
Polyamine Oxidase Expression Is Downregulated by 17β-Estradiol via Estrogen Receptor 2 in Human MCF-7 Breast Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
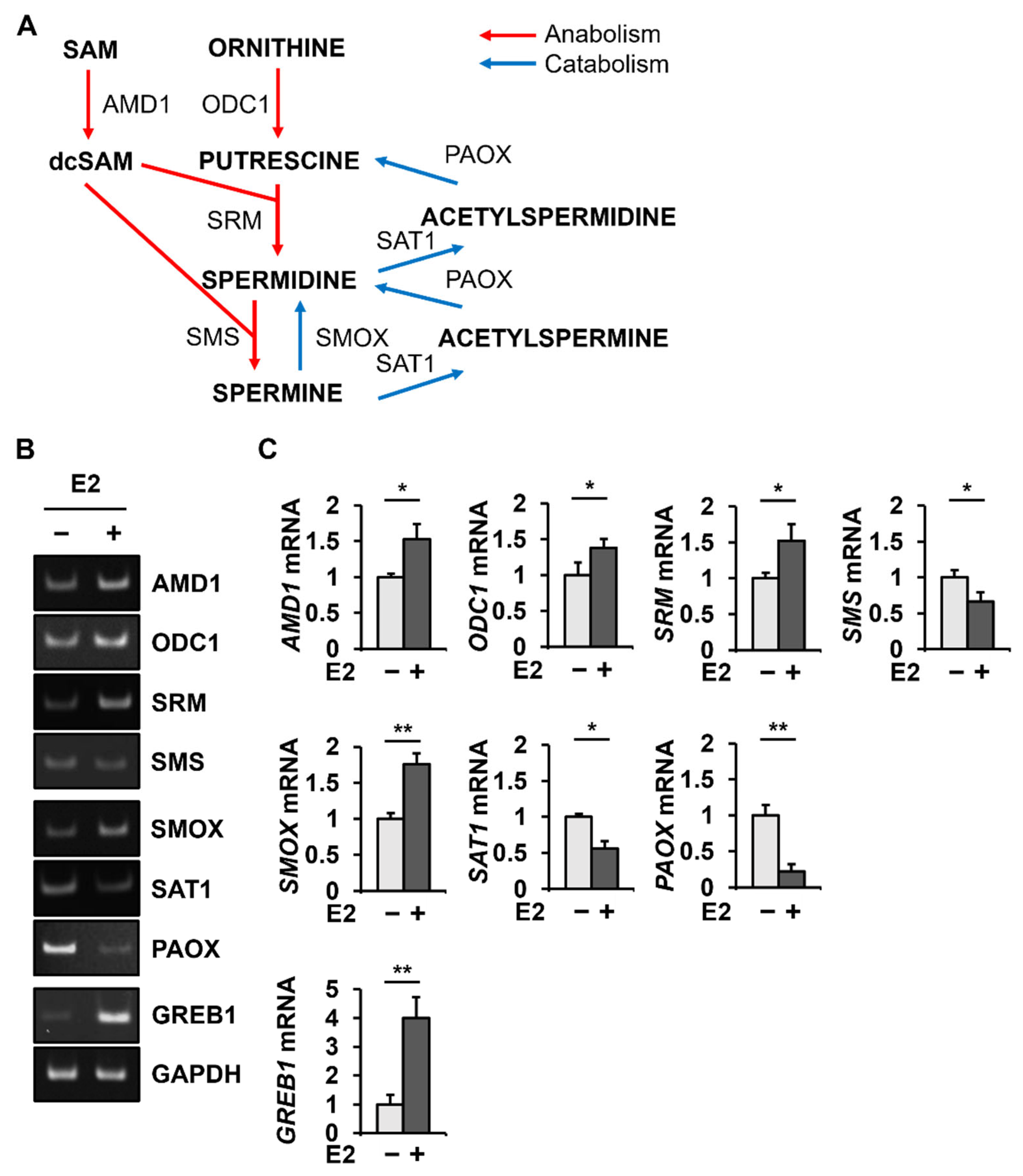
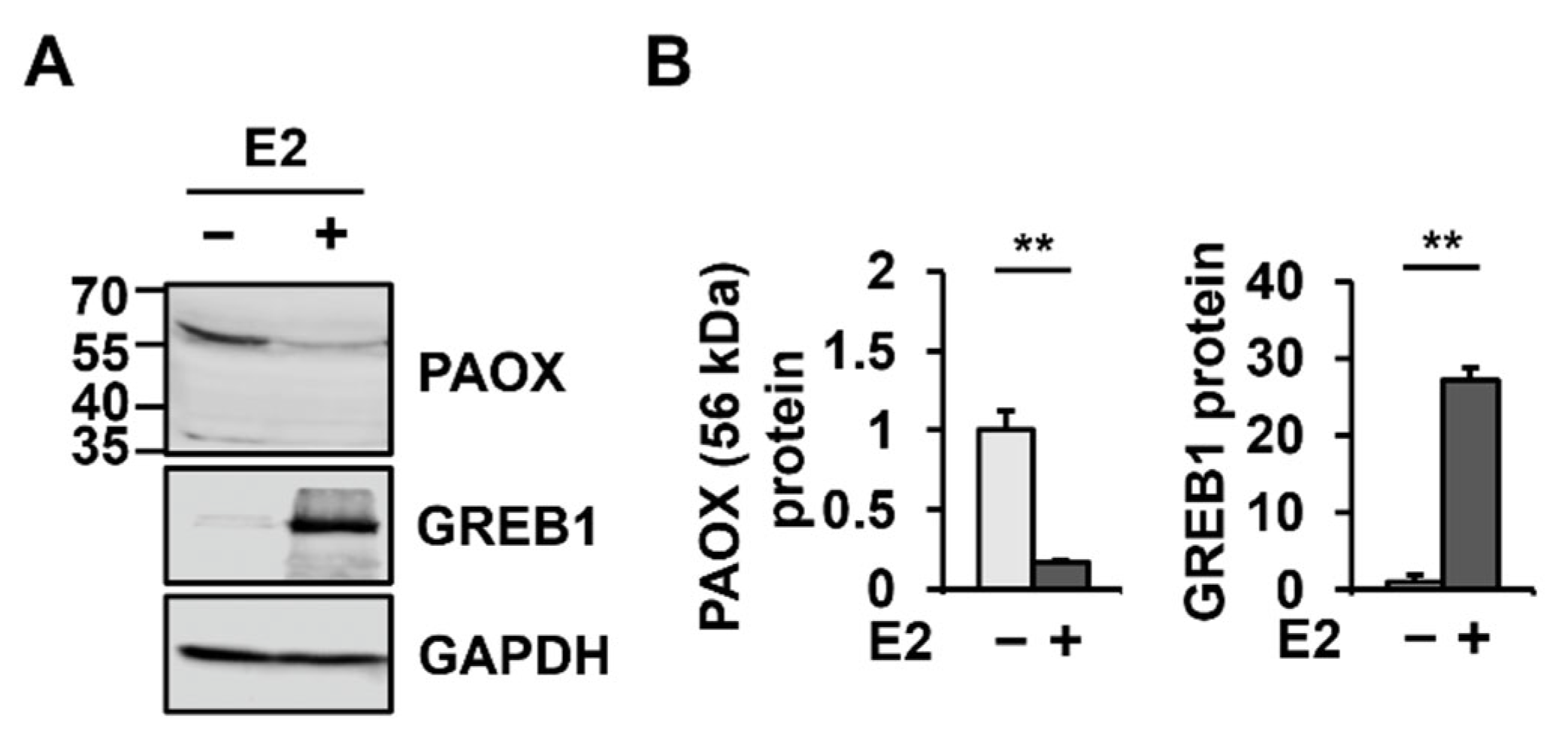
2.1. E2 Changes PAOX mRNA Levels Most among the Genes Involved in Polyamine Metabolism
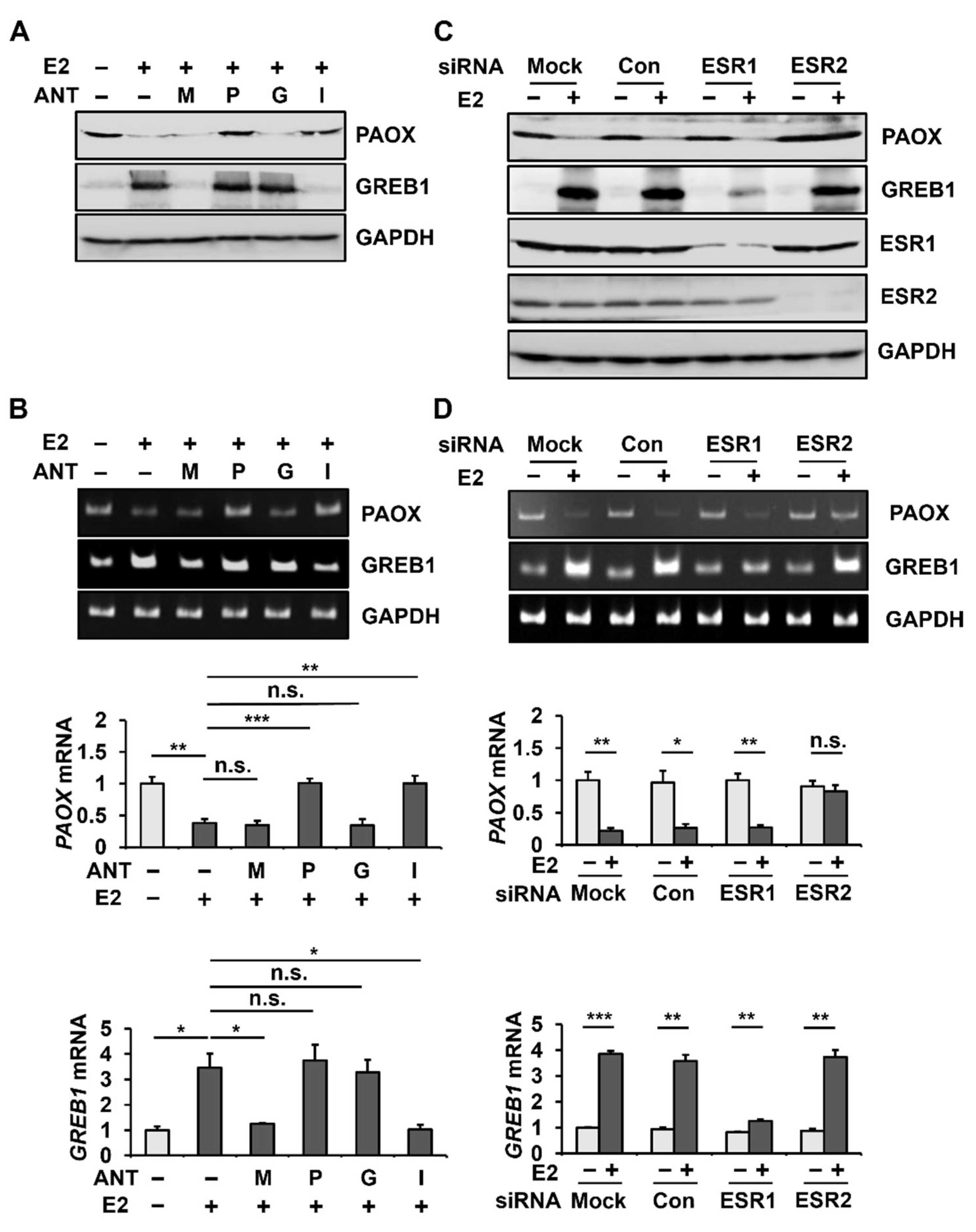
2.2. E2 Decreases PAOX Expression via the ESR2
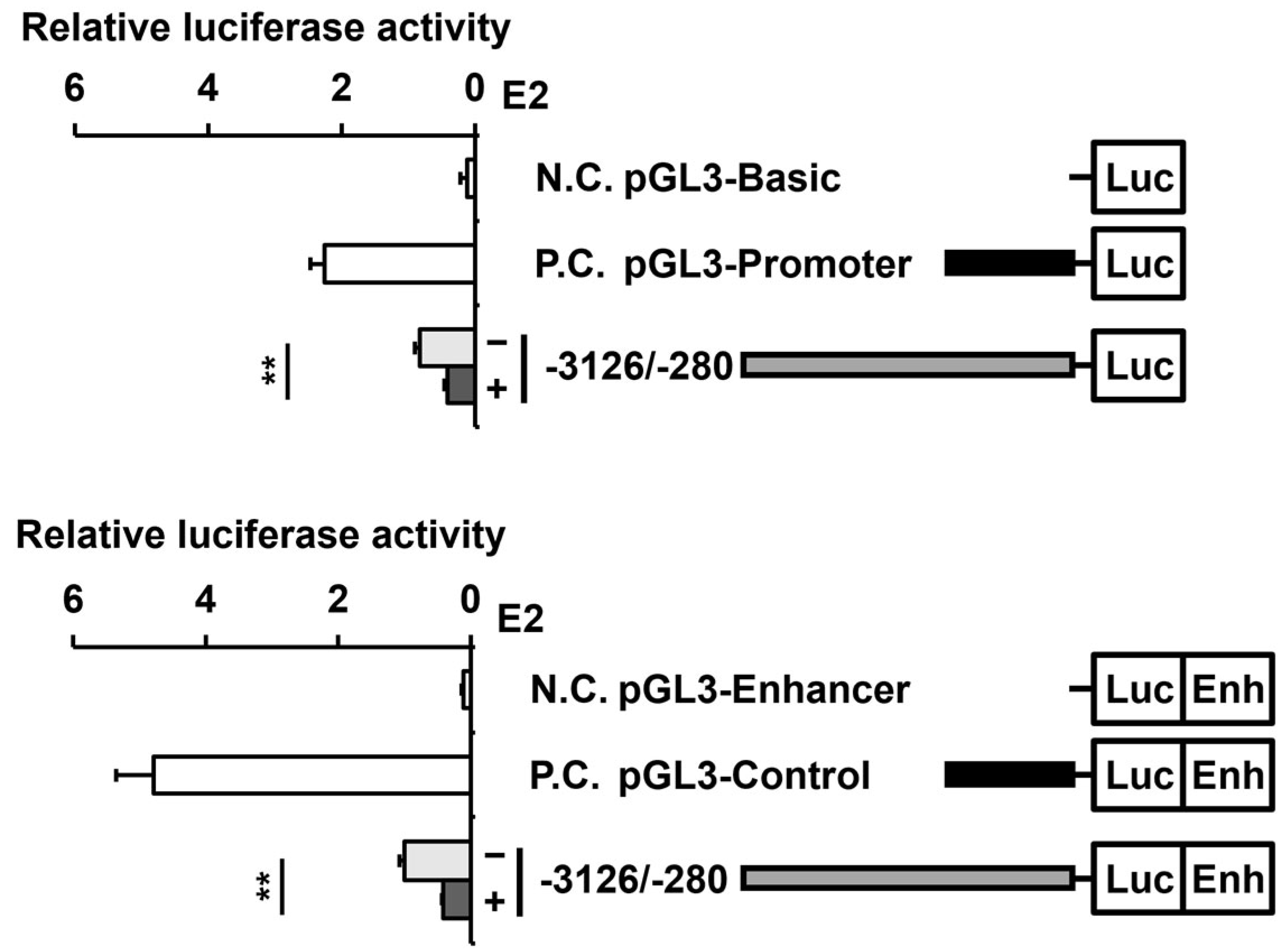
2.3. E2 Reduces the Activity of the PAOX Promoter in an ESR2-Dependent Manner
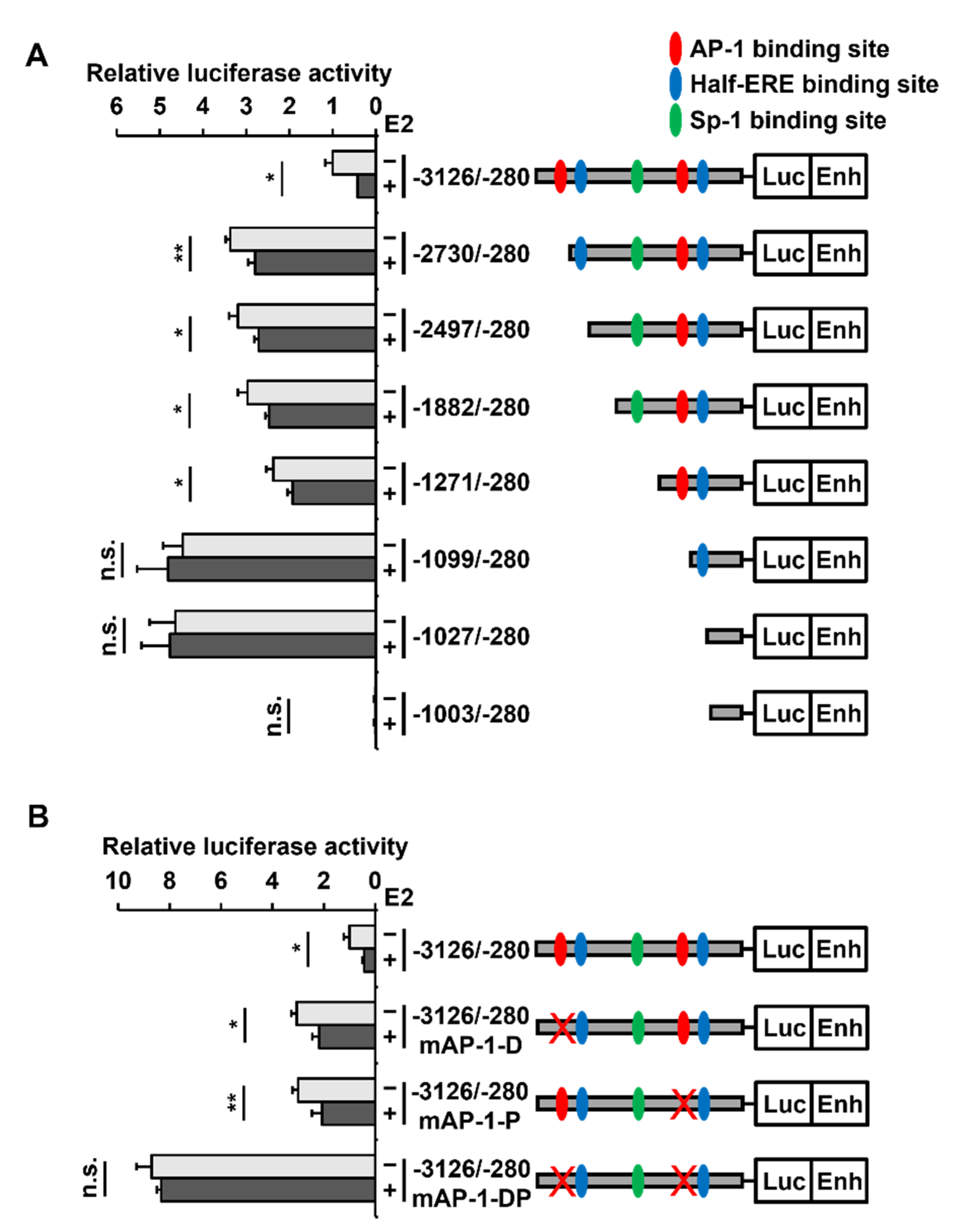
2.4. Two AP-1 Sites within the PAOX Promoter Are Involved in the E2-Mediated PAOX Repression
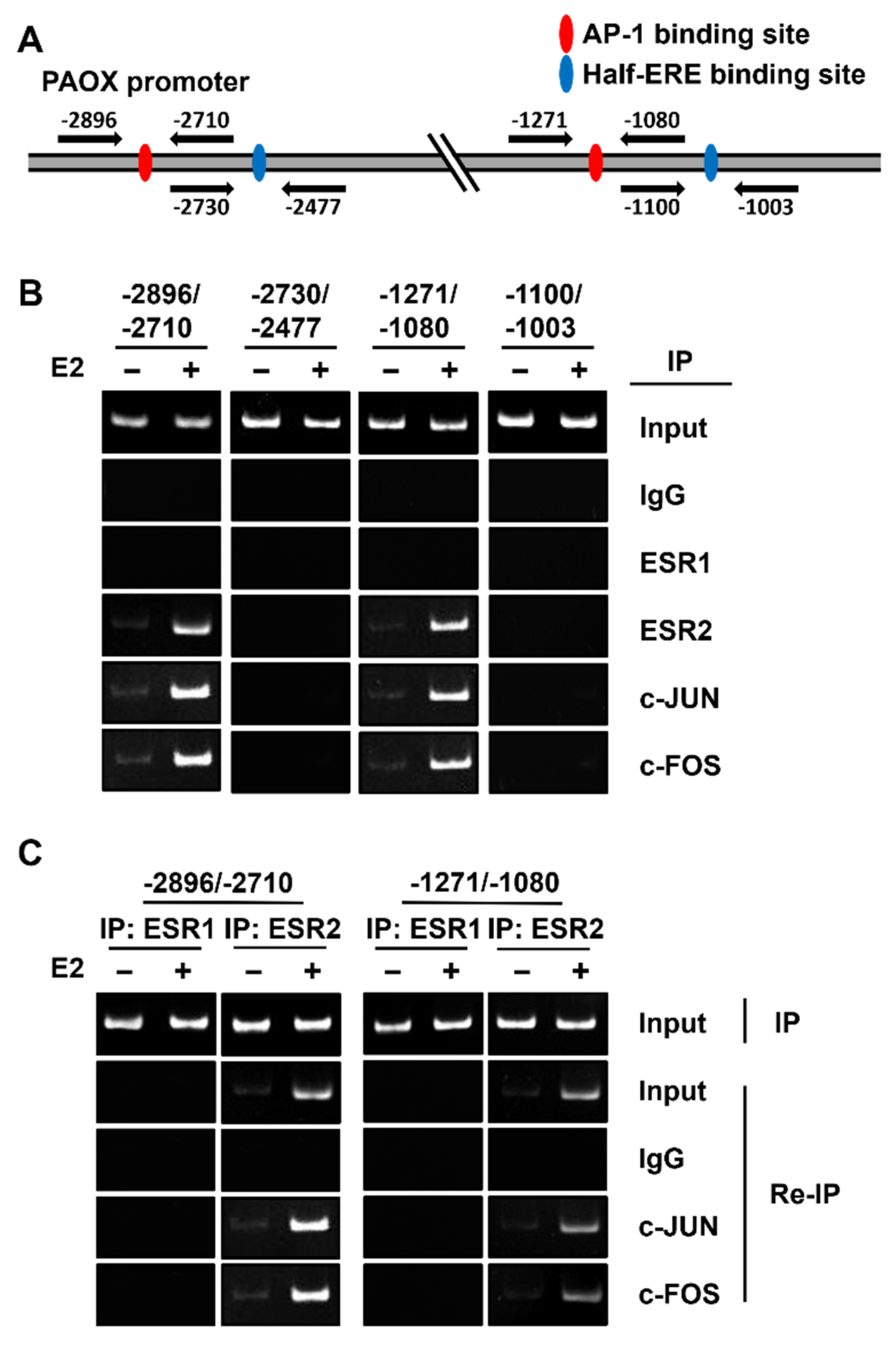
2.5. ESR2 Binds to the AP-1 Binding Sites of the PAOX Promoter
3. Discussion
4. Methods and Materials
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. RNA Isolation and PCR
4.4. Western Blotting
4.5. Treatment of E2 to MCF-7 Cells
4.6. Transfection of siRNAs
4.7. Prediction of Transcription Factor-Binding Site
4.8. Cloning of the Human PAOX Promoter in Luciferase Reporter Vectors
4.9. Dual-Luciferase Reporter Assay
4.10. Site-Directed Mutagenesis (SDM)
4.11. ChIP and Re-IP
4.12. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Battaglia, V.; DeStefano Shields, C.; Murray-Stewart, T.; Casero, R.A., Jr. Polyamine catabolism in carcinogenesis: Potential targets for chemotherapy and chemoprevention. Amino Acids 2014, 46, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urdiales, J.L.; Medina, M.A.; Sánchez-Jiménez, F. Polyamine metabolism revisited. Eur. J. Gastroenterol. Hepatol. 2001, 13, 1015–1019. [Google Scholar] [CrossRef] [PubMed]
- Bae, D.H.; Lane, D.J.R.; Jansson, P.J.; Richardson, D.R. The old and new biochemistry of polyamines. Biochim. Biophys. Acta. Gen. Subj. 2018, 1862, 2053–2068. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A., Jr.; Murray Stewart, T.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Mammalian polyamine metabolism and function. IUBMB Life 2009, 61, 880–894. [Google Scholar] [CrossRef] [PubMed]
- Sandusky-Beltran, L.A.; Kovalenko, A.; Ma, C.; Calahatian, J.I.T.; Placides, D.S.; Watler, M.D.; Hunt, J.B.; Darling, A.L.; Baker, J.D.; Blair, L.J.; et al. Spermidine/spermine-N(1)-acetyltransferase ablation impacts tauopathy-induced polyamine stress response. Alzheimers Res. Ther. 2019, 11, 58. [Google Scholar] [CrossRef]
- Shin, V.Y.; Liu, E.S.; Koo, M.W.; Wang, J.Y.; Matsui, H.; Cho, C.H. Cigarette smoke extracts delay wound healing in the stomach: Involvement of polyamine synthesis. Exp. Biol. Med. 2002, 227, 114–124. [Google Scholar] [CrossRef]
- Kubo, S.; Tamori, A.; Nishiguchi, S.; Kinoshita, H.; Hirohashi, K.; Kuroki, T.; Omura, T.; Otani, S. Effect of alcohol abuse on polyamine metabolism in hepatocellular carcinoma and noncancerous hepatic tissue. Surgery 1998, 123, 205–211. [Google Scholar] [CrossRef]
- Igarashi, K.; Ueda, S.; Yoshida, K.; Kashiwagi, K. Polyamines in renal failure. Amino Acids 2006, 31, 477–483. [Google Scholar] [CrossRef]
- Zwighaft, Z.; Aviram, R.; Shalev, M.; Rousso-Noori, L.; Kraut-Cohen, J.; Golik, M.; Brandis, A.; Reinke, H.; Aharoni, A.; Kahana, C.; et al. Circadian Clock Control by Polyamine Levels through a Mechanism that Declines with Age. Cell Metab. 2015, 22, 874–885. [Google Scholar] [CrossRef] [Green Version]
- Das, R.; Kanungo, M.S. Activity and modulation of ornithine decarboxylase and concentrations of polyamines in various tissues of rats as a function of age. Exp. Gerontol. 1982, 17, 95–103. [Google Scholar] [CrossRef]
- Minois, N.; Carmona-Gutierrez, D.; Madeo, F. Polyamines in aging and disease. Aging 2011, 3, 716–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, T.A.; Johnson, K.M. Menopause. Med. Clin. North. Am. 2015, 99, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Hoga, L.; Rodolpho, J.; Gonçalves, B.; Quirino, B. Women’s experience of menopause: A systematic review of qualitative evidence. JBI Database System Rev. Implement. Rep. 2015, 13, 250–337. [Google Scholar] [CrossRef]
- Hall, G.; Phillips, T.J. Estrogen and skin: The effects of estrogen, menopause, and hormone replacement therapy on the skin. J. Am. Acad. Dermatol. 2005, 53, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Hodis, H.N.; Mack, W.J.; Henderson, V.W.; Shoupe, D.; Budoff, M.J.; Hwang-Levine, J.; Li, Y.; Feng, M.; Dustin, L.; Kono, N.; et al. Vascular Effects of Early versus Late Postmenopausal Treatment with Estradiol. N. Engl. J. Med. 2016, 374, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Goluch-Koniuszy, Z.S. Nutrition of women with hair loss problem during the period of menopause. Prz. Menopauzalny 2016, 15, 56–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, V.W. Menopause and disorders of the central nervous system. Minerva Ginecol. 2005, 57, 579–592. [Google Scholar]
- Management of osteoporosis in postmenopausal women: The 2021 position statement of The North American Menopause Society. Menopause 2021, 28, 973–997. [CrossRef]
- Yamamoto, T.; Hinoi, E.; Fujita, H.; Iezaki, T.; Takahata, Y.; Takamori, M.; Yoneda, Y. The natural polyamines spermidine and spermine prevent bone loss through preferential disruption of osteoclastic activation in ovariectomized mice. Br. J. Pharmacol. 2012, 166, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Byun, J.A.; Choi, M.H.; Moon, M.H.; Kong, G.; Chul Chung, B. Serum polyamines in pre- and post-operative patients with breast cancer corrected by menopausal status. Cancer Lett. 2009, 273, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Gray, H.E.; Jasper, T.W.; Luttge, W.G.; Shukla, J.B.; Rennert, O.M. Estrogen increases hypothalamic and pituitary polyamine levels in ovariectomized rats. J. Neurochem. 1980, 34, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Lew, B.L.; Sim, W.Y.; Lee, J.; Hong, J.; Chung, B.C. Altered polyamine profiling in the hair of patients with androgenic alopecia and alopecia areata. J. Dermatol. 2019, 46, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Ramot, Y.; Pietilä, M.; Giuliani, G.; Rinaldi, F.; Alhonen, L.; Paus, R. Polyamines and hair: A couple in search of perfection. Exp. Dermatol. 2010, 19, 784–790. [Google Scholar] [CrossRef]
- Takano, K.; Ogura, M.; Nakamura, Y.; Yoneda, Y. Neuronal and glial responses to polyamines in the ischemic brain. Curr. Neurovasc. Res. 2005, 2, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Polis, B.; Karasik, D.; Samson, A.O. Alzheimer’s disease as a chronic maladaptive polyamine stress response. Aging 2021, 13, 10770–10795. [Google Scholar] [CrossRef]
- Iezaki, T.; Hinoi, E.; Yamamoto, T.; Ishiura, R.; Ogawa, S.; Yoneda, Y. Amelioration by the natural polyamine spermine of cartilage and bone destruction in rats with collagen-induced arthritis. J. Pharmacol. Sci. 2012, 119, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevalier, C.; Kieser, S.; Çolakoğlu, M.; Hadadi, N.; Brun, J.; Rigo, D.; Suárez-Zamorano, N.; Spiljar, M.; Fabbiano, S.; Busse, B.; et al. Warmth Prevents Bone Loss Through the Gut Microbiota. Cell Metab. 2020, 32, 575–590. [Google Scholar] [CrossRef]
- Thomas, T.; Thomas, T.J. Estradiol control of ornithine decarboxylase mRNA, enzyme activity, and polyamine levels in MCF-7 breast cancer cells: Therapeutic implications. Breast Cancer Res. Treat. 1994, 29, 189–201. [Google Scholar] [CrossRef]
- Cohen, S.; O’Malley, B.W.; Stastny, M. Estrogenic induction of ornithine decarboxylase in vivo and in vitro. Science 1970, 170, 336–338. [Google Scholar] [CrossRef]
- Hodgkinson, K.; Forrest, L.A.; Vuong, N.; Garson, K.; Djordjevic, B.; Vanderhyden, B.C. GREB1 is an estrogen receptor-regulated tumour promoter that is frequently expressed in ovarian cancer. Oncogene 2018, 37, 5873–5886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deschênes, J.; Bourdeau, V.; White, J.H.; Mader, S. Regulation of GREB1 transcription by estrogen receptor alpha through a multipartite enhancer spread over 20 kb of upstream flanking sequences. J. Biol. Chem. 2007, 282, 17335–17339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray Stewart, T.; Dunston, T.T.; Woster, P.M.; Casero, R.A., Jr. Polyamine catabolism and oxidative damage. J. Biol. Chem. 2018, 293, 18736–18745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnova, O.A.; Bartosch, B.; Zakirova, N.F.; Kochetkov, S.N.; Ivanov, A.V. Polyamine Metabolism and Oxidative Protein Folding in the ER as ROS-Producing Systems Neglected in Virology. Int. J. Mol. Sci. 2018, 19, 1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrtačnik, P.; Ostanek, B.; Mencej-Bedrač, S.; Marc, J. The many faces of estrogen signaling. Biochem. Med. 2014, 24, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Gong, X.; Yang, X.; Shang, X.; Du, Q.; Liao, Q.; Xie, R.; Chen, Y.; Xu, J. The roles of estrogen and estrogen receptors in gastrointestinal disease. Oncol. Lett. 2019, 18, 5673–5680. [Google Scholar] [CrossRef] [Green Version]
- Weatherman, R.V. Untangling the estrogen receptor web. Nat. Chem. Biol. 2006, 2, 175–176. [Google Scholar] [CrossRef]
- Powell, E.; Xu, W. Intermolecular interactions identify ligand-selective activity of estrogen receptor alpha/beta dimers. Proc. Natl. Acad. Sci. USA 2008, 105, 19012–19017. [Google Scholar] [CrossRef] [Green Version]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar]
- Nilsson, S.; Mäkelä, S.; Treuter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.A. Mechanisms of estrogen action. Physiol. Rev. 2001, 81, 1535–1565. [Google Scholar] [CrossRef]
- Safe, S.; Kim, K. Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J. Mol. Endocrinol. 2008, 41, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, S.; Demay, F.; Ferrière, F.; Pakdel, F. Phytochemicals Targeting Estrogen Receptors: Beneficial Rather Than Adverse Effects? Int. J. Mol. Sci. 2017, 18, 1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouhimoghadam, M.; Lu, A.S.; Salem, A.K.; Filardo, E.J. Therapeutic Perspectives on the Modulation of G-Protein Coupled Estrogen Receptor, GPER, Function. Front Endocrinol. 2020, 11, 591217. [Google Scholar] [CrossRef] [PubMed]
- DeLeon, C.; Wang, D.Q.; Arnatt, C.K. G Protein-Coupled Estrogen Receptor, GPER1, Offers a Novel Target for the Treatment of Digestive Diseases. Front Endocrinol. 2020, 11, 578536. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lin, R.J.; Xie, W.; Wilpitz, D.; Evans, R.M. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell 1999, 98, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Stossi, F.; Madak-Erdogan, Z.; Katzenellenbogen, B.S. Estrogen receptor alpha represses transcription of early target genes via p300 and CtBP1. Mol. Cell Biol. 2009, 29, 1749–1759. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; Sanda, N.; Miyawaki, Y.; Fujimori, Y.; Yamada, T.; Takagi, A.; Murate, T.; Saito, H.; Kojima, T. Down-regulation of PROS1 gene expression by 17beta-estradiol via estrogen receptor alpha (ERalpha)-Sp1 interaction recruiting receptor-interacting protein 140 and the corepressor-HDAC3 complex. J. Biol. Chem. 2010, 285, 13444–13453. [Google Scholar] [CrossRef] [Green Version]
- Frasor, J.; Danes, J.M.; Komm, B.; Chang, K.C.; Lyttle, C.R.; Katzenellenbogen, B.S. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: Insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology 2003, 144, 4562–4574. [Google Scholar] [CrossRef]
- Grober, O.M.; Mutarelli, M.; Giurato, G.; Ravo, M.; Cicatiello, L.; De Filippo, M.R.; Ferraro, L.; Nassa, G.; Papa, M.F.; Paris, O.; et al. Global analysis of estrogen receptor beta binding to breast cancer cell genome reveals an extensive interplay with estrogen receptor alpha for target gene regulation. BMC Genom. 2011, 12, 36. [Google Scholar] [CrossRef]
- Bunjobpol, W.; Dulloo, I.; Igarashi, K.; Concin, N.; Matsuo, K.; Sabapathy, K. Suppression of acetylpolyamine oxidase by selected AP-1 members regulates DNp73 abundance: Mechanistic insights for overcoming DNp73-mediated resistance to chemotherapeutic drugs. Cell Death Differ. 2014, 21, 1240–1249. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yang, L.; Zhao, Y.; Zeng, G.; Wu, Y.; Chen, Y.; Zhang, J.; Zeng, Q. Estrogen is a novel regulator of Tnfaip1 in mouse hippocampus. Int. J. Mol. Med. 2014, 34, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Bartella, V.; Rizza, P.; Barone, I.; Zito, D.; Giordano, F.; Giordano, C.; Catalano, S.; Mauro, L.; Sisci, D.; Panno, M.L.; et al. Estrogen receptor beta binds Sp1 and recruits a corepressor complex to the estrogen receptor alpha gene promoter. Breast Cancer Res. Treat. 2012, 134, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayanathan, V.; Venkiteswaran, S.; Nair, S.K.; Verma, A.; Thomas, T.J.; Zhu, B.T.; Thomas, T. Physiologic levels of 2-methoxyestradiol interfere with nongenomic signaling of 17beta-estradiol in human breast cancer cells. Clin. Cancer Res. 2006, 12, 2038–2048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 26, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.J.; Thomas, T.; John, S.; Hsu, H.C.; Yang, P.; Keinänen, T.A.; Hyvönen, M.T. Tamoxifen metabolite endoxifen interferes with the polyamine pathway in breast cancer. Amino Acids 2016, 48, 2293–2302. [Google Scholar] [CrossRef] [PubMed]
- Lima, G.; Shiu, R.P. Role of polyamines in estradiol-induced growth of human breast cancer cells. Cancer Res. 1985, 45, 2466–2470. [Google Scholar] [PubMed]
- Manni, A.; Wright, C. Polyamines as mediators of estrogen action on the growth of experimental breast cancer in rats. J. Natl. Cancer Inst. 1984, 73, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Manni, A.; Wechter, R.; Grove, R.; Wei, L.; Martel, J.; Demers, L. Polyamine profiles and growth properties of ornithine decarboxylase overexpressing MCF-7 breast cancer cells in culture. Breast Cancer Res. Treat. 1995, 34, 45–53. [Google Scholar] [CrossRef]
- Mirza, F.S.; Prestwood, K.M. Bone health and aging: Implications for menopause. Endocrinol. Metab. Clin. North Am. 2004, 33, 741–759. [Google Scholar] [CrossRef]
- Ahlborg, H.G.; Johnell, O.; Turner, C.H.; Rannevik, G.; Karlsson, M.K. Bone loss and bone size after menopause. N. Engl. J. Med. 2003, 349, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Recker, R.R.; Lappe, J.M.; Davies, M.; Kimmel, D. Perimenopausal bone histomorphometry before and after menopause. Bone 2018, 108, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Reginster, J.Y.; Burlet, N. Osteoporosis: A still increasing prevalence. Bone 2006, 38, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Kyrilli, A.; Gacquer, D.; Detours, V.; Lefort, A.; Libert, F.; Twyffels, L.; Van Den Eeckhaute, L.; Strickaert, A.; Maenhaut, C.; De Deken, X.; et al. Dissecting the Role of Thyrotropin in the DNA Damage Response in Human Thyrocytes after 131I, γ Radiation and H2O2. J. Clin. Endocrinol. Metab. 2020, 105, 839–853. [Google Scholar] [CrossRef]
- Choi, Y.E.; Song, M.J.; Hara, M.; Imanaka-Yoshida, K.; Lee, D.H.; Chung, J.H.; Lee, S.-T. Effects of tenascin C on the integrity of extracellular matrix and skin aging. Int. J. Mol. Sci. 2020, 21, 8693. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jeong, H.D.; Song, M.J.; Lee, D.H.; Chung, J.H.; Lee, S.-T. SOD3 Suppresses the Expression of MMP-1 and Increases the Integrity of Extracellular Matrix in Fibroblasts. Antioxidants 2022, 11, 928. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.-S.; Park, M.-K.; Kim, J.H.; Oh, S.W.; Jang, J.-Y.; Lee, H.; Lee, S.-T. PTK7, a Catalytically inactive receptor tyrosine kinase, increases oncogenic phenotypes in xenograft tumors of esophageal squamous cell carcinoma KYSE-30 Cells. Int. J. Mol. Sci. 2022, 23, 2391. [Google Scholar] [CrossRef] [PubMed]
- Gearing, L.J.; Cumming, H.E.; Chapman, R.; Finkel, A.M.; Woodhouse, I.B.; Luu, K.; Gould, J.A.; Forster, S.C.; Hertzog, P.J. CiiiDER: A tool for predicting and analysing transcription factor binding sites. PLoS ONE 2019, 14, e0215495. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Seo, E.K.; Lee, S.-T. Skullcapflavone II inhibits degradation of type I collagen by suppressing MMP-1 transcription in human skin fibroblasts. Int. J. Mol. Sci. 2019, 20, 2734. [Google Scholar] [CrossRef] [Green Version]
- Barish, G.D.; Tangirala, R.K. Chromatin immunoprecipitation. Methods Mol. Biol. 2013, 1027, 327–342. [Google Scholar]
- Truax, A.D.; Greer, S.F. ChIP and Re-ChIP assays: Investigating interactions between regulatory proteins, histone modifications, and the DNA sequences to which they bind. Methods Mol. Biol. 2012, 809, 175–188. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.H.; Lee, S.-T. Polyamine Oxidase Expression Is Downregulated by 17β-Estradiol via Estrogen Receptor 2 in Human MCF-7 Breast Cancer Cells. Int. J. Mol. Sci. 2022, 23, 7521. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23147521
Kim JH, Lee S-T. Polyamine Oxidase Expression Is Downregulated by 17β-Estradiol via Estrogen Receptor 2 in Human MCF-7 Breast Cancer Cells. International Journal of Molecular Sciences. 2022; 23(14):7521. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23147521
Chicago/Turabian StyleKim, Jin Hyung, and Seung-Taek Lee. 2022. "Polyamine Oxidase Expression Is Downregulated by 17β-Estradiol via Estrogen Receptor 2 in Human MCF-7 Breast Cancer Cells" International Journal of Molecular Sciences 23, no. 14: 7521. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23147521