
Increased Acetylcholine Levels and Other Brain Effects in 5XFAD Mice after Treatment with 8,14-Dihydroxy Metabolite of Efavirenz


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Brain Sterols
2.2. Brain Aβ Content
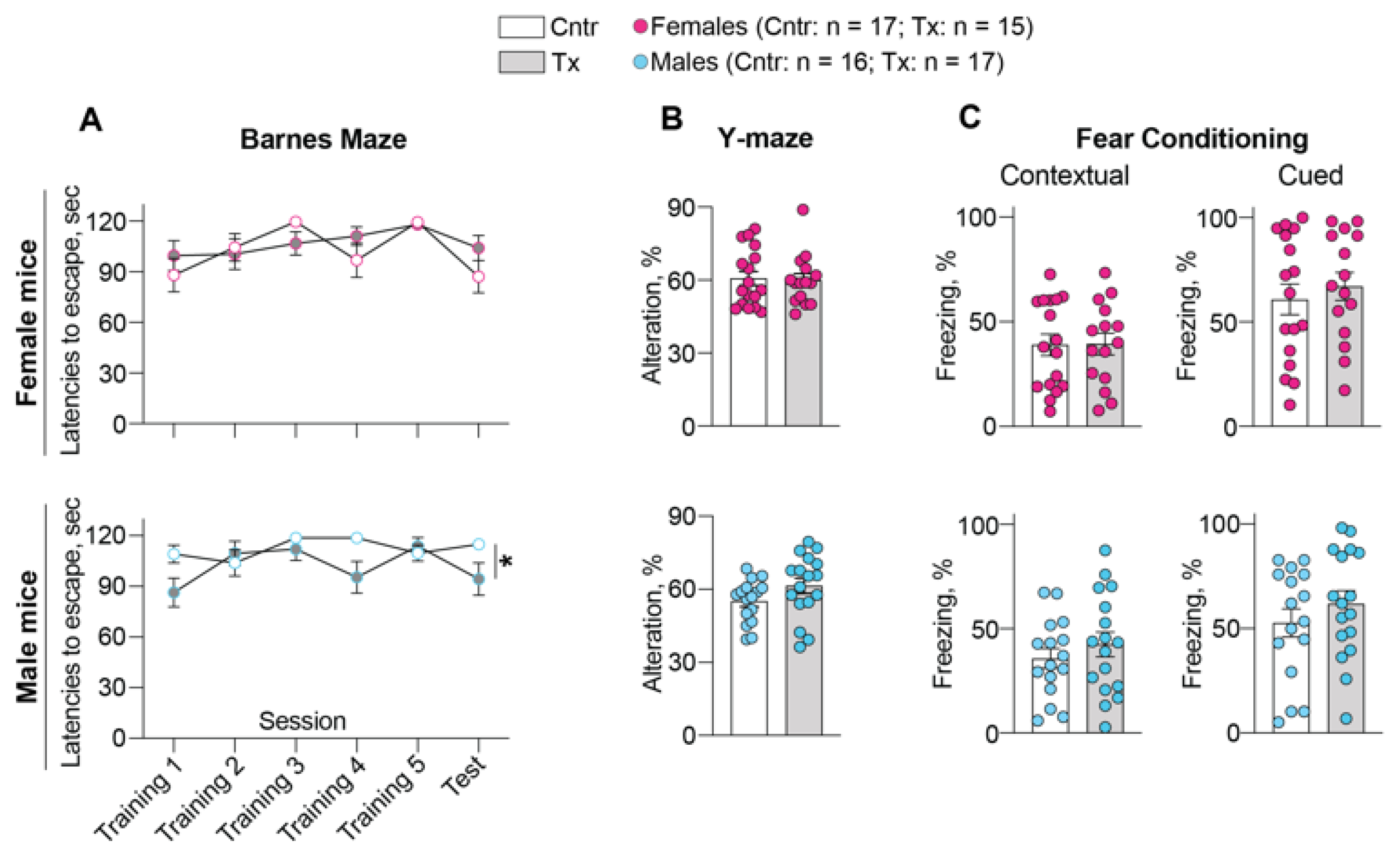
2.3. Behavioral Assessments
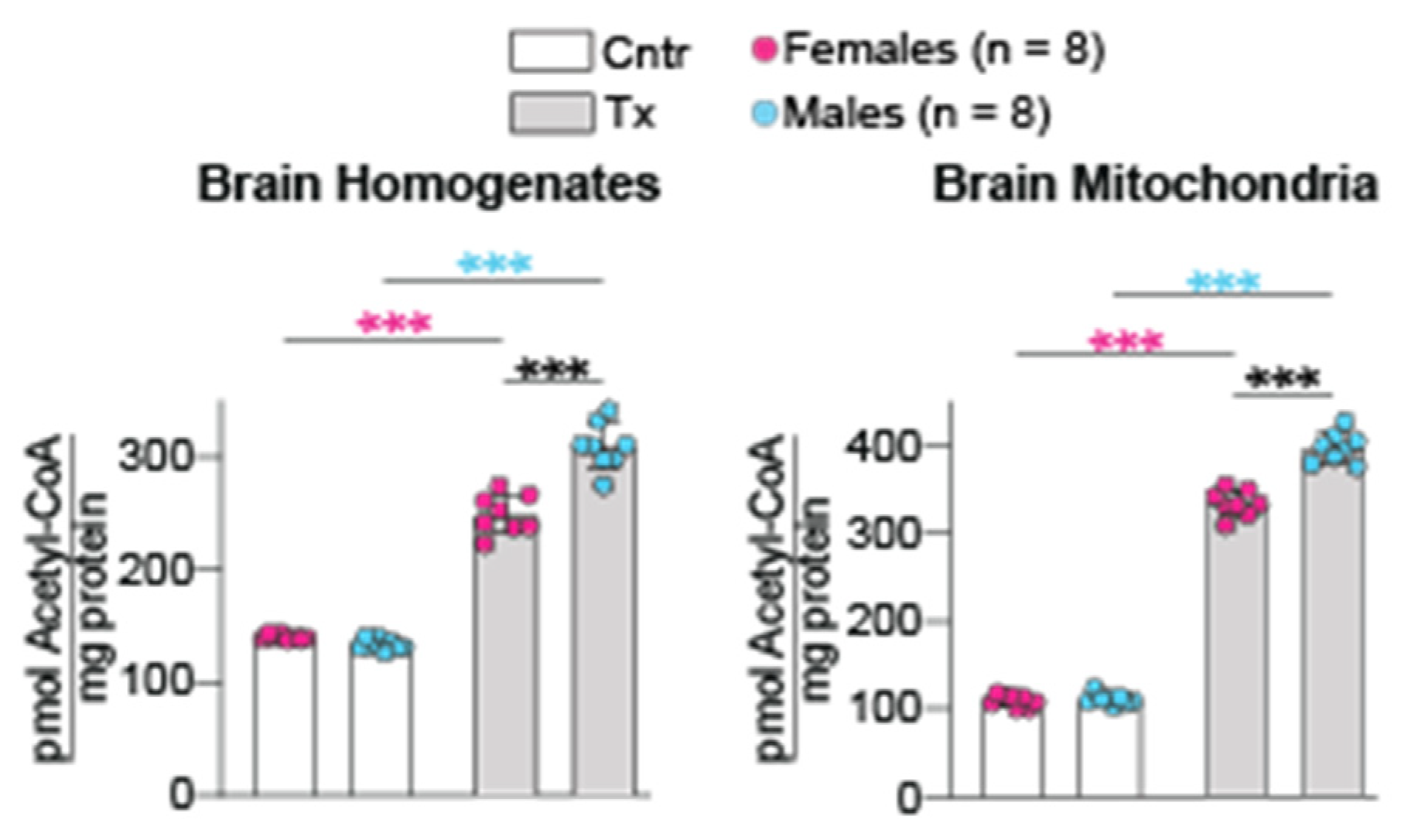
2.4. Brain Acetyl-CoA Levels
2.5. Brain Ach Levels
2.6. Brain Expresion of Ach-Related Genes
2.7. CYP46A1 Activation In Vitro
2.8. Brain Levels of Various Marker Proteins and CYP46A1
3. Discussion
4. Materials and Methods
4.1. Animals and Treatment
4.2. Behavioral Assessments
4.3. Brain Processing
4.4. Quantitative Studies
4.5. CYP46A1 Activation In Vitro
4.6. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pikuleva, I.A.; Cartier, N. Cholesterol Hydroxylating Cytochrome P450 46A1: From Mechanisms of Action to Clinical Applications. Front. Aging Neurosci. 2021, 13, 696778. [Google Scholar] [CrossRef] [PubMed]
- Pikuleva, I.A. Targeting cytochrome P450 46A1 and brain cholesterol 24-hydroxylation to treat neurodegenerative diseases. Explor. Neuroprotective Ther. 2021, 1, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Hudry, E.; Van Dam, D.; Kulik, W.; De Deyn, P.P.; Stet, F.S.; Ahouansou, O.; Benraiss, A.; Delacourte, A.; Bougneres, P.; Aubourg, P.; et al. Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer’s disease. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Burlot, M.A.; Braudeau, J.; Michaelsen-Preusse, K.; Potier, B.; Ayciriex, S.; Varin, J.; Gautier, B.; Djelti, F.; Audrain, M.; Dauphinot, L.; et al. Cholesterol 24-hydroxylase defect is implicated in memory impairments associated with Alzheimer-like Tau pathology. Hum. Mol. Genet. 2015, 24, 5965–5976. [Google Scholar] [CrossRef]
- Boussicault, L.; Alves, S.; Lamaziere, A.; Planques, A.; Heck, N.; Moumne, L.; Despres, G.; Bolte, S.; Hu, A.; Pages, C.; et al. CYP46A1, the rate-limiting enzyme for cholesterol degradation, is neuroprotective in Huntington’s disease. Brain 2016, 139 Pt 3, 953–970. [Google Scholar] [CrossRef] [Green Version]
- Mast, N.; Saadane, A.; Valencia-Olvera, A.; Constans, J.; Maxfield, E.; Arakawa, H.; Li, Y.; Landreth, G.; Pikuleva, I.A. Cholesterol-metabolizing enzyme cytochrome P450 46A1 as a pharmacologic target for Alzheimer’s disease. Neuropharmacology 2017, 123, 465–476. [Google Scholar] [CrossRef]
- Petrov, A.M.; Lam, M.; Mast, N.; Moon, J.; Li, Y.; Maxfield, E.; Pikuleva, I.A. CYP46A1 Activation by Efavirenz Leads to Behavioral Improvement without Significant Changes in Amyloid Plaque Load in the Brain of 5XFAD Mice. Neurother. J. Am. Soc. Exp. NeuroTherapeutics 2019, 16, 710–724. [Google Scholar] [CrossRef]
- Patel, T.K.; Patel, V.B.; Rana, D.G. Possible anti-depressant effect of efavirenz and pro-depressive-like effect of voriconazole in specified doses in various experimental models of depression in mice. Pharmacol. Rep. 2017, 69, 1082–1087. [Google Scholar] [CrossRef]
- Kacher, R.; Lamaziere, A.; Heck, N.; Kappes, V.; Mounier, C.; Despres, G.; Dembitskaya, Y.; Perrin, E.; Christaller, W.; Sasidharan Nair, S.; et al. CYP46A1 gene therapy deciphers the role of brain cholesterol metabolism in Huntington’s disease. Brain 2019, 142, 2432–2450. [Google Scholar] [CrossRef]
- Mitroi, D.N.; Pereyra-Gomez, G.; Soto-Huelin, B.; Senovilla, F.; Kobayashi, T.; Esteban, J.A.; Ledesma, M.D. NPC1 enables cholesterol mobilization during long-term potentiation that can be restored in Niemann-Pick disease type C by CYP46A1 activation. EMBO Rep. 2019, 20, e48143. [Google Scholar] [CrossRef]
- Nobrega, C.; Mendonca, L.; Marcelo, A.; Lamaziere, A.; Tome, S.; Despres, G.; Matos, C.A.; Mechmet, F.; Langui, D.; den Dunnen, W.; et al. Restoring brain cholesterol turnover improves autophagy and has therapeutic potential in mouse models of spinocerebellar ataxia. Acta Neuropathol. 2019, 135, 837–858. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Wang, S.; Yang, N.; Wang, X.; Zhao, W.; Saed, H.S.; Daubon, T.; Huang, B.; Chen, A.; Li, G.; et al. Therapeutic implications of altered cholesterol homeostasis mediated by loss of CYP46A1 in human glioblastoma. EMBO Mol. Med. 2020, 12, e10924. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Hannaoui, S.; Nemani, S.; Tahir, W.; Zemlyankina, I.; Cherry, P.; Shim, S.Y.; Sim, V.; Schaetzl, H.M.; Gilch, S. Oral administration of repurposed drug targeting Cyp46A1 increases survival times of prion infected mice. Acta Neuropathol. Commun. 2021, 9, 58. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Petrov, A.M.; Mast, N.; Li, Y.; Pikuleva, I.A. The key genes, phosphoproteins, processes, and pathways affected by efavirenz-activated CYP46A1 in the amyloid-decreasing paradigm of efavirenz treatment. FASEB J. 2019, 33, 8782–8798. [Google Scholar] [CrossRef]
- Mast, N.; El-Darzi, N.; Petrov, A.M.; Li, Y.; Pikuleva, I.A. CYP46A1-dependent and independent effects of efavirenz treatment. Brain Commun. 2020, 2, fcaa180. [Google Scholar] [CrossRef] [PubMed]
- Mast, N.; Li, Y.; Linger, M.; Clark, M.; Wiseman, J.; Pikuleva, I.A. Pharmacologic stimulation of cytochrome P450 46A1 and cerebral cholesterol turnover in mice. J. Biol. Chem. 2014, 289, 3529–3538. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.W.; Mast, N.; Hudgens, J.W.; Lin, J.B.; Turko, I.V.; Pikuleva, I.A. Mapping of the allosteric site in cholesterol hydroxylase CYP46A1 for efavirenz, a drug that stimulates enzyme activity. J. Biol. Chem. 2016, 291, 11876–11886. [Google Scholar] [CrossRef] [Green Version]
- Mast, N.; Anderson, K.W.; Johnson, K.M.; Phan, T.T.N.; Guengerich, F.P.; Pikuleva, I.A. In Vitro cytochrome P450 46A1 (CYP46A1) activation by neuroactive compounds. J. Biol. Chem. 2017, 292, 12934–12946. [Google Scholar] [CrossRef] [Green Version]
- Avery, L.B.; VanAusdall, J.L.; Hendrix, C.W.; Bumpus, N.N. Compartmentalization and antiviral effect of efavirenz metabolites in blood plasma, seminal plasma, and cerebrospinal fluid. Drug Metab. Dispos. 2013, 41, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Ward, B.A.; Gorski, J.C.; Jones, D.R.; Hall, S.D.; Flockhart, D.A.; Desta, Z. The cytochrome P450 2B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: Implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J. Pharmacol. Exp. Ther. 2003, 306, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Ogburn, E.T.; Jones, D.R.; Masters, A.R.; Xu, C.; Guo, Y.; Desta, Z. Efavirenz primary and secondary metabolism in vitro and in vivo: Identification of novel metabolic pathways and cytochrome P450 2A6 as the principal catalyst of efavirenz 7-hydroxylation. Drug Metab. Dispos. 2010, 38, 1218–1229. [Google Scholar] [CrossRef] [PubMed]
- Bumpus, N.N.; Kent, U.M.; Hollenberg, P.F. Metabolism of efavirenz and 8-hydroxyefavirenz by P450 2B6 leads to inactivation by two distinct mechanisms. J. Pharmacol. Exp. Ther. 2006, 318, 345–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avery, L.B.; Sacktor, N.; McArthur, J.C.; Hendrix, C.W. Protein-free efavirenz concentrations in cerebrospinal fluid and blood plasma are equivalent: Applying the law of mass action to predict protein-free drug concentration. Antimicrob. Agents Chemother. 2013, 57, 1409–1414. [Google Scholar] [CrossRef] [Green Version]
- Mast, N.; Fotinich, A.; Pikuleva, I.A. The hydroxylation position rather than chirality determines how efavirenz metabolites activate CYP46A1 In Vitro. Drug Metab Dispos 2022, 50, 923–930. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Turley, S.D. Cholesterol metabolism in the brain. Curr. Opin. Lipidol. 2001, 12, 105–112. [Google Scholar] [CrossRef]
- Lund, E.G.; Xie, C.; Kotti, T.; Turley, S.D.; Dietschy, J.M.; Russell, D.W. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J. Biol. Chem. 2003, 278, 22980–22988. [Google Scholar] [CrossRef] [Green Version]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 2011, 50, 357–371. [Google Scholar] [CrossRef]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell. Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab 2015, 21, 805–821. [Google Scholar] [CrossRef] [Green Version]
- Mast, N.; Petrov, A.M.; Prendergast, E.; Bederman, I.; Pikuleva, I.A. Brain Acetyl-CoA Production and Phosphorylation of Cytoskeletal Proteins Are Targets of CYP46A1 Activity Modulation and Altered Sterol Flux. Neurother. J. Am. Soc. Exp. NeuroTherapeutics 2021, 18, 2040–2060. [Google Scholar] [CrossRef] [PubMed]
- Tucek, S. Short-term control of the synthesis of acetylcholine. Prog. Biophys. Mol. Biol. 1993, 60, 59–69. [Google Scholar] [CrossRef]
- Chen, Z.R.; Huang, J.B.; Yang, S.L.; Hong, F.F. Role of Cholinergic Signaling in Alzheimer’s Disease. Molecules 2022, 27, 1816. [Google Scholar] [CrossRef] [PubMed]
- Terry, A.V., Jr.; Buccafusco, J.J. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: Recent challenges and their implications for novel drug development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef]
- Okuda, T.; Haga, T. Functional characterization of the human high-affinity choline transporter. FEBS Lett. 2000, 484, 92–97. [Google Scholar] [CrossRef] [Green Version]
- Okuda, T.; Haga, T.; Kanai, Y.; Endou, H.; Ishihara, T.; Katsura, I. Identification and characterization of the high-affinity choline transporter. Nat. Neurosci. 2000, 3, 120–125. [Google Scholar] [CrossRef]
- Augustinsson, K.B.; Nachmansohn, D. Distinction between Acetylcholine-Esterase and Other Choline Ester-splitting Enzymes. Science 1949, 110, 98–99. [Google Scholar] [CrossRef]
- Ahmed, T.; Zahid, S.; Mahboob, A.; Farhat, S.M. Cholinergic System and Post-translational Modifications: An Insight on the Role in Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 480–494. [Google Scholar] [CrossRef]
- Erickson, J.D.; Varoqui, H.; Schafer, M.K.; Modi, W.; Diebler, M.F.; Weihe, E.; Rand, J.; Eiden, L.E.; Bonner, T.I.; Usdin, T.B. Functional identification of a vesicular acetylcholine transporter and its expression from a "cholinergic" gene locus. J. Biol. Chem. 1994, 269, 21929–21932. [Google Scholar] [CrossRef]
- Maeda, S.; Qu, Q.; Robertson, M.J.; Skiniotis, G.; Kobilka, B.K. Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science 2019, 364, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Perrier, A.L.; Massoulie, J.; Krejci, E. PRiMA: The membrane anchor of acetylcholinesterase in the brain. Neuron 2002, 33, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Oda, Y. Choline acetyltransferase: The structure, distribution and pathologic changes in the central nervous system. Pathol. Int. 1999, 49, 921–937. [Google Scholar] [CrossRef]
- Kwon, S.E.; Chapman, E.R. Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron 2011, 70, 847–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nithianantharajah, J.; Levis, H.; Murphy, M. Environmental enrichment results in cortical and subcortical changes in levels of synaptophysin and PSD-95 proteins. Neurobiol. Learn. Mem. 2004, 81, 200–210. [Google Scholar] [CrossRef]
- Essrich, C.; Lorez, M.; Benson, J.A.; Fritschy, J.-M.; Lüscher, B. Postsynaptic clustering of major GABAA receptor subtypes requires the γ2 subunit and gephyrin. Nat. Neurosci. 1998, 1, 563–571. [Google Scholar] [CrossRef]
- Holtmaat, A.; Svoboda, K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci. 2009, 10, 647–658. [Google Scholar] [CrossRef]
- Jurga, A.M.; Paleczna, M.; Kadluczka, J.; Kuter, K.Z. Beyond the GFAP-Astrocyte Protein Markers in the Brain. Biomolecules 2021, 11, 1361. [Google Scholar] [CrossRef]
- Schwabenland, M.; Brück, W.; Priller, J.; Stadelmann, C.; Lassmann, H.; Prinz, M. Analyzing microglial phenotypes across neuropathologies: A practical guide. Acta Neuropathol. 2021, 142, 923–936. [Google Scholar] [CrossRef]
- Mast, N.; Verwilst, P.; Wilkey, C.J.; Guengerich, F.P.; Pikuleva, I.A. In Vitro Activation of Cytochrome P450 46A1 (CYP46A1) by Efavirenz-Related Compounds. J. Med. Chem. 2020, 63, 6477–6488. [Google Scholar] [CrossRef] [Green Version]
- Hitchcock, S.A.; Pennington, L.D. Structure-brain exposure relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef] [PubMed]
- Bach, M.E.; Hawkins, R.D.; Osman, M.; Kandel, E.R.; Mayford, M. Impairment of spatial but not contextual memory in CaMKII mutant mice with a selective loss of hippocampal LTP in the range of the theta frequency. Cell 1995, 81, 905–915. [Google Scholar] [CrossRef] [Green Version]
- Spann, N.J.; Garmire, L.X.; McDonald, J.G.; Myers, D.S.; Milne, S.B.; Shibata, N.; Reichart, D.; Fox, J.N.; Shaked, I.; Heudobler, D.; et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell 2012, 151, 138–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calkin, A.C.; Tontonoz, P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell. Biol. 2012, 13, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Jakobsson, T.; Treuter, E.; Gustafsson, J.A.; Steffensen, K.R. Liver X receptor biology and pharmacology: New pathways, challenges and opportunities. Trends Pharmacol. Sci. 2012, 33, 394–404. [Google Scholar] [CrossRef]
- Ronowska, A.; Szutowicz, A.; Bielarczyk, H.; Gul-Hinc, S.; Klimaszewska-Łata, J.; Dyś, A.; Zyśk, M.; Jankowska-Kulawy, A. The Regulatory Effects of Acetyl-CoA Distribution in the Healthy and Diseased Brain. Front. Cell. Neurosci. 2018, 12, 169. [Google Scholar] [CrossRef] [Green Version]
- Coyle, J.T.; Price, D.L.; DeLong, M.R. Alzheimer’s disease: A disorder of cortical cholinergic innervation. Science 1983, 219, 1184–1190. [Google Scholar] [CrossRef]
- Li, X.T. Alzheimer’s disease therapy based on acetylcholinesterase inhibitor/blocker effects on voltage-gated potassium channels. Metab. Brain Dis. 2022, 37, 581–587. [Google Scholar] [CrossRef]
- Petrov, A.M.; Mast, N.; Li, Y.; Denker, J.; Pikuleva, I.A. Brain sterol flux mediated by cytochrome P450 46A1 affects membrane properties and membrane-dependent processes. Brain Commun. 2020, 2, fcaa043. [Google Scholar] [CrossRef] [Green Version]
- Ovsepian, S.V.; O’Leary, V.B.; Zaborszky, L. Cholinergic Mechanisms in the Cerebral Cortex: Beyond Synaptic Transmission. Neuroscientist 2016, 22, 238–251. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.; Hawes, N.L.; Hurd, R.E.; Davisson, M.T.; Nusinowitz, S.; Heckenlively, J.R. Retinal degeneration mutants in the mouse. Vision Res. 2002, 42, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Petrov, A.M.; Pikuleva, I.A. Cholesterol 24-Hydroxylation by CYP46A1: Benefits of Modulation for Brain Diseases. Neurother. J. Am. Soc. Exp. NeuroTherapeutics 2019, 16, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mast, N.; Reem, R.; Bederman, I.; Huang, S.; DiPatre, P.L.; Björkhem, I.; Pikuleva, I.A. Cholestenoic acid is an important elimination product of cholesterol in the retina: Comparison of retinal cholesterol metabolism with that in the brain. Investig. Ophthalmol. Vis. Sci. 2011, 52, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.R.; Anderson, M.F. Isolation of mitochondria from rat brain using Percoll density gradient centrifugation. Nat. Protoc. 2008, 3, 1228–1239. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- White, M.A.; Mast, N.; Björkhem, I.; Johnson, E.F.; Stout, C.D.; Pikuleva, I.A. Use of complementary cation and anion heavy-atom salt derivatives to solve the structure of cytochrome P450 46A1. Acta Crystallogr. D Biol. Crystallogr. 2008, 64 Pt 5, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Hanna, I.H.; Teiber, J.F.; Kokones, K.L.; Hollenberg, P.F. Role of the alanine at position 363 of cytochrome P450 2B2 in influencing the NADPH- and hydroperoxide-supported activities. Arch. Biochem. Biophys. 1998, 350, 324–332. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mast, N.; Li, Y.; Pikuleva, I.A. Increased Acetylcholine Levels and Other Brain Effects in 5XFAD Mice after Treatment with 8,14-Dihydroxy Metabolite of Efavirenz. Int. J. Mol. Sci. 2022, 23, 7669. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23147669
Mast N, Li Y, Pikuleva IA. Increased Acetylcholine Levels and Other Brain Effects in 5XFAD Mice after Treatment with 8,14-Dihydroxy Metabolite of Efavirenz. International Journal of Molecular Sciences. 2022; 23(14):7669. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23147669
Chicago/Turabian StyleMast, Natalia, Yong Li, and Irina A. Pikuleva. 2022. "Increased Acetylcholine Levels and Other Brain Effects in 5XFAD Mice after Treatment with 8,14-Dihydroxy Metabolite of Efavirenz" International Journal of Molecular Sciences 23, no. 14: 7669. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23147669