
HDAC Class I Inhibitor Domatinostat Preferentially Targets Glioma Stem Cells over Their Differentiated Progeny

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Preferential Inhibition of Glioma Stem Cell Growth by Domatinostat
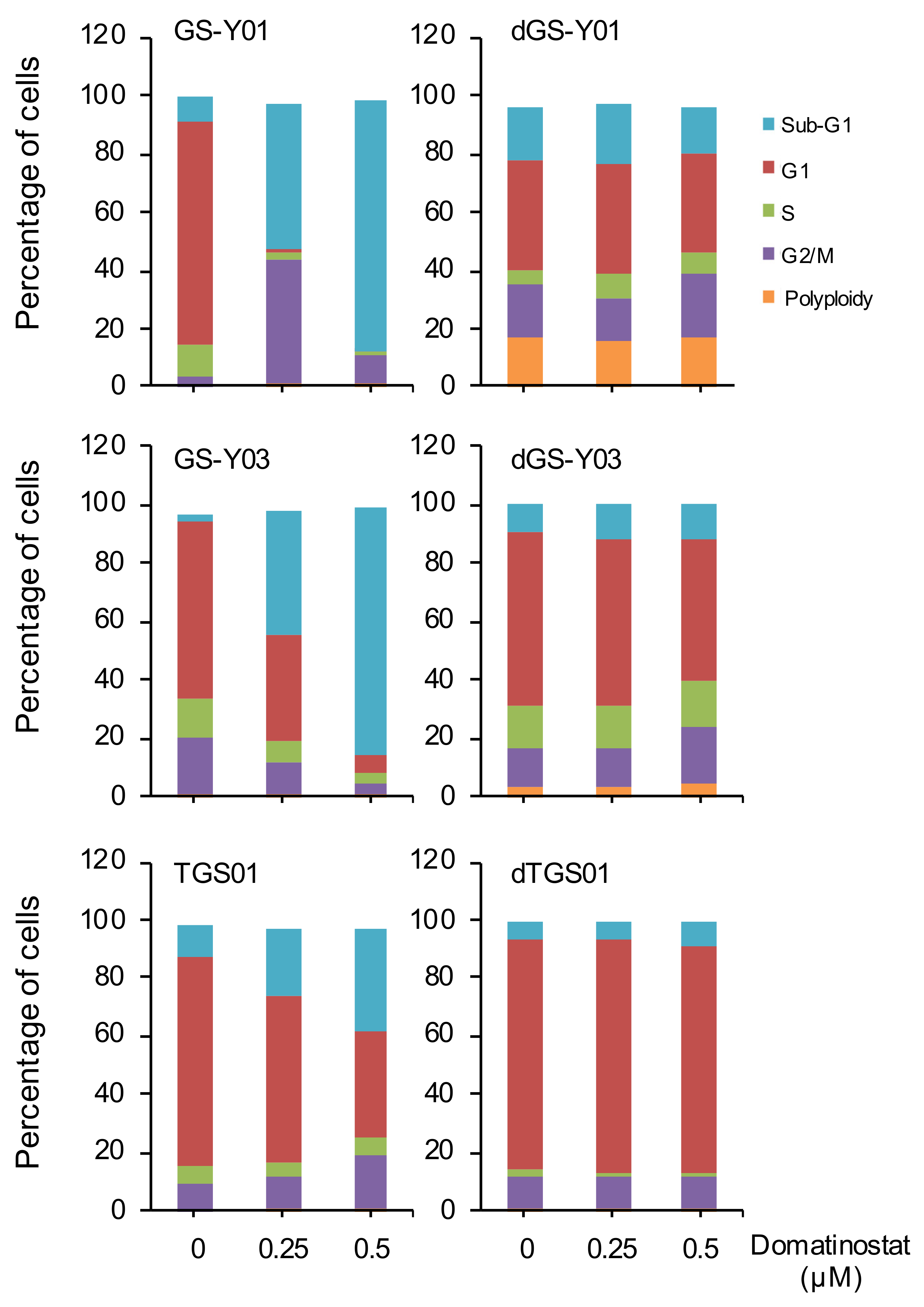
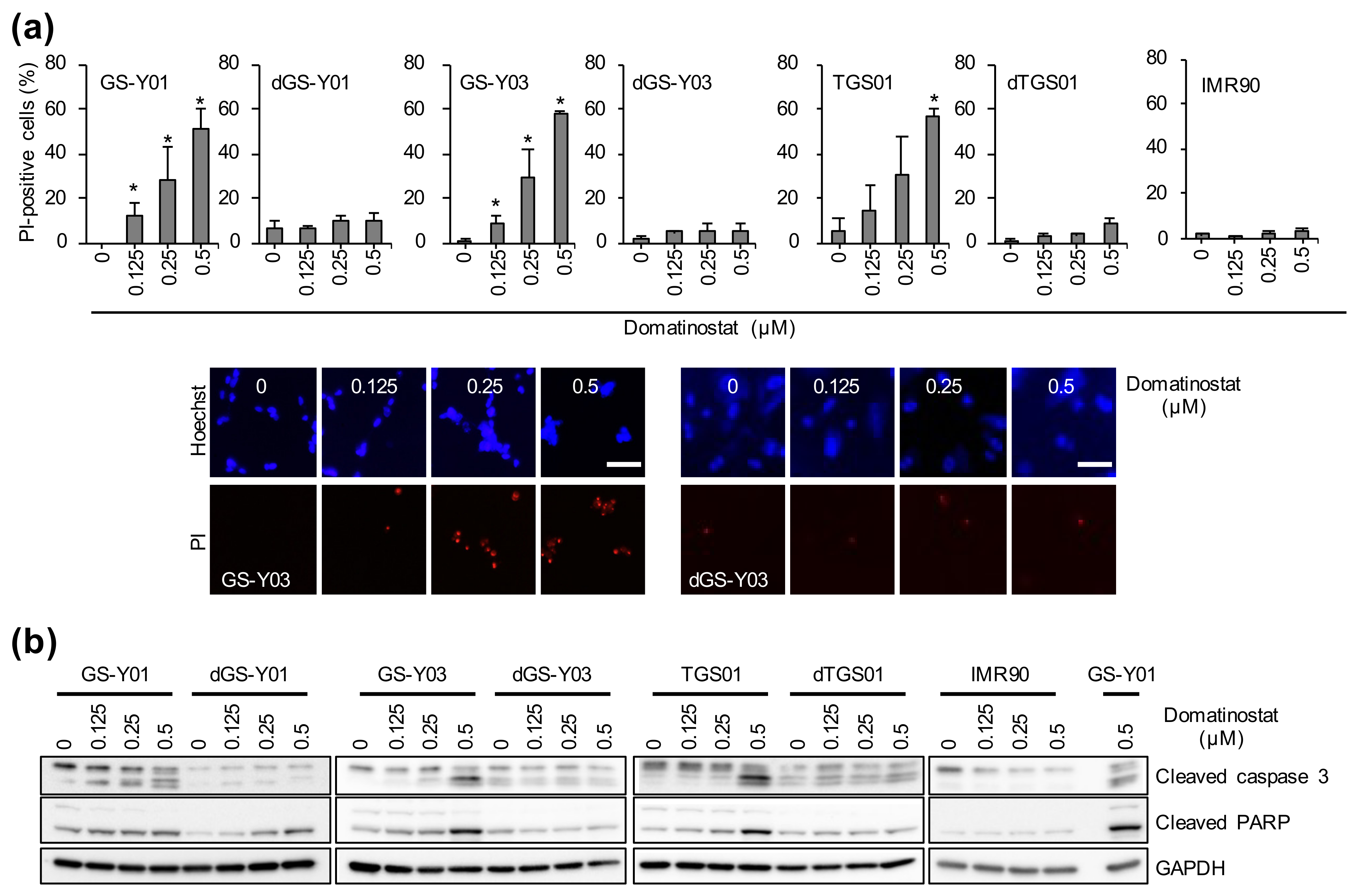
2.2. Domatinostat Activates the Caspase Pathway and Induces Cell Death in GSCs
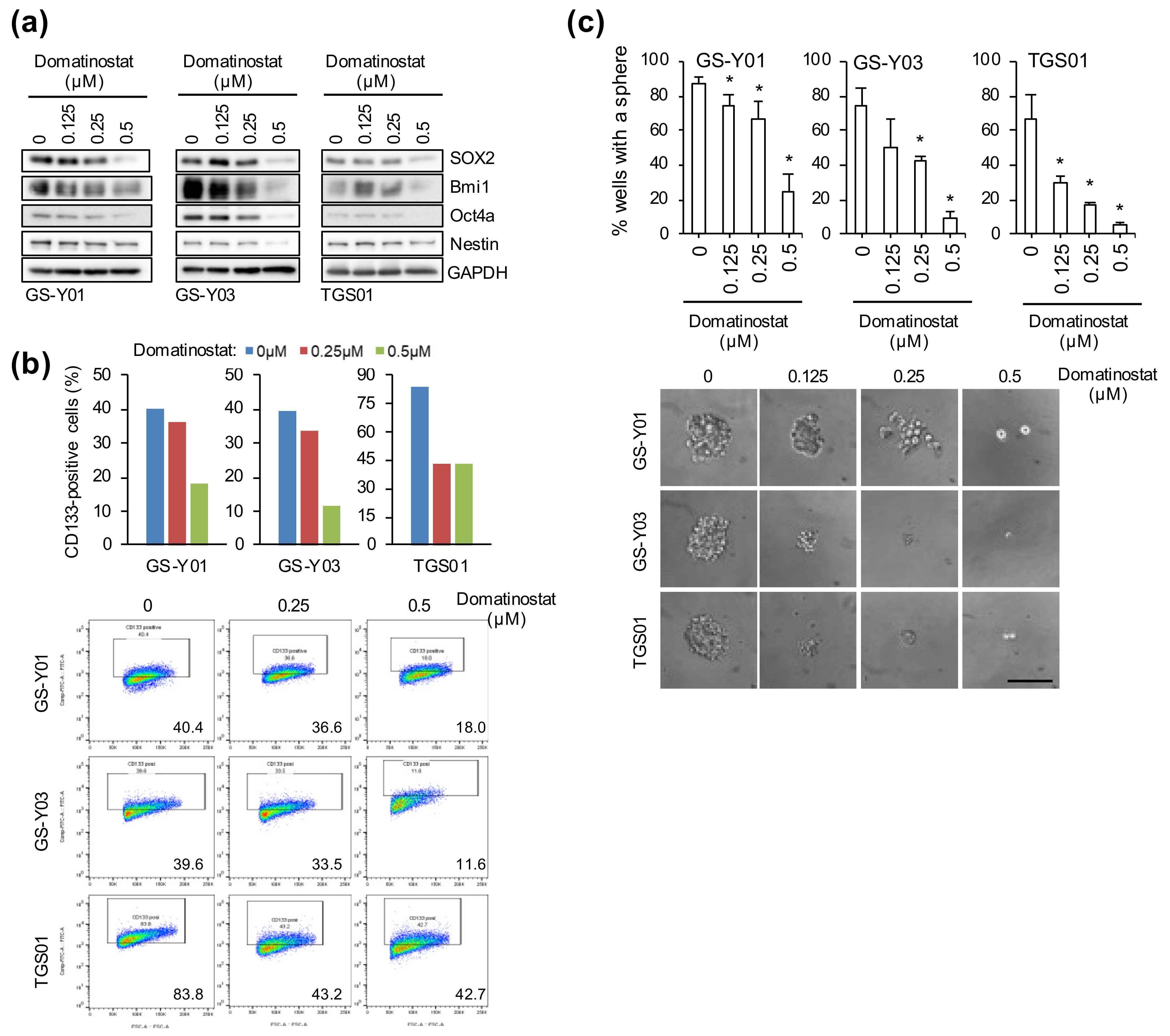
2.3. Self-Renewal Capacity Is Lost in GSCs That Survived Domatinostat Treatment
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Trypan Blue Dye Exclusion Assay
4.5. Propidium Iodide Incorporation Assay
4.6. Flow Cytometric Analysis
4.7. Western Blot Analysis
4.8. Sphere Formation Assay
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mathew, E.N.; Berry, B.C.; Yang, H.W.; Carroll, R.S.; Johnson, M.D. Delivering Therapeutics to Glioblastoma: Overcoming Biological Constraints. Int. J. Mol. Sci. 2022, 23, 1711. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Colak, S.; Medema, J.P. Cancer stem cells—Important players in tumor therapy resistance. FEBS J. 2014, 281, 4779–4791. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Fang, X.; Luo, Q.; Ouyang, G. Cancer stem cells: A potential target for cancer therapy. Cell. Mol. Life Sci. 2015, 72, 3411–3424. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuramoto, K.; Suzuki, S.; Sakaki, H.; Takeda, H.; Sanomachi, T.; Seino, S.; Narita, Y.; Kayama, T.; Kitanaka, C.; Okada, M. Licochalcone A specifically induces cell death in glioma stem cells via mitochondrial dysfunction. FEBS Open Bio 2017, 7, 835–844. [Google Scholar] [CrossRef]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C.; Okada, M. Verteporfin inhibits oxidative phosphorylation and induces cell death specifically in glioma stem cells. FEBS J. 2020, 287, 2023–2036. [Google Scholar] [CrossRef]
- Okada, M.; Suzuki, S.; Togashi, K.; Sugai, A.; Yamamoto, M.; Kitanaka, C. Targeting Folate Metabolism Is Selectively Cytotoxic to Glioma Stem Cells and Effectively Cooperates with Differentiation Therapy to Eliminate Tumor-Initiating Cells in Glioma Xenografts. Int. J. Mol. Sci. 2021, 22, 11633. [Google Scholar] [CrossRef]
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, L.J.Y.; Prager, B.C.; Flavahan, W.A.; Liu, X.; et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat. Neurosci. 2017, 20, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Tashima, T.; Murata, H.; Kodama, H. Design and synthesis of novel and highly-active pan-histone deacetylase (pan-HDAC) inhibitors. Bioorg. Med. Chem. 2014, 22, 3720–3731. [Google Scholar] [CrossRef]
- Fu, M.; Wan, F.; Li, Z.; Zhang, F. 4SC-202 activates ASK1-dependent mitochondrial apoptosis pathway to inhibit hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2016, 471, 267–273. [Google Scholar] [CrossRef]
- Pinkerneil, M.; Hoffmann, M.J.; Kohlhof, H.; Schulz, W.A.; Niegisch, G. Evaluation of the Therapeutic Potential of the Novel Isotype Specific HDAC Inhibitor 4SC-202 in Urothelial Carcinoma Cell Lines. Target. Oncol. 2016, 11, 783–798. [Google Scholar] [CrossRef] [Green Version]
- Zhijun, H.; Shusheng, W.; Han, M.; Jianping, L.; Li-Sen, Q.; Dechun, L. Pre-clinical characterization of 4SC-202, a novel class I HDAC inhibitor, against colorectal cancer cells. Tumor Biol. 2016, 37, 10257–10267. [Google Scholar] [CrossRef]
- Torres, H.M.; VanCleave, A.M.; Vollmer, M.; Callahan, D.L.; Smithback, A.; Conn, J.M.; Rodezno-Antunes, T.; Gao, Z.; Cao, Y.; Afeworki, Y.; et al. Selective Targeting of Class I Histone Deacetylases in a Model of Human Osteosarcoma. Cancers 2021, 13, 4199. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Z.; Kuang, X.; Ma, D.; Xiong, J.; Lu, T.; Zhang, Y.; Yu, K.; Zhang, S.; Wang, J.; et al. 4SC-202 induces apoptosis in myelodysplastic syndromes and the underlying mechanism. Am. J. Transl. Res. 2020, 12, 2968–2983. [Google Scholar]
- Wobser, M.; Weber, A.; Glunz, A.; Tauch, S.; Seitz, K.; Butelmann, T.; Hesbacher, S.; Goebeler, M.; Bartz, R.; Kohlhof, H.; et al. Elucidating the mechanism of action of domatinostat (4SC-202) in cutaneous T cell lymphoma cells. J. Hematol. Oncol. 2019, 12, 30. [Google Scholar] [CrossRef] [Green Version]
- Gruber, W.; Peer, E.; Elmer, D.P.; Sternberg, C.; Tesanovic, S.; Del Burgo, P.; Coni, S.; Canettieri, G.; Neureiter, D.; Bartz, R.; et al. Targeting class I histone deacetylases by the novel small molecule inhibitor 4SC-202 blocks oncogenic hedgehog-GLI signaling and overcomes smoothened inhibitor resistance. Int. J. Cancer 2018, 142, 968–975. [Google Scholar] [CrossRef]
- Messerli, S.M.; Hoffman, M.M.; Gnimpieba, E.Z.; Kohlhof, H.; Bhardwaj, R.D. 4SC-202 as a Potential Treatment for the Pediatric Brain Tumor Medulloblastoma. Brain Sci. 2017, 7, 147. [Google Scholar] [CrossRef] [Green Version]
- Mishra, V.K.; Wegwitz, F.; Kosinsky, R.L.; Sen, M.; Baumgartner, R.; Wulff, T.; Siveke, J.T.; Schildhaus, H.U.; Najafova, Z.; Kari, V.; et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic Acids Res. 2017, 45, 6334–6349. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Bretz, A.C.; Gravemeyer, J.; Spassova, I.; Muminova, S.; Gambichler, T.; Sriram, A.; Ferrone, S.; Becker, J.C. The HDAC Inhibitor Domatinostat Promotes Cell-Cycle Arrest, Induces Apoptosis, and Increases Immunogenicity of Merkel Cell Carcinoma Cells. J. Investig. Dermatol. 2021, 141, 903–912.e4. [Google Scholar] [CrossRef]
- Hoffman, M.M.; Zylla, J.S.; Bhattacharya, S.; Calar, K.; Hartman, T.W.; Bhardwaj, R.D.; Miskimins, W.K.; de la Puente, P.; Gnimpieba, E.Z.; Messerli, S.M. Analysis of Dual Class I Histone Deacetylase and Lysine Demethylase Inhibitor Domatinostat (4SC-202) on Growth and Cellular and Genomic Landscape of Atypical Teratoid/Rhabdoid. Cancers 2020, 12, 756. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Deng, M.; Zhang, C.; Ping, F.; Wang, H.; Wang, Y.; Fan, Z.; Ren, X.; Tao, X.; Wu, T.; et al. Combined class I histone deacetylase and mTORC1/C2 inhibition suppresses the initiation and recurrence of oral squamous cell carcinomas by repressing SOX2. Cancer Lett. 2019, 454, 108–119. [Google Scholar] [CrossRef]
- Roca, M.S.; Moccia, T.; Iannelli, F.; Testa, C.; Vitagliano, C.; Minopoli, M.; Camerlingo, R.; De Riso, G.; De Cecio, R.; Bruzzese, F.; et al. HDAC class I inhibitor domatinostat sensitizes pancreatic cancer to chemotherapy by targeting cancer stem cell compartment via FOXM1 modulation. J. Exp. Clin. Cancer Res. 2022, 41, 83. [Google Scholar] [CrossRef]
- Zylla, J.L.S.; Hoffman, M.M.; Plesselova, S.; Bhattacharya, S.; Calar, K.; Afeworki, Y.; de la Puente, P.; Gnimpieba, E.Z.; Miskimins, W.K.; Messerli, S.M. Reduction of Metastasis via Epigenetic Modulation in a Murine Model of Metastatic Triple Negative Breast Cancer (TNBC). Cancers 2022, 14, 1753. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bögler, O.; et al. m(6)A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 2017, 31, 591–606.e6. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Zhang, M.; Zhou, Y.; Guo, W.; Yi, M.; Zhang, Z.; Ding, Y.; Wang, Y. The application of histone deacetylases inhibitors in glioblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 138. [Google Scholar] [CrossRef]
- Kunadis, E.; Lakiotaki, E.; Korkolopoulou, P.; Piperi, C. Targeting post-translational histone modifying enzymes in glioblastoma. Pharmacol. Ther. 2021, 220, 107721. [Google Scholar] [CrossRef]
- Reddy, R.G.; Bhat, U.A.; Chakravarty, S.; Kumar, A. Advances in histone deacetylase inhibitors in targeting glioblastoma stem cells. Cancer Chemother. Pharmacol. 2020, 86, 165–179. [Google Scholar] [CrossRef]
- Hervouet, E. The Promising Role of New Generation HDACis in Anti-Cancer Therapies. EBioMedicine 2018, 32, 6–7. [Google Scholar] [CrossRef]
- Ruzic, D.; Djoković, N.; Srdić-Rajić, T.; Echeverria, C.; Nikolic, K.; Santibanez, J.F. Targeting Histone Deacetylases: Opportunities for Cancer Treatment and Chemoprevention. Pharmaceutics 2022, 14, 209. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, T.C.; El-Osta, A. Will broad-spectrum histone deacetylase inhibitors be superseded by more specific compounds? Leukemia 2007, 21, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Bangert, A.; Häcker, S.; Cristofanon, S.; Debatin, K.M.; Fulda, S. Chemosensitization of glioblastoma cells by the histone deacetylase inhibitor MS275. Anticancer Drugs 2011, 22, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Essien, E.I.; Hofer, T.P.; Atkinson, M.J.; Anastasov, N. Combining HDAC and MEK Inhibitors with Radiation against Glioblastoma-Derived Spheres. Cells 2022, 11, 775. [Google Scholar] [CrossRef] [PubMed]
- von Tresckow, B.; Sayehli, C.; Aulitzky, W.E.; Goebeler, M.E.; Schwab, M.; Braz, E.; Krauss, B.; Krauss, R.; Hermann, F.; Bartz, R.; et al. Phase I study of domatinostat (4SC-202), a class I histone deacetylase inhibitor in patients with advanced hematological malignancies. Eur. J. Haematol. 2019, 102, 163–173. [Google Scholar] [CrossRef]
- Tashima, T. Brain Cancer Chemotherapy through a Delivery System across the Blood-Brain Barrier into the Brain Based on Receptor-Mediated Transcytosis Using Monoclonal Antibody Conjugates. Biomedicines 2022, 10, 1597. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, W.J.; Park, J.A.; Choi, Y.K.; Kwon, Y.W.; Kim, K.W. Blood-brain barrier interfaces and brain tumors. Arch. Pharm. Res. 2006, 29, 265–275. [Google Scholar] [CrossRef]
- Leten, C.; Struys, T.; Dresselaers, T.; Himmelreich, U. In vivo and ex vivo assessment of the blood brain barrier integrity in different glioblastoma animal models. J. Neurooncol. 2014, 119, 297–306. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Matsuda, K.; Sato, A.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci. Rep. 2012, 2, 516. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Okada, M.; Sanomachi, T.; Togashi, K.; Seino, S.; Sato, A.; Yamamoto, M.; Kitanaka, C. Therapeutic targeting of pancreatic cancer stem cells by dexamethasone modulation of the MKP-1–JNK axis. J. Biol. Chem. 2020, 295, 18328–18342. [Google Scholar] [CrossRef]
- Okada, M.; Sato, A.; Shibuya, K.; Watanabe, E.; Seino, S.; Suzuki, S.; Seino, M.; Narita, Y.; Shibui, S.; Kayama, T.; et al. JNK contributes to temozolomide resistance of stem-like glioblastoma cells via regulation of MGMT expression. Int. J. Oncol. 2014, 44, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Takeda, H.; Sakaki, H.; Kuramoto, K.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C. Repositioning CEP-1347, a chemical agent originally developed for the treatment of Parkinson’s disease, as an anti-cancer stem cell drug. Oncotarget 2017, 8, 94872–94882. [Google Scholar] [CrossRef] [Green Version]
- Okada, M.; Jang, S.W.; Ye, K. Ebp1 association with nucleophosmin/B23 is essential for regulating cell proliferation and suppressing apoptosis. J. Biol. Chem. 2007, 282, 36744–36754. [Google Scholar] [CrossRef] [Green Version]
- Watarai, H.; Okada, M.; Kuramoto, K.; Takeda, H.; Sakaki, H.; Suzuki, S.; Seino, S.; Oizumi, H.; Sadahiro, M.; Kitanaka, C. Impact of H3K27 Demethylase Inhibitor GSKJ4 on NSCLC Cells Alone and in Combination with Metformin. Anticancer Res. 2016, 36, 6083–6092. [Google Scholar] [CrossRef] [Green Version]
- Kuramoto, K.; Yamamoto, M.; Suzuki, S.; Togashi, K.; Sanomachi, T.; Kitanaka, C.; Okada, M. Inhibition of the Lipid Droplet–Peroxisome Proliferator-Activated Receptor α Axis Suppresses Cancer Stem Cell Properties. Genes 2021, 12, 99. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakagawa-Saito, Y.; Saitoh, S.; Mitobe, Y.; Sugai, A.; Togashi, K.; Suzuki, S.; Kitanaka, C.; Okada, M. HDAC Class I Inhibitor Domatinostat Preferentially Targets Glioma Stem Cells over Their Differentiated Progeny. Int. J. Mol. Sci. 2022, 23, 8084. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158084
Nakagawa-Saito Y, Saitoh S, Mitobe Y, Sugai A, Togashi K, Suzuki S, Kitanaka C, Okada M. HDAC Class I Inhibitor Domatinostat Preferentially Targets Glioma Stem Cells over Their Differentiated Progeny. International Journal of Molecular Sciences. 2022; 23(15):8084. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158084
Chicago/Turabian StyleNakagawa-Saito, Yurika, Shinichi Saitoh, Yuta Mitobe, Asuka Sugai, Keita Togashi, Shuhei Suzuki, Chifumi Kitanaka, and Masashi Okada. 2022. "HDAC Class I Inhibitor Domatinostat Preferentially Targets Glioma Stem Cells over Their Differentiated Progeny" International Journal of Molecular Sciences 23, no. 15: 8084. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158084