ferroptosis has been linked to the pathophysiology of multiple disorders of ischemia-reperfusion, including intestinal ischemia [6], myocardial infarction [7], and ischemic stroke [8]. Ferroptosis was recently identified as a key mediator of cortical mitochondrial damage [9] and hippocampal neuronal death [10] after neonatal HIBI. Despite the known involvement of ferroptosis in these conditions, the endogenous mechanisms remain poorly understood.
The timing of intervention is likely as important as the target, because neonatal HIBI is a triphasic injury [11]. The first phase initiates with the hypoxic-ischemic insult, the primary energy failure and hypoperfusion resulting in necrotic cell death in the minutes to hours after the restoration of normoxia and adequate perfusion. The secondary phase starts between 6 and 12 h after injury and continues until about 72 h after injury. It is characterized by deteriorating mitochondrial function and an acute inflammatory response, with apoptosis being the hallmark cell death process during this phase, though this is also the timing where ferroptotic changes have been observed (24–72 h [9,10]). After the secondary phase is the tertiary phase, which includes partial recovery and continued inflammation and gliosis [12]. When possible, this review will note the timing of the mechanisms and interventions studied to attempt to better understand the temporal changes in ferroptosis after neonatal HIBI.
Overall, this manuscript will review ferroptosis pathways and the role ferroptosis may play in HIBI-related cell death as well as potential therapeutic interventions to target neonatal HIBI. A recent review provided a general discussion of ferroptosis in neonatal brain injury (including preterm injury such as intraventricular hemorrhage) [13]; here, we will update the literature and focus on a more in-depth description of neonatal HIBI, including a comprehensive discussion of current investigative therapies that may target ferroptosis after neonatal HIBI.
2. Ferroptosis Pathways
Although oxygen-glucose deprivation has been the primary method of modeling hypoxia-ischemia in vitro, much like the clinical pathophysiology of HIBI, oxygen-glucose deprivation results in the activation of several parallel cell death mechanisms [14,15]. As such, the majority of the research described in this section will focus on pure ferroptosis models. These are usually generated by inducing ferroptosis through the inhibition of cystine transport (through the system Xc- pathway) by administering erastin or extracellular glutamate or the direct inhibition of glutathione peroxidase 4 (GPX4) with the chemical (1S,3R)-RSL3.
2.1. Phospholipid Peroxidation and Ferroptosis
Oxidative stress, lipid peroxidation, and cell death have long been implicated in many (patho)physiological conditions, including cancer, neurodegeneration, and ischemia-reperfusion injuries [16,17,18,19,20]. The specific association between lipid peroxidation and cell death was described in early studies linking the hepatotoxicity of carbon tetrachloride (CCl4) to lipid peroxidation [21]. Lipids with one or more double bond can undergo lipid peroxidation, but polyunsaturated species (more than one double bond) are more susceptible to this process. Polyunsaturated fatty acids (PUFAs) present in phospholipids (e.g., phosphatidylcholine, phosphatidylethanolamine, phosphatidylinositol, cardiolipin, etc.) are among the major targets of free radical-mediated lipid peroxidation. The biochemistry of lipid peroxidation has been reviewed in detail many times [3,22], and discussing these mechanisms is beyond the scope of the present review. It is important to note, however, that the peroxidation of lipids leads to the formation of highly reactive species, including epoxides, hydroperoxides, and aldehydes. The generation and accumulation of these compounds combined with the cellular inability to detoxify them are key factors in the ferroptotic cell death cascade. This is especially true in newborns who possess immature and incomplete antioxidant defenses as well as high concentrations of PUFAs accumulated in the brain during early life to support brain development [23,24].
Within the last decade, the association between lipid peroxidation and cell death has become more relevant, with the description of a regulated necrotic cell death modality characterized by increased phospholipid peroxidation [4,25,26,27]. The use of different small molecules and genetic animal models then led to the understanding that this method of cell death was iron-dependent [28], leading to its name: ferroptosis. Although phospholipid oxidation products are increased during the ferroptosis cascade [27], cardiolipin (a species found exclusively in the mitochondria) products are not increased. It is not clear yet whether they are not formed or they are rapidly consumed during the process [29]. There are multiple ferroptosis pathways, many of which revolve around iron, acyl-CoA, and glutathione (GSH) metabolism (particularly GSH synthesis and the activity of GPX4). These pathways are demonstrated in Figure 1, and some of the key pathways are further described below.
2.2. The Cystine/Glutamate Antiporter (Xc-)
GSH is among the most important small-molecule antioxidants in cells, where it is found in the range of 1–10 mM [30]. It is a tripeptide synthesized from glutamate, glycine and cysteine. The antioxidant activity of GSH relies on its ability to quench highly reactive species, including hydroxyl radicals, peroxyl radicals, and alcoxyl radicals [31]. The oxidation of GSH generates glutathione disulfide (GSSG), which consists of two glutathione molecules linked by a disulfide bond. GSH is also used in the elimination of electrophilic peroxidation products (e.g., 4-hydroxynonenal [4HNE]) in reactions catalyzed by glutathione-S-transferase enzymes (GSTs) [32,33]. GSH is a substrate for glutathione peroxidase enzymes (eight isoforms, GPX1-GPX8; see Section 2.3 for more details), which reduce peroxides to their corresponding alcohols. The GSH/GSSG ratio can be used to estimate the overall redox status of a cell [34]. As cellular oxidative stress increases, GSH is consumed and GSSG is generated, decreasing the GSH/GSSG ratio [34]. Decreases in this ratio have been described in many (patho)physiological processes, including neurodegeneration, ischemia-reperfusion injuries and during the ferroptotic cell death cascade [35,36].
The rate-limiting step in the endogenous synthesis of GSH is the uptake of cysteine to the cytosol. Cysteine is obtained by the cells via the cystine/glutamate antiporter (Xc-) system, which relies on importing cystine (the oxidized form of cysteine) at the expense of a glutamate in a 1:1 ratio [37]. Once inside the cell, cystine is rapidly reduced to cysteine and then utilized for GSH synthesis [38]. In 2012, Dixon et al. utilized erastin, an irreversible inhibitor of the Xc- system, to demonstrate that cells died by ferroptosis when deprived of their cystine uptake mechanism [28]. The cystine deprivation halted the synthesis of GSH resulting in increased lipid peroxidation and cell death [28]. The synthesis of GSH can also be inhibited by buthionine sulfoximine (BSO), sulfasalazine, glutamate, and sorafenib, and several studies have demonstrated that the administration of these compounds increases the cellular susceptibility to ferroptosis [26,39,40]. Inversely, attempts have been made to boost the synthesis of GSH by using the non-toxic precursor N-acetylcysteine (NAC), which could modulate ferroptosis and have therapeutic applications [41,42].
2.3. The Glutathione Peroxidase 4 (GPX4) Pathway
One of the main mechanisms to detoxify phospholipid hydroperoxides is GPX4. GPX4 is a seleno-cysteine enzyme that reduces phospholipid hydroperoxides to their corresponding hydroxides at the expense of GSH and NADPH [43]. Removing or decreasing the cellular GPX4 activity (either by genetic manipulation or chemical inhibition) leads to increased lipid peroxidation and accumulation of phospholipid hydroperoxides [44]. Selectively deleting GPX4 in neurons results in mortality in the neonatal period [45,46], while conditionally deleting this enzyme in adult animals leads to neurodegeneration, mitochondrial oxidative damage and ferroptosis [47]. Similarly, the use of RSL3, a specific inhibitor of GPX4, leads to increased lipid peroxidation and associated ferroptosis [48,49]. It is worth mentioning that the inhibition of GSH synthesis described in the previous section also affects the GPX4 activity, as GSH is a critical co-substrate that allows for the reduction of phospholipid hydroperoxides [3]. Hence, compromising the cysteine-GSH-GPX4 system leads to the accumulation of lipid peroxidation products and associated ferroptotic cell death.
2.4. The Roles of ACSL4 and LPCAT3 in Ferroptosis Sensitivity
The enzyme acyl-CoA synthetase long-chain family member 4 (ACSL4) also plays a central role in the regulation of ferroptosis. ACSL4 is required for activation of ω-6 PUFAs, especially arachidonic acid (AA) and adrenic acid (AdA) to their acyl-CoA derivatives, allowing for their incorporation into phospholipids [50]. AA and AdA are highly oxidizable fatty acids, and in a scenario where lipid peroxidation is increased, they become primary targets for oxidation [27]. In 2016, Doll et al. demonstrated that modulating ACSL4′s activity affects the cellular sensitivity to ferroptosis [51]. As described in Section 2.3, knocking out GPX4 in cells led to increased lipid peroxidation and ferroptosis. The introduction of a double knockout (both ACSL4 and GPX4) in the cells translated into an increased resistance to ferroptosis [51]. Since ACSL4 allows for the esterification of highly oxidizable PUFAs for incorporation into the cell membrane, its knockout depletes the membrane of PUFAs, resulting in a less oxidizable membrane and decreased phospholipid peroxidation and ferroptosis. These findings suggest that manipulating ACSL4 activity can be a potential therapeutic approach for conditions where ferroptosis is a contributing factor (e.g., HIBI). Notably, increasing the levels of monosaturated fatty acids (MUFAs) translates into increased resistance to ferroptosis [52]. The supplementation with MUFAs changed the cellular lipidome, decreasing the incorporation of PUFAs in the membrane and, therefore, suppressing ferroptosis.
The incorporation of PUFAs in the cell membrane requires two steps. ACSL4 first activates the fatty acid by adding a CoA unit, as described above. The next step is catalyzed by lysophosphatidylcholine acyltransferase 3 (LPCAT3), an enzyme that takes the PUFA-CoA (AA-CoA and AdA-CoA) and synthesizes the corresponding phospholipid-PUFA [27,53]. LPCAT3 catalyzes such reactions with phosphatidylcholine (PC) and phosphatidylethanolamine (PE). It has been shown that modulating LPCAT3 activity affects resistance to ferroptosis [27]. Suppression of LPCAT3 activity translates into increase resistance to ferroptosis, which can be attributed to the lower abundance of highly oxidizable PUFAs in the membrane; less oxidizable substrate translates to less phospholipid peroxidation and suppression of ferroptosis.
2.5. The Role of Hypoxia-Inducible Factors (HIFs) in Ferroptosis Sensitivity
HIF1α is the 3′ enhancer of the erythropoietin gene and contains three domains: the N-terminal domain, the C-terminal transactivation domain which primarily regulates gene transcription activity, and an oxygen-dependent degradation domain which mediates oxygen-dependent stability [54]. HIF1α suppresses ACSL4 after adult ischemic stroke, thereby attenuating neuronal death and inhibiting microglial cytokine production [55]. HIF1α has also been shown to promote intracellular lipid storage in lipid droplets, reducing fatty acid oxidation and protecting cells from ferroptosis [56]. Though less studied than the 1α isoform, HIF2α also likely plays an inverse but similarly important role in ferroptosis. HIF2α has been shown to selectively enrich lipids containing polyunsaturated fatty acyl side chains, resulting in a more ferroptosis-sensitive cell state [57].
The HIF-related EGLNs are oxygen- and iron-dependent enzymes that may also alter cellular ferroptosis. EGLNs are proline hydroxylases (PHDs) that hydroxylate proline residues on HIF1α, resulting in increased HIF1α degradation and increased susceptibility to ferroptosis [58]. EGLN knockdown by hypoxia or chemical inhibition results in increased HIF1α expression [56] and increased ferroptosis resistance.
2.6. FSP1-CoQ10-NADPH Pathway
Ferroptosis suppressor protein 1 (FSP1)—previously known as apoptosis-inducing factor mitochondria-2 (AIFM2)—is a key GPX4-independent modulator of lipid peroxidation. FSP1 catalyzes the NAD(P)H-dependent reduction of coenzyme Q10 (CoQ10, also known as ubiquinone) to ubiquinol (CoQ10H2), which is a potent antioxidant that attenuates lipid peroxidation [59,60,61]. CoQ10 is a lipophilic molecule present in every cell membrane, especially in the inner mitochondrial membrane, where it is responsible for transporting electrons from complexes I and II to complex III [62,63]. CoQ10 deficiency has been linked to many diseases, including encephalomyopathy, cerebellar ataxia, ischemic heart disease, and metabolic syndrome [64,65].
In addition to its role in oxidative phosphorylation, CoQ10 is a potent antioxidant [64,66,67,68]. CoQ10 rapidly reacts with alcoxyl and peroxyl radicals, thus preventing the propagation of lipid peroxidation [69]. Studies have shown that deficiency in CoQ10 leads to increased lipid peroxidation markers, including malondialdehyde (MDA) [70]. More recently, it has been shown that altering CoQ10 metabolism modulates ferroptosis [71].
2.7. The Role of Lipoxygenases in Ferroptosis
Lipid peroxidation occurs via two main mechanisms: metal-mediated peroxyl radical formation and an enzyme-mediated pathway catalyzed by lipoxygenases (LOXs) [72,73,74]. Two specific LOXs have been associated with ferroptotic cell death: 15-lipoxygenase (15-LOX-1) and 12-lipoxygenase (12-LOX) [72,73,74]. In 2017, Wenzel and co-workers described the pro-ferroptotic effects of 15-LOX-1. The authors used phosphatidylethanolamine-binding protein 1 (PEB1), a protein inhibitor that complexes with 15-LOX and decreases its activity, to demonstrate that the inhibition of 15-LOX-1 suppressed ferroptosis [74]. Moreover, while the pharmacological inhibition of 15-LOX-1 increased resistance to ferroptosis in gpx4−/− mice [75], the genetic manipulation of alox15 (gene that encodes 15-LOX-1) did not affect the course of ferroptosis in gpx4−/− mice [76]. More recently, Shah and their co-workers utilized cell culture models to characterize the contributions of 5-LOX, 12-LOX and 15-LOX-1 [73]. The overexpression of the three LOX isoforms increased the sensitivity of cells to ferroptosis. Notably, LOX-mediated ferroptosis was suppressed by liproxstatin-1 and other antioxidants, indicating that progression of the lipid peroxidation cascade is vital for cell death [73]. Pharmacological inhibitors of 5-LOX failed to protect cells from ferroptosis, however, suggesting that not all LOX isoforms contribute equally to the process [73]. In their conclusion, the authors proposed that LOXs contribute to the overall pool of hydroperoxides, but the main contributor to the ferroptotic cell death is the uncontrolled progression of lipid peroxidation.
3. Mechanisms and Therapeutic Targeting of Ferroptosis in HIBI
Many of the ferroptosis pathways have been demonstrated to be altered after neonatal HIBI, and most of them have at least one published study demonstrating an attempt to alter the pathway for neuroprotection (Figure 2, Table 1).
3.1. Oxidative Stress and Lipid Peroxidation
3.1.1. Lipid Peroxidation and HIBI
It is widely accepted that oxidative stress is a key component of neonatal HIBI; a concept that is supported by studies demonstrating alterations in markers of oxidative stress such as MDA and GSH. Blood MDA concentrations have been shown to be increased in cord blood and at 48 h of life in infants with perinatal asphyxia compared to healthy controls [85]. Attenuation of oxidative stress is thought to be one of the mechanisms of action of therapeutic hypothermia, as 72 h of hypothermia reduced MDA levels compared to normothermic controls [86].
MDA and GSH are nonspecific biomarkers for cell injury and protection, however, and other assays are more specific for the lipid peroxidation that occurs during ferroptosis compared to other etiologies of oxidative stress. For instance, one study has assessed lipid peroxidation urinary biomarkers in the first 6 h of life and at 12, 24, 48, 72, and 96 h after initiation of hypothermia [87]. The investigators demonstrated that urine isoprostanes increased over time and prostaglandins decreased. Similarly, lipid peroxidation (LPO) concentrations measured by ELISA may correlate with severity of encephalopathy, Apgar score, and initial blood pH. Additionally, LPO concentrations were higher in infants with asphyxia who died compared to those who survived [88]. LPO elevation had 89% sensitivity, 96% specificity, 96% PPV, and 90% NPV for the diagnosis of neonatal hypoxic-ischemic encephalopathy [89].
3.1.2. Interventions Targeting Lipid Peroxidation after HIBI
Several current and potential interventions non-specifically affect or specifically target oxidative stress and lipid peroxidation. One of the mechanisms by which therapeutic hypothermia—the only proven effective therapy in neonatal HIBI—is thought to confer neuroprotection is in part through reduction of free radical production [90,91]. Other investigative therapies include metformin, which suppresses mitochondrial complex I, thereby restricting mitochondrial respiration [92].
Other scavengers of oxygen free radicals and general antioxidants that have been tested in neonatal HIBI include allopurinol [77], melatonin [83], and N-acetylcysteine [93,94]. Vitamin D has also been used with and without NAC to improve the cellular redox state after oxidative brain injury [95,96,97]. Similarly, vitamin E (α-tocopherol) is a well-studied antioxidant that when administered prior to injury in a piglet model of hypoxic brain injury may decrease lipid peroxidation [98]. The antioxidant action of vitamin E is supported further by administration of vitamin C, which facilitates the return of the harmful oxidized α-tocopherol radical back to α-tocopherol [99,100]. In addition to several other potential mechanisms of action in neonatal HIBI, erythropoietin may also exert its neuroprotective effects on lipid peroxidation [101,102].
Although lipid peroxidation is a key mechanism of ferroptotic cell death, it is not specific to ferroptosis alone. The remainder of this review will focus on the data surrounding the involvement of ferroptosis-specific pathways in neonatal HIBI pathophysiology.
3.2. Fenton Reaction
3.2.1. Fenton Reaction and HIBI
The iron-based Fenton reaction is one of the extrinsic or transporter-dependent ferroptosis pathways that has been associated with neonatal HIBI. Newborns with severe asphyxia have been shown to have significantly elevated levels of free (not protein-bound) iron and lipid peroxidation in the plasma throughout the first 24 h of life. Additionally, those infants with severe asphyxia that went on to have abnormal neurodevelopment at 1 year of age were also more likely to have detectable levels of free iron in the plasma compared to those with normal development [103]. This was subsequently confirmed in the newborn lamb model of HIBI. In the animal model, the elevation in plasma free iron was also associated with increased MDA concentrations and decreased alpha-tocopherol and ascorbic acid/dehydroascorbic acid concentrations [104]. Neither of the previous studies assessed brain concentrations of iron products, but preliminary investigations in brain tissue have been performed in the rat model. At 72 h after neonatal HIBI in the rat model, the ipsilateral brain demonstrated significantly increased expression of transferrin receptor, ferritin heavy chain, and ferritin light chain [9]. It is important to note, however, that there is a balance that must be achieved with regards to iron concentrations in the brain, as too little iron can also be injurious and also leads to adverse neurodevelopmental outcomes [105].
3.2.2. Interventions Targeting Fenton Reaction after HIBI
Investigations into targeting this pathway have included attempts to decrease free iron production or scavenge free iron products to prevent the production of free radicals, or to attenuate the injury caused by iron radicals that have already been formed. One study used N-omega-nitro-L-arginine to inhibit nitric oxide formation and showed that administration immediately after HIBI resulted in significantly lower plasma concentrations of free iron as well as lower MDA [104]. Iron chelators such as deferoxamine have also been used in multiple studies of neonatal HIBI to attempt to decrease free iron products in the blood and brain [106,107,108,109,110], though no human studies have yet been reported. Similarly, administration of exogenous erythropoietin may inhibit Fenton chemistry, potentially through the iron scavenging process of erythropoiesis [80,81]. Lastly, vitamin D—specifically 1,25-hydroxyvitamin D—also has the potential to decrease iron-induced lipid peroxidation and neuronal injury [111,112].
3.3. GPX4 and System Xc-
3.3.1. GPX4 and System Xc- Pathway
GPX4 activity is a key regulator of ferroptosis and has been shown to play an important role in neonatal HIBI pathophysiology. Although the cumulative level of all GPX proteins does not appear to be altered after neonatal HIBI in mice [113], the GPX4 isoform protein concentration is decreased [10] along with decreases in GSH and 4-HNE in the ipsilateral hippocampus after HIBI compared to sham controls [84]. GPX concentrations were found to be elevated in the cerebrospinal fluid of neonates with HIE, correlating with encephalopathy severity (measured by modified Sarnat staging), arterial cord blood pH, and neurodevelopment at one year as measured by the Denver Developmental Screening Test [114]. These results are complicated somewhat by the inclusion of preterm infants (as low as 32 weeks of gestation) in the study and the lack of specific GPX4 measurements, however.
GPX overexpression in a transgenic mouse model resulted in significantly decreased histologic brain injury scores after neonatal HIBI [113,115]. This upregulation of GPX results in increased FLICE inhibitory protein (FLIP) which is a key regulator in the Fas death receptor pathway and inhibits cleavage of caspase-8 into its active form [116]. The endogenous upregulation of GPX after hypoxic-preconditioning may be in part responsible for the neuroprotective effects of the preconditioning against HIBI, an effect that was not replicated by increasing GSH concentrations alone [115]. Although GPX4 has been the most thoroughly studied GPX isoform, GPX1 knockout was also shown to increase injury after ischemia/reperfusion in adult brains [117,118].
For the System Xc-, hypoxia alone results in increased expression of SLC3A2 immediately after injury in the forebrain of a mouse model [119]. This increase in system Xc- activity and the resulting increase in cystine uptake has also been associated with hypoxic preconditioning in neural stem cells [120]. Conversely, after HIBI, decreased SLC7A11 protein expression is seen in the ipsilateral hemisphere at both 24 and 72 h after injury and is associated with decreased GSH and GPX4 [9,10]. The difference between the increased SLC3A2 in the first study and the decreased SLC7A11 in the latter study could be due to the different pathophysiology in hypoxia alone versus hypoxia-ischemia, but also could be related to the timing of evaluation given the multiphasic nature of hypoxic-ischemic injury.
3.3.2. Interventions Targeting GPX4 Pathway after HIBI
Several interventions have been shown to target or directly alter GPX4 signaling. For example, targeted manipulation of GPX4 may be possible through the use of microRNA antagonism (resulting in increased GPX4 expression), as was demonstrated in renal ischemia and reperfusion where investigators demonstrated decreased renal injury with silencing of miR-182-5p and miR-378a-3p [121]. Intraperitoneal injection of adaptaquin, which is a hypoxia-inducible factor prolyl 4-hydrolase (HIF-PHD)-inhibitor, to the neonatal mouse model of HIBI also resulted in increased GPX4 mRNA expression in the cortex. This increase was associated with decreased cell death in the cortex and overall reduction in infarct volume, though the tissue-sparing effect was more pronounced in males than females [122].
A less targeted but more clinically feasible approach at this time would be to use an FDA-approved medication to alter GPX4 expression. For instance, melatonin administration after neonatal HIBI attenuated GPX4, GSH, and 4-HNE changes, with subsequent improvement in behavioral outcomes [84]. Erythropoietin also may be able to increase GPX expression. The only study to date to address this question, however, measured general GPX expression rather than the specific sub-types [82]. This is an important distinction because although multiple members of the GPX family can detoxify H2O2 and fatty acid hydroperoxides, GPX1 and others are not able to reduce hydroperoxides in membrane lipids to the extent of GPX4 [123]. Further studies will be necessary to confirm the specific mechanism by which erythropoietin attenuates lipid peroxidation.
3.4. FSP1-CoQ10-NADPH Pathway
Knockout of Aifm2/Fsp1 resulted in greater brain injury at 72 h after HIBI, associated with increased expression level of AIF1 [124]. Additionally, CoQ10 supplementation has shown some promise as a neuroprotective intervention in models of adult brain ischemia [125,126]. Although CoQ10 administration has not yet been explored in neonates, the mitochondria-targeted ubiquinone compound mitoquinol has been trialed in the rat model of neonatal HIBI. The group only assessed the number of medium-spiny neurons in the striatum, however, and found no difference between mitoquinol and vehicle control [127].
One key factor in the modulation of CoQ10 homeostasis relies on the manipulation of its biosynthesis. CoQ10 is synthesized de novo using isoprene units as building blocks [128,129]. The isoprene units are provided by the mevalonate pathway, and therefore can be manipulated by the use of statins [128,130,131]. Indeed, a meta-analysis study shows that statin treatment decreased circulating CoQ10 levels [132], and other studies suggest that CoQ10 supplementation can increase its circulating levels in patients taking statins [131,133]. All together, these observations suggest that ferroptosis can be modulated by statins and that many of the statin-associated myopathies could be related to lipid peroxidation and associated ferroptosis, which should be the topic of future investigations.
3.5. Downstream Pathways
The role of TLR4 in ferroptosis is complex. Although ferroptotic regulation of 4-HNE and oxidized phospholipids results in activation of TLR4 and subsequent induction of proinflammatory cytokines through NF-κB activation [134], it has also been hypothesized that neonatal HIBI increases ferroptosis by direct activation of TLR4. To support this hypothesis, Zhu, et al. demonstrated that HIBI increased TLR4 expression, and inhibition of TLR4 using TAK-242 resulted in decreased hippocampal expression of ferroptosis-related genes ATP5G3, PTGS2, CS, IREB2, and RPL8, and increased SLC7A11 and GPX4 in both cell culture and rat models of neonatal HIBI [10]. These data suggest that, at least in neonatal HIBI, TLR4 may also provide upstream regulation of ferroptosis in addition to being upregulated downstream from ferroptosis.
The ferroptosis-related release of danger-associated molecular patterns (DAMPs) such as high-mobility group box 1 (HMGB1) and the reactive aldehydes 4-hydroxynonenal (4-HNE) and MDA has been used to develop biomarkers of ferroptotic injury. These chemicals are also in themselves pro-inflammatory, however, so may additionally provide targets for therapeutic intervention. For example, HMGB1 is a transcription factor involved in chromatin remodeling and inflammation signaling. HMGB1 is released from injured cells and contributes to brain injury after neonatal HIBI in part through microglial regulation [135,136]. It was previously shown in cancer cells that knock down HMGB1 decreased erastin-induced ferroptosis through the RAS-JNK/p38 pathway [137], and a very recent study demonstrated that HMGB1 inhibition by glycyrrhizin administration decreased neuronal ferroptosis in both a cell culture oxygen-glucose deprivation model and after neonatal HIBI in rats in vivo [138].
4. Conclusions
Ferroptotic cell death can result from the activation of multiple pathways that result in uncontrolled lipid peroxidation. Recent studies have suggested that ferroptosis occurs after neonatal HIBI and that attenuating ferroptosis-related changes after injury may be neuroprotective. Despite the promising early studies, several significant questions remain unexamined in the current literature. These include outstanding questions regarding several of the specific mechanisms of ferroptosis, such as the role of other ferroptosis-associated pathways that have not yet been demonstrated in HIBI (such as KRAS [139]) and the role of stress granule formation [140]. Additional HIBI-specific questions include the temporal nature of the cellular changes after HIBI and whether ferroptosis follows the timing of the three canonical phases of injury. Lastly, many unanswered questions remain surrounding the use of the interventions listed in Figure 2 (and others not yet investigated), including whether the suppression of ferroptotic cell death simply redirects the injury to one or more of the other cell death pathways known to affect cells after neonatal HIBI. Ultimately, however, the current literature suggests that there is considerable promise that attenuating ferroptosis could decrease brain injury and improve outcomes in this high-risk population.
Author Contributions
Conceptualization, E.S.P. and T.C.G.-M.; writing—original draft preparation, E.S.P. and T.C.G.-M.; writing—review and editing, E.S.P. and T.C.G.-M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by the National Institute of Mental Health, grant number MH110636. The APC was funded by the Child Health Research Institute.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jacobs, S.E.; Berg, M.; Hunt, R.; Tarnow-Mordi, W.O.; Inder, T.E.; Davis, P.G. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst. Rev. 2013, 2013, CD003311. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.C.; Kozuki, N.; Blencowe, H.; Vos, T.; Bahalim, A.; Darmstadt, G.L.; Niermeyer, S.; Ellis, M.; Robertson, N.J.; Cousens, S.; et al. Intrapartum-related neonatal encephalopathy incidence and impairment at regional and global levels for 2010 with trends from 1990. Pediatric Res. 2013, 74 (Suppl. S1), 50–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat. Chem. Biol 2019, 15, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, Y.; Wu, J.; Yang, J.; Li, M.; Chen, Q. The Potential Value of Targeting Ferroptosis in Early Brain Injury After Acute CNS Disease. Front. Mol. Neurosci. 2020, 13, 110. [Google Scholar] [CrossRef]
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019, 26, 2284–2299. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Li, Y.; Zhang, S.; Zhou, X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics 2021, 11, 3052–3059. [Google Scholar] [CrossRef]
- Bu, Z.-Q.; Yu, H.-Y.; Wang, J.; He, X.; Cui, Y.-R.; Feng, J.-C.; Feng, J. Emerging Role of Ferroptosis in the Pathogenesis of Ischemic Stroke: A New Therapeutic Target? ASN Neuro 2021, 13, 17590914211037505. [Google Scholar] [CrossRef]
- Lin, W.; Zhang, T.; Zheng, J.; Zhou, Y.; Lin, Z.; Fu, X. Ferroptosis is Involved in Hypoxic-ischemic Brain Damage in Neonatal Rats. Neuroscience 2022, 487, 131–142. [Google Scholar] [CrossRef]
- Zhu, K.; Zhu, X.; Sun, S.; Yang, W.; Liu, S.; Tang, Z.; Zhang, R.; Li, J.; Shen, T.; Hei, M. Inhibition of TLR4 prevents hippocampal hypoxic-ischemic injury by regulating ferroptosis in neonatal rats. Exp. Neurol. 2021, 345, 113828. [Google Scholar] [CrossRef]
- Hassell, K.J.; Ezzati, M.; Alonso-Alconada, D.; Hausenloy, D.J.; Robertson, N.J. New horizons for newborn brain protection: Enhancing endogenous neuroprotection. Arch. Dis. Child.-Fetal Neonatal Ed. 2015, 100, F541–F552. [Google Scholar] [CrossRef] [PubMed]
- Volpe, J.J.; Inder, T.E.; Darras, B.T.; de Vries, L.S.; du Plessis, A.J.; Neil, J.; Perlman, J.M. Volpe’s Neurology of the Newborn e-Book; Elsevier Health Sciences: Philadelphia, PA, USA, 2017. [Google Scholar]
- Wu, Y.; Song, J.; Wang, Y.; Wang, X.; Culmsee, C.; Zhu, C. The Potential Role of Ferroptosis in Neonatal Brain Injury. Front. Neurosci. 2019, 13, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, A.; Delgado-Esteban, M.; Bolaños, J.P.; Medina, J.M. Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J. Neurochem. 2002, 81, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Juntunen, M.; Hagman, S.; Moisan, A.; Narkilahti, S.; Miettinen, S. In Vitro Oxygen-Glucose Deprivation-Induced Stroke Models with Human Neuroblastoma Cell- and Induced Pluripotent Stem Cell-Derived Neurons. Stem Cells Int. 2020, 2020, 8841026. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef] [Green Version]
- Porter, N.A.; Xu, L.; Pratt, D.A. Reactive Sterol Electrophiles: Mechanisms of Formation and Reactions with Proteins and Amino Acid Nucleophiles. Chemistry 2020, 2, 390–417. [Google Scholar] [CrossRef]
- Reed, T.T. Lipid peroxidation and neurodegenerative disease. Free Radic. Biol. Med. 2011, 51, 1302–1319. [Google Scholar] [CrossRef]
- Perluigi, M.; Coccia, R.; Butterfield, D.A. 4-Hydroxy-2-nonenal, a reactive product of lipid peroxidation, and neurodegenerative diseases: A toxic combination illuminated by redox proteomics studies. Antioxid. Redox Signal. 2012, 17, 1590–1609. [Google Scholar] [CrossRef] [Green Version]
- Recknagel, R.O. Carbon tetrachloride hepatotoxicity. Pharmacol. Rev. 1967, 19, 145–208. [Google Scholar]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Georgeson, G.D.; Szőny, B.J.; Streitman, K.; Varga, I.S.; Kovács, A.; Kovács, L.; László, A. Antioxidant enzyme activities are decreased in preterm infants and in neonates born via caesarean section. Eur. J. Obstet. Gynecol. Reprod. Biol. 2002, 103, 136–139. [Google Scholar] [CrossRef]
- Martinez, M. Tissue levels of polyunsaturated fatty acids during early human development. J. Pediatr. 1992, 120, S129–S138. [Google Scholar] [CrossRef]
- Angeli, J.P.F.; Freitas, F.P.; Nepachalovich, P.; Puentes, L.; Zilka, O.; Inague, A.; Lorenz, S.; Kunz, V.; Nehring, H.; da Silva, T.N.X. 7-Dehydrocholesterol Is an Endogenous Suppressor of Ferroptosis. Res. Sq. 2021; preprint. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Alary, J.; Guéraud, F.; Cravedi, J.P. Fate of 4-hydroxynonenal in vivo: Disposition and metabolic pathways. Mol. Asp. Med. 2003, 24, 177–187. [Google Scholar] [CrossRef]
- Boyland, E.; Chasseaud, L.F. The role of glutathione and glutathione S-transferases in mercapturic acid biosynthesis. Adv. Enzym. Relat. Areas Mol. Biol. 1969, 32, 173–219. [Google Scholar] [CrossRef]
- Jones, D.P. Redox potential of GSH/GSSG couple: Assay and biological significance. Methods Enzym. 2002, 348, 93–112. [Google Scholar] [CrossRef]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Dhalla, N.S.; Elmoselhi, A.B.; Hata, T.; Makino, N. Status of myocardial antioxidants in ischemia-reperfusion injury. Cardiovasc. Res. 2000, 47, 446–456. [Google Scholar] [CrossRef]
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System xc− cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2012, 165, 20–34. [Google Scholar] [CrossRef] [Green Version]
- Mandal, P.K.; Seiler, A.; Perisic, T.; Kölle, P.; Banjac Canak, A.; Förster, H.; Weiss, N.; Kremmer, E.; Lieberman, M.W.; Bannai, S.; et al. System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J. Biol. Chem. 2010, 285, 22244–22253. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef] [Green Version]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Karuppagounder, S.S.; Alin, L.; Chen, Y.; Brand, D.; Bourassa, M.W.; Dietrich, K.; Wilkinson, C.M.; Nadeau, C.A.; Kumar, A.; Perry, S.; et al. N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E(2) to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann. Neurol. 2018, 84, 854–872. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Zhang, Y.; Ma, S.; Li, J.; Wang, X.; Liang, M.; Sferruzzi-Perri, A.N.; Wu, X.; Ma, H.; Brännström, M.; et al. Suppression of uterine and placental ferroptosis by N-acetylcysteine in a rat model of polycystic ovary syndrome. Mol. Hum. Reprod. 2021, 27, gaab067. [Google Scholar] [CrossRef]
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid. Redox Signal. 2018, 29, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yant, L.J.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic. Biol. Med. 2003, 34, 496–502. [Google Scholar] [CrossRef]
- Imai, H.; Hirao, F.; Sakamoto, T.; Sekine, K.; Mizukura, Y.; Saito, M.; Kitamoto, T.; Hayasaka, M.; Hanaoka, K.; Nakagawa, Y. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem. Biophys. Res. Commun. 2003, 305, 278–286. [Google Scholar] [CrossRef]
- Chen, L.; Hambright, W.S.; Na, R.; Ran, Q. Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. J. Biol. Chem. 2015, 290, 28097–28106. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef] [Green Version]
- Hirschhorn, T.; Stockwell, B.R. The development of the concept of ferroptosis. Free. Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e429. [Google Scholar] [CrossRef]
- Hashidate-Yoshida, T.; Harayama, T.; Hishikawa, D.; Morimoto, R.; Hamano, F.; Tokuoka, S.M.; Eto, M.; Tamura-Nakano, M.; Yanobu-Takanashi, R.; Mukumoto, Y.; et al. Fatty acid remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport. eLife 2015, 4, e06328. [Google Scholar] [CrossRef]
- Fan, X.; Heijnen, C.J.; van der Kooij, M.A.; Groenendaal, F.; van Bel, F. The role and regulation of hypoxia-inducible factor-1α expression in brain development and neonatal hypoxic–ischemic brain injury. Brain Res. Rev. 2009, 62, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, Y.; Zhao, X.; Shao, L.; Liu, G.; Sun, C.; Xu, R.; Zhang, Z. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav. Immun. 2021, 93, 312–321. [Google Scholar] [CrossRef]
- Yang, M.; Chen, P.; Liu, J.; Zhu, S.; Kroemer, G.; Klionsky, D.J.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci. Adv. 2019, 5, eaaw2238. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.-Y.; Deasy, R.; Kost-Alimova, M.; Dančík, V.; et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 2019, 10, 1617. [Google Scholar] [CrossRef] [PubMed]
- Appelhoff, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Camacho, J.D.; Bernier, M.; López-Lluch, G.; Navas, P. Coenzyme Q(10) Supplementation in Aging and Disease. Front. Physiol. 2018, 9, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcázar-Fabra, M.; Navas, P.; Brea-Calvo, G. Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim. Biophys. Acta 2016, 1857, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Acosta, M.J.; Vazquez Fonseca, L.; Desbats, M.A.; Cerqua, C.; Zordan, R.; Trevisson, E.; Salviati, L. Coenzyme Q biosynthesis in health and disease. Biochim. Biophys. Acta 2016, 1857, 1079–1085. [Google Scholar] [CrossRef]
- Enriquez, J.A.; Lenaz, G. Coenzyme q and the respiratory chain: Coenzyme q pool and mitochondrial supercomplexes. Mol. Syndromol. 2014, 5, 119–140. [Google Scholar] [CrossRef] [Green Version]
- Sohal, R.S.; Kamzalov, S.; Sumien, N.; Ferguson, M.; Rebrin, I.; Heinrich, K.R.; Forster, M.J. Effect of coenzyme Q10 intake on endogenous coenzyme Q content, mitochondrial electron transport chain, antioxidative defenses, and life span of mice. Free Radic. Biol. Med. 2006, 40, 480–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zozina, V.I.; Covantev, S.; Goroshko, O.A.; Krasnykh, L.M.; Kukes, V.G. Coenzyme Q10 in Cardiovascular and Metabolic Diseases: Current State of the Problem. Curr. Cardiol. Rev. 2018, 14, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Mohr, D.; Stocker, R. Radical-mediated oxidation of isolated human very-low-density lipoprotein. Arter. Thromb. 1994, 14, 1186–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.J.; Lin, Y.C.; Huang, Y.C.; Ko, Y.W.; Hsia, S.; Lin, P.T. The relationship between coenzyme Q10, oxidative stress, and antioxidant enzymes activities and coronary artery disease. Sci. World J. 2012, 2012, 792756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aejmelaeus, R.; Metsä-Ketelä, T.; Laippala, P.; Solakivi, T.; Alho, H. Ubiquinol-10 and total peroxyl radical trapping capacity of LDL lipoproteins during aging: The effects of Q-10 supplementation. Mol. Asp. Med. 1997, 18, S113–S120. [Google Scholar] [CrossRef]
- Littarru, G.P.; Tiano, L. Bioenergetic and antioxidant properties of coenzyme Q10: Recent developments. Mol. Biotechnol. 2007, 37, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Do, Q.; Lee, D.D.; Dinh, A.N.; Seguin, R.P.; Zhang, R.; Xu, L. Development and Application of a Peroxyl Radical Clock Approach for Measuring Both Hydrogen-Atom Transfer and Peroxyl Radical Addition Rate Constants. J. Org. Chem. 2021, 86, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Kebbati, A.H.; Zhang, Y.; Tang, Y.; Okello, E.; Huang, C. Effect of coenzyme Q10 on the incidence of atrial fibrillation in patients with heart failure. J. Investig. Med. 2015, 63, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Brütsch, S.H.; Wang, C.C.; Li, L.; Stender, H.; Neziroglu, N.; Richter, C.; Kuhn, H.; Borchert, A. Expression of inactive glutathione peroxidase 4 leads to embryonic lethality, and inactivation of the Alox15 gene does not rescue such knock-in mice. Antioxid. Redox Signal. 2015, 22, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, T.; McGuire, W. Allopurinol for preventing mortality and morbidity in newborn infants with hypoxic-ischaemic encephalopathy. Cochrane Database Syst. Rev. 2012, 7, Cd006817. [Google Scholar] [CrossRef] [PubMed]
- León-Carmona, J.R.; Galano, A. Is Caffeine a Good Scavenger of Oxygenated Free Radicals? J. Phys. Chem. B 2011, 115, 4538–4546. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F.; Dempsey, R.J. Phospholipase A2, hydroxyl radicals, and lipid peroxidation in transient cerebral ischemia. Antioxid. Redox Signal. 2003, 5, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Bailey, D.M.; Lundby, C.; Berg, R.M.G.; Taudorf, S.; Rahmouni, H.; Gutowski, M.; Mulholland, C.; Sullivan, J.; Swenson, E.R.; McEneny, J. On the antioxidant properties of erythropoietin and its association with the oxidative–nitrosative stress response to hypoxia in humans. Acta Physiol. 2014, 212, 175–187. [Google Scholar] [CrossRef]
- Bany-Mohammed, F.M.; Slivka, S.; Hallman, M. Recombinant human erythropoietin: Possible role as an antioxidant in premature rabbits. Pediatr. Res. 1996, 40, 381–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genc, S.; Akhisaroglu, M.; Kuralay, F.; Genc, K. Erythropoietin restores glutathione peroxidase activity in 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine-induced neurotoxicity in C57BL mice and stimulates murine astroglial glutathione peroxidase production in vitro. Neurosci. Lett. 2002, 321, 73–76. [Google Scholar] [CrossRef]
- Alonso-Alconada, D.; Álvarez, A.; Arteaga, O.; Martínez-Ibargüen, A.; Hilario, E. Neuroprotective effect of melatonin: A novel therapy against perinatal hypoxia-ischemia. Int. J. Mol. Sci. 2013, 14, 9379–9395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gou, Z.; Su, X.; Hu, X.; Zhou, Y.; Huang, L.; Fan, Y.; Li, J.; Lu, L. Melatonin improves hypoxic-ischemic brain damage through the Akt/Nrf2/Gpx4 signaling pathway. Brain Res. Bull. 2020, 163, 40–48. [Google Scholar] [CrossRef] [PubMed]
- El Bana, S.M.; Maher, S.E.; Gaber, A.F.; Aly, S.S. Serum and Urinary Malondialdehyde (MDA), Uric acid, and Protein as markers of perinatal asphyxia. Electron. Physician 2016, 8, 2614–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joy, R.; Pournami, F.; Bethou, A.; Bhat, V.B.; Bobby, Z. Effect of therapeutic hypothermia on oxidative stress and outcome in term neonates with perinatal asphyxia: A randomized controlled trial. J. Trop. Pediatrics 2013, 59, 17–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cascant-Vilaplana, M.M.; Sánchez-Illana, Á.; Piñeiro-Ramos, J.D.; Llorens-Salvador, R.; Quintás, G.; Oger, C.; Galano, J.-M.; Vigor, C.; Durand, T.; Kuligowski, J. Do Levels of Lipid Peroxidation Biomarkers Reflect the Degree of Brain Injury in Newborns? Antioxid. Redox Signal. 2021, 35, 1467–1475. [Google Scholar] [CrossRef]
- Ramy, N.; Al Sharany, W.; Mohamed, M.A.; Madani, H.; Saleh, E.; Aly, H. Lipid peroxides in the serum of asphyxiated neonates. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2016, 36, 849–852. [Google Scholar] [CrossRef] [PubMed]
- Barrera-de León, J.C.; Cervantes-Munguía, R.; Vásquez, C.; Higareda-Almaraz, M.A.; Bravo-Cuellar, A.; González-López, L. Usefulness of serum lipid peroxide as a diagnostic test for hypoxic ischemic encephalopathy in the full-term neonate. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2013, 33, 15–20. [Google Scholar] [CrossRef] [PubMed]
- McManus, T.; Sadgrove, M.; Pringle, A.K.; Chad, J.E.; Sundstrom, L.E. Intraischaemic hypothermia reduces free radical production and protects against ischaemic insults in cultured hippocampal slices. J. Neurochem. 2004, 91, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Thoresen, M.; Satas, S.; Puka-Sundvall, M.; Whitelaw, A.; Hallström, A.; Løberg, E.M.; Ungerstedt, U.; Steen, P.A.; Hagberg, H. Post-hypoxic hypothermia reduces cerebrocortical release of NO and excitotoxins. Neuroreport 1997, 8, 3359–3362. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Jiang, H.; Ye, L.; Cai, C.; Hu, Y.; Pan, S.; Li, P.; Xiao, J.; Lin, Z. Metformin treatment after the hypoxia-ischemia attenuates brain injury in newborn rats. Oncotarget 2017, 8, 75308–75325. [Google Scholar] [CrossRef] [Green Version]
- Park, D.; Shin, K.; Choi, E.K.; Choi, Y.; Jang, J.Y.; Kim, J.; Jeong, H.S.; Lee, W.; Lee, Y.B.; Kim, S.U.; et al. Protective effects of N-acetyl-L-cysteine in human oligodendrocyte progenitor cells and restoration of motor function in neonatal rats with hypoxic-ischemic encephalopathy. Evid.-Based Complement. Altern. Med. 2015, 2015, 764251. [Google Scholar] [CrossRef] [Green Version]
- Jatana, M.; Singh, I.; Singh, A.K.; Jenkins, D. Combination of systemic hypothermia and N-acetylcysteine attenuates hypoxic-ischemic brain injury in neonatal rats. Pediatr. Res. 2006, 59, 684–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, L.E.; Moss, H.G.; Lowe, D.W.; Brown, T.; Wiest, D.B.; Hollis, B.W.; Singh, I.; Jenkins, D.D. NAC and Vitamin D Restore CNS Glutathione in Endotoxin-Sensitized Neonatal Hypoxic-Ischemic Rats. Antioxidants 2021, 10, 489. [Google Scholar] [CrossRef] [PubMed]
- Ibi, M.; Sawada, H.; Nakanishi, M.; Kume, T.; Katsuki, H.; Kaneko, S.; Shimohama, S.; Akaike, A. Protective effects of 1 alpha,25-(OH)(2)D(3) against the neurotoxicity of glutamate and reactive oxygen species in mesencephalic culture. Neuropharmacology 2001, 40, 761–771. [Google Scholar] [CrossRef]
- Jenkins, D.D.; Moss, H.G.; Brown, T.R.; Yazdani, M.; Thayyil, S.; Montaldo, P.; Vento, M.; Kuligowski, J.; Wagner, C.; Hollis, B.W.; et al. NAC and Vitamin D Improve CNS and Plasma Oxidative Stress in Neonatal HIE and Are Associated with Favorable Long-Term Outcomes. Antioxidants 2021, 10, 1344. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.M.; Razdan, B.; Mishra, O.P.; Johnson, L.; Delivoria-Papadopoulos, M. Protective effect of alpha-tocopherol on brain cell membrane function during cerebral cortical hypoxia in newborn piglets. Brain Res. 1994, 653, 45–50. [Google Scholar] [CrossRef]
- Beyer, R.E. The role of ascorbate in antioxidant protection of biomembranes: Interaction with vitamin E and coenzyme Q. J. Bioenerg. Biomembr. 1994, 26, 349–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakai, A.; Shibazaki, Y.; Taniuchi, Y.; Oya, A.; Asakura, H.; Koshino, T.; Araki, T. Vitamins ameliorate secondary mitochondrial failure in neonatal rat brain. Pediatr. Neurol. 2002, 27, 30–35. [Google Scholar] [CrossRef]
- Kumral, A.; Gonenc, S.; Acikgoz, O.; Sonmez, A.; Genc, K.; Yilmaz, O.; Gokmen, N.; Duman, N.; Ozkan, H. Erythropoietin increases glutathione peroxidase enzyme activity and decreases lipid peroxidation levels in hypoxic-ischemic brain injury in neonatal rats. Biol. Neonate 2005, 87, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Akisu, M.; Tuzun, S.; Arslanoglu, S.; Yalaz, M.; Kultursay, N. Effect of recombinant human erythropoietin administration on lipid peroxidation and antioxidant enzyme (s) activities in preterm infants. Acta Med. Okayama 2001, 55, 357–362. [Google Scholar] [PubMed]
- Dorrepaal, C.A.; Berger, H.M.; Benders, M.J.; van Zoeren-Grobben, D.; Van de Bor, M.; Van Bel, F. Nonprotein-bound iron in postasphyxial reperfusion injury of the newborn. Pediatrics 1996, 98, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Dorrepaal, C.A.; van Bel, F.; Moison, R.M.; Shadid, M.; van de Bor, M.; Steendijk, P.; Berger, H.M. Oxidative stress during post-hypoxic-ischemic reperfusion in the newborn lamb: The effect of nitric oxide synthesis inhibition. Pediatr. Res. 1997, 41, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozoff, B.; Clark, K.M.; Jing, Y.; Armony-Sivan, R.; Angelilli, M.L.; Jacobson, S.W. Dose-response relationships between iron deficiency with or without anemia and infant social-emotional behavior. J. Pediatr. 2008, 152, 696–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters-Scholte, C.; Braun, K.; Koster, J.; Kops, N.; Blomgren, K.; Buonocore, G.; van Buul-Offers, S.; Hagberg, H.; Nicolay, K.; van Bel, F.; et al. Effects of allopurinol and deferoxamine on reperfusion injury of the brain in newborn piglets after neonatal hypoxia-ischemia. Pediatr. Res. 2003, 54, 516–522. [Google Scholar] [CrossRef] [Green Version]
- Palmer, C.; Roberts, R.L.; Bero, C. Deferoxamine posttreatment reduces ischemic brain injury in neonatal rats. Stroke A J. Cereb. Circ. 1994, 25, 1039–1045. [Google Scholar] [CrossRef] [Green Version]
- Groenendaal, F.; Shadid, M.; McGowan, J.E.; Mishra, O.P.; van Bel, F. Effects of deferoxamine, a chelator of free iron, on NA(+), K(+)-ATPase activity of cortical brain cell membrane during early reperfusion after hypoxia-ischemia in newborn lambs. Pediatr. Res. 2000, 48, 560–564. [Google Scholar] [CrossRef] [Green Version]
- Papazisis, G.; Pourzitaki, C.; Sardeli, C.; Lallas, A.; Amaniti, E.; Kouvelas, D. Deferoxamine decreases the excitatory amino acid levels and improves the histological outcome in the hippocampus of neonatal rats after hypoxia-ischemia. Pharmacol. Res. 2008, 57, 73–78. [Google Scholar] [CrossRef]
- Shadid, M.; Buonocore, G.; Groenendaal, F.; Moison, R.; Ferrali, M.; Berger, H.M.; van Bel, F. Effect of deferoxamine and allopurinol on non-protein-bound iron concentrations in plasma and cortical brain tissue of newborn lambs following hypoxia-ischemia. Neurosci. Lett. 1998, 248, 5–8. [Google Scholar] [CrossRef]
- Chen, K.B.; Lin, A.M.; Chiu, T.H. Systemic vitamin D3 attenuated oxidative injuries in the locus coeruleus of rat brain. Ann. N. Y. Acad. Sci. 2003, 993, 313–324; discussion 319–345. [Google Scholar] [CrossRef]
- Uberti, F.; Morsanuto, V.; Bardelli, C.; Molinari, C. Protective effects of 1alpha,25-Dihydroxyvitamin D3 on cultured neural cells exposed to catalytic iron. Physiol. Rep. 2016, 4, e12769. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, R.A.; Jiang, X.; Francisco, C.; Christen, S.; Vexler, Z.S.; Täuber, M.G.; Ferriero, D.M. Manipulation of Antioxidant Pathways in Neonatal Murine Brain. Pediatr. Res. 2004, 56, 656–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasiljević, B.; Maglajlić-Djukić, S.; Gojnić, M.; Stanković, S. The role of oxidative stress in perinatal hypoxic-ischemic brain injury. Srp. Arh. Celok. Lek. 2012, 140, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; Aminoff, A.; Lee, C.L.; Christen, S.; Ferriero, D.M. Hypoxic Preconditioning Reverses Protection After Neonatal Hypoxia-Ischemia in Glutathione Peroxidase Transgenic Murine Brain. Pediatr. Res. 2007, 61, 666–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payton, K.S.; Sheldon, R.A.; Mack, D.W.; Zhu, C.; Blomgren, K.; Ferriero, D.M.; Northington, F.J. Antioxidant status alters levels of Fas-associated death domain-like IL-1B-converting enzyme inhibitory protein following neonatal hypoxia-ischemia. Dev. Neurosci. 2007, 29, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Crack, P.J.; Taylor, J.M.; Flentjar, N.J.; de Haan, J.; Hertzog, P.; Iannello, R.C.; Kola, I. Increased infarct size and exacerbated apoptosis in the glutathione peroxidase-1 (Gpx-1) knockout mouse brain in response to ischemia/reperfusion injury. J. Neurochem. 2001, 78, 1389–1399. [Google Scholar] [CrossRef]
- Crack, P.J.; Taylor, J.M.; de Haan, J.B.; Kola, I.; Hertzog, P.; Iannello, R.C. Glutathione peroxidase-1 contributes to the neuroprotection seen in the superoxide dismutase-1 transgenic mouse in response to ischemia/reperfusion injury. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2003, 23, 19–22. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, E.; Roberts, J.; Tennant, D.A.; Boardman, J.P.; Drake, A.J. Metabolic adaptations to hypoxia in the neonatal mouse forebrain can occur independently of the transporters SLC7A5 and SLC3A2. Sci. Rep. 2021, 11, 9092. [Google Scholar] [CrossRef]
- Sims, B.; Clarke, M.; Francillion, L.; Kindred, E.; Hopkins, E.S.; Sontheimer, H. Hypoxic preconditioning involves system Xc- regulation in mouse neural stem cells. Stem Cell Res. 2012, 8, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Ding, C.; Ding, X.; Zheng, J.; Wang, B.; Li, Y.; Xiang, H.; Dou, M.; Qiao, Y.; Tian, P.; Xue, W. miR-182-5p and miR-378a-3p regulate ferroptosis in I/R-induced renal injury. Cell Death Dis. 2020, 11, 929. [Google Scholar] [CrossRef]
- Li, K.; Li, T.; Wang, Y.; Xu, Y.; Zhang, S.; Culmsee, C.; Wang, X.; Zhu, C. Sex differences in neonatal mouse brain injury after hypoxia-ischemia and adaptaquin treatment. J. Neurochem. 2019, 150, 759–775. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.; Zhang, Y.; Li, T.; Xie, C.; Sun, Y.; Xu, Y.; Zhou, K.; Huo, K.; Wang, Y.; Wang, X.; et al. Lack of the brain-specific isoform of apoptosis-inducing factor aggravates cerebral damage in a model of neonatal hypoxia-ischemia. Cell Death Dis. 2018, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Belousova, M.; Tokareva, O.G.; Gorodetskaya, E.; Kalenikova, E.I.; Medvedev, O.S. Intravenous Treatment With Coenzyme Q10 Improves Neurological Outcome and Reduces Infarct Volume After Transient Focal Brain Ischemia in Rats. J. Cardiovasc. Pharmacol. 2016, 67, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Aal, S.A.; Abd El-Fattah, M.A.; El-Abhar, H.S. CoQ10 Augments Rosuvastatin Neuroprotective Effect in a Model of Global Ischemia via Inhibition of NF-κB/JNK3/Bax and Activation of Akt/FOXO3A/Bim Cues. Front. Pharmacol. 2017, 8, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, C.E.; Murphy, M.P.; Smith, R.A.; Oorschot, D.E. Neonatal rat hypoxia-ischemia: Effect of the anti-oxidant mitoquinol, and S-PBN. Pediatr. Int. Off. J. Jpn. Pediatric Soc. 2008, 50, 481–488. [Google Scholar] [CrossRef]
- Deichmann, R.; Lavie, C.; Andrews, S. Coenzyme q10 and statin-induced mitochondrial dysfunction. Ochsner J. 2010, 10, 16–21. [Google Scholar] [PubMed]
- Stefely, J.A.; Pagliarini, D.J. Biochemistry of Mitochondrial Coenzyme Q Biosynthesis. Trends Biochem. Sci 2017, 42, 824–843. [Google Scholar] [CrossRef]
- Shukla, S.; Dubey, K.K. CoQ10 a super-vitamin: Review on application and biosynthesis. 3 Biotech 2018, 8, 249. [Google Scholar] [CrossRef]
- Folkers, K.; Langsjoen, P.; Willis, R.; Richardson, P.; Xia, L.J.; Ye, C.Q.; Tamagawa, H. Lovastatin decreases coenzyme Q levels in humans. Proc. Natl. Acad. Sci. USA 1990, 87, 8931–8934. [Google Scholar] [CrossRef] [Green Version]
- Qu, H.; Meng, Y.Y.; Chai, H.; Liang, F.; Zhang, J.Y.; Gao, Z.Y.; Shi, D.Z. The effect of statin treatment on circulating coenzyme Q10 concentrations: An updated meta-analysis of randomized controlled trials. Eur. J. Med. Res. 2018, 23, 57. [Google Scholar] [CrossRef]
- Marcoff, L.; Thompson, P.D. The role of coenzyme Q10 in statin-associated myopathy: A systematic review. J. Am. Coll. Cardiol. 2007, 49, 2231–2237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Ferroptosis in infection, inflammation, and immunity. J. Exp. Med. 2021, 218, e20210518. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, J.; Kim, B.; Jaitpal, S.; Meng, S.S.; Adjepong, K.; Imamura, S.; Wake, H.; Nishibori, M.; Stopa, E.G. High-mobility group box-1 translocation and release after hypoxic ischemic brain injury in neonatal rats. Exp. Neurol. 2019, 311, 1–14. [Google Scholar] [CrossRef]
- Sun, Y.; Hei, M.; Fang, Z.; Tang, Z.; Wang, B.; Hu, N. High-mobility group box 1 contributes to cerebral cortex injury in a neonatal hypoxic-ischemic rat model by regulating the phenotypic polarization of microglia. Front. Cell. Neurosci. 2019, 13, 506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, F.; Chai, W.; Xie, M.; Yang, M.; Yu, Y.; Cao, L.; Yang, L. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRAS(Q61L) cells. Am. J. Cancer Res. 2019, 9, 730–739. [Google Scholar] [PubMed]
- Zhu, K.; Zhu, X.; Liu, S.; Yu, J.; Wu, S.; Hei, M. Glycyrrhizin Attenuates Hypoxic-Ischemic Brain Damage by Inhibiting Ferroptosis and Neuroinflammation in Neonatal Rats via the HMGB1/GPX4 Pathway. Oxid. Med. Cell. Longev. 2022, 2022, 8438528. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from biology to therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef]
- He, Z.; Yang, J.; Sui, C.; Zhang, P.; Wang, T.; Mou, T.; Sun, K.; Wang, Y.; Xu, Z.; Li, G.; et al. FAM98A promotes resistance to 5-fluorouracil in colorectal cancer by suppressing ferroptosis. Arch. Biochem. Biophys. 2022, 722, 109216. [Google Scholar] [CrossRef]
Figure 1.
Pathways involved in ferroptotic cell death. (A) The cysteine-GSH-GPX4 system plays a central role in the cellular antioxidant mechanism and ferroptosis prevention. Inhibition of the Xc- system by erastin deprives the cells of cysteine, thus depriving them of GSH, triggering ferroptosis. Inhibition of GPX4 by RSL3 deprives the cells from the enzyme that reduces phospholipid peroxides (LOOH) to their corresponding alcohols (LOH), increasing lipid peroxidation and associated ferroptosis. Both erastin and RSL3 have been extensively used to induce ferroptosis in cell culture models. (B) The enzyme acyl-CoA synthetase long-chain family member 4 (ACSL4) activates highly oxidizable PUFAs, which is a critical step in their incorporation into phospholipids and into the cell membrane. Depleting the cells of ACSL4 increases the cellular resistance to ferroptosis. LPCAT3 catalyzes the incorporation of PUFA-CoA into phospholipids. Suppressing LPCAT3 activity increases resistance to ferroptosis. (C) FSP1 catalyzes the regeneration of CoQ10 at the expense of NAD(P)H. CoQ10-H2 (reduced form) is an effective lipid-soluble antioxidant, acting by directly reacting with free radical species in the membrane and preventing lipid peroxidation. CR, cystine reductase; GS, glutathione synthetase; GSH, reduced glutathione; GSSG, glutathione disulfide; GR, glutathione reductase; FSP-1, ferroptosis suppressor protein 1; CoQ10, coenzyme Q10; HO-1, heme oxygenase 1; LPCAT3, lysophosphatidylcholine acyltransferase 3; PUFA, polyunsaturated fatty acid.
Figure 1.
Pathways involved in ferroptotic cell death. (A) The cysteine-GSH-GPX4 system plays a central role in the cellular antioxidant mechanism and ferroptosis prevention. Inhibition of the Xc- system by erastin deprives the cells of cysteine, thus depriving them of GSH, triggering ferroptosis. Inhibition of GPX4 by RSL3 deprives the cells from the enzyme that reduces phospholipid peroxides (LOOH) to their corresponding alcohols (LOH), increasing lipid peroxidation and associated ferroptosis. Both erastin and RSL3 have been extensively used to induce ferroptosis in cell culture models. (B) The enzyme acyl-CoA synthetase long-chain family member 4 (ACSL4) activates highly oxidizable PUFAs, which is a critical step in their incorporation into phospholipids and into the cell membrane. Depleting the cells of ACSL4 increases the cellular resistance to ferroptosis. LPCAT3 catalyzes the incorporation of PUFA-CoA into phospholipids. Suppressing LPCAT3 activity increases resistance to ferroptosis. (C) FSP1 catalyzes the regeneration of CoQ10 at the expense of NAD(P)H. CoQ10-H2 (reduced form) is an effective lipid-soluble antioxidant, acting by directly reacting with free radical species in the membrane and preventing lipid peroxidation. CR, cystine reductase; GS, glutathione synthetase; GSH, reduced glutathione; GSSG, glutathione disulfide; GR, glutathione reductase; FSP-1, ferroptosis suppressor protein 1; CoQ10, coenzyme Q10; HO-1, heme oxygenase 1; LPCAT3, lysophosphatidylcholine acyltransferase 3; PUFA, polyunsaturated fatty acid.

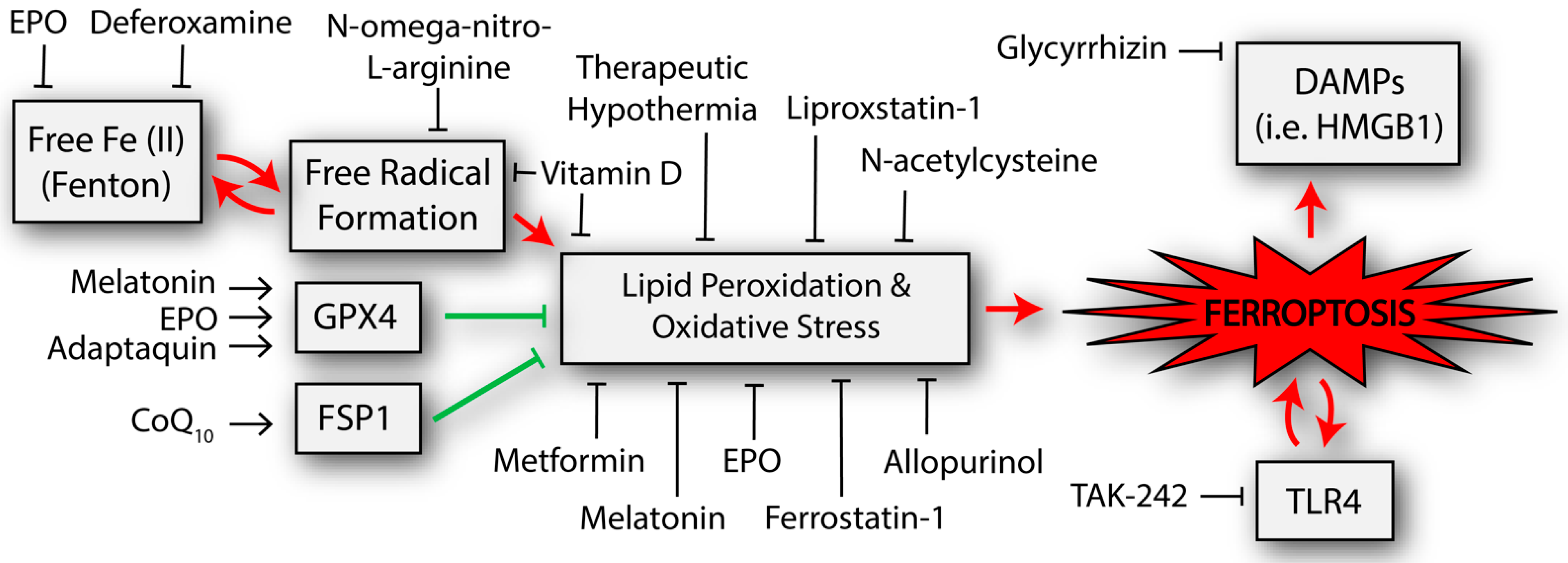
Figure 2.
Potential neuroprotective interventions that have been shown to affect the ferroptosis-related pathways in studies of neonatal hypoxic-ischemic brain injury. Other interventions that have been trialed in adult populations but not yet neonates are described in the text but not included in the figure. The double arrows represent byproducts that can further potentiate the initial process (e.g., TLR4 is increased from the process of ferroptosis but TLR4 appears to also activate ferroptosis). CoQ10, coenzyme Q10; DAMPs, damage associated molecular patterns; EPO, erythropoietin; FSP1, ferroptosis suppressor protein 1; GPX4, glutathione peroxidase 4; HMGB1, high mobility group box 1; TLR4, toll-like receptor 4.
Figure 2.
Potential neuroprotective interventions that have been shown to affect the ferroptosis-related pathways in studies of neonatal hypoxic-ischemic brain injury. Other interventions that have been trialed in adult populations but not yet neonates are described in the text but not included in the figure. The double arrows represent byproducts that can further potentiate the initial process (e.g., TLR4 is increased from the process of ferroptosis but TLR4 appears to also activate ferroptosis). CoQ10, coenzyme Q10; DAMPs, damage associated molecular patterns; EPO, erythropoietin; FSP1, ferroptosis suppressor protein 1; GPX4, glutathione peroxidase 4; HMGB1, high mobility group box 1; TLR4, toll-like receptor 4.


{kind=link}
{kind=link}
Table 1.
Ongoing clinical trials (or completed trials without published results) assessing interventions that may affect ferroptosis in neonatal hypoxic-ischemic brain injury. List reflects studies registered with Clinicaltrials.gov as of June 2022.
Table 1.
Ongoing clinical trials (or completed trials without published results) assessing interventions that may affect ferroptosis in neonatal hypoxic-ischemic brain injury. List reflects studies registered with Clinicaltrials.gov as of June 2022.
| Intervention | Potential Mechanism | Identifier * | Status |
|---|---|---|---|
| Allopurinol | Scavenges free radicals [77] | NCT03162653 | Recruiting |
| Caffeine | Scavenges free radicals [78] | NCT03913221 | Recruiting |
| NCT05295784 | Not yet recruiting | ||
| Citicoline | Alters phosphatidylcholine synthesis, resulting in attenuation of lipid peroxidation [79] | NCT03181646 | Unknown |
| Darbepoetin/ Erythropoietin | Inhibits Fenton chemistry, scavenges iron through erythropoiesis [80,81], increases GPX expression [82] | NCT00719407 | Completed |
| NCT03071861 | Recruiting | ||
| NCT03079167 | Active | ||
| NCT03163589 | Unknown | ||
| Melatonin | Scavenges free radicals [83], attenuates GPX4, GSH, and 4-HNE changes [84] | NCT02621944 | Recruiting |
| NCT03806816 | Recruiting |
* Clinicaltrials.gov identifier.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Peeples, E.S.; Genaro-Mattos, T.C. Ferroptosis: A Promising Therapeutic Target for Neonatal Hypoxic-Ischemic Brain Injury. Int. J. Mol. Sci. 2022, 23, 7420. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137420
AMA Style
Peeples ES, Genaro-Mattos TC. Ferroptosis: A Promising Therapeutic Target for Neonatal Hypoxic-Ischemic Brain Injury. International Journal of Molecular Sciences. 2022; 23(13):7420. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137420
Chicago/Turabian StylePeeples, Eric S., and Thiago C. Genaro-Mattos. 2022. "Ferroptosis: A Promising Therapeutic Target for Neonatal Hypoxic-Ischemic Brain Injury" International Journal of Molecular Sciences 23, no. 13: 7420. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137420
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.