2.1. Spontaneous Aggregation of Human Serum Albumin
The high sensitivity of fluorescence methods allows studying the protein structural changes and the process of their aggregation. The aromatic amino acids with intrinsic fluorescence properties: tyrosine (Tyr)-18, phenylalanine (Phe)-31 and only one tryptophan (Trp214) are responsible for the absorption and fluorescence of HSA in the near UV range. Although the fluorescence quantum yields of tryptophan and tyrosine are similar and several times higher than phenylalanine, the Trp214 residue plays a decisive role in the fluorescence of HSA. Both Tyr and Trp have the absorption maximum at 278 nm, but tryptophan, unlike tyrosine, shows significant absorbance at around 295 nm. For a wavelength of 295 nm, Trp214 can be excited without interference from the tyrosine. The contribution of Phe to the intrinsic fluorescence of HSA is negligible [
22].
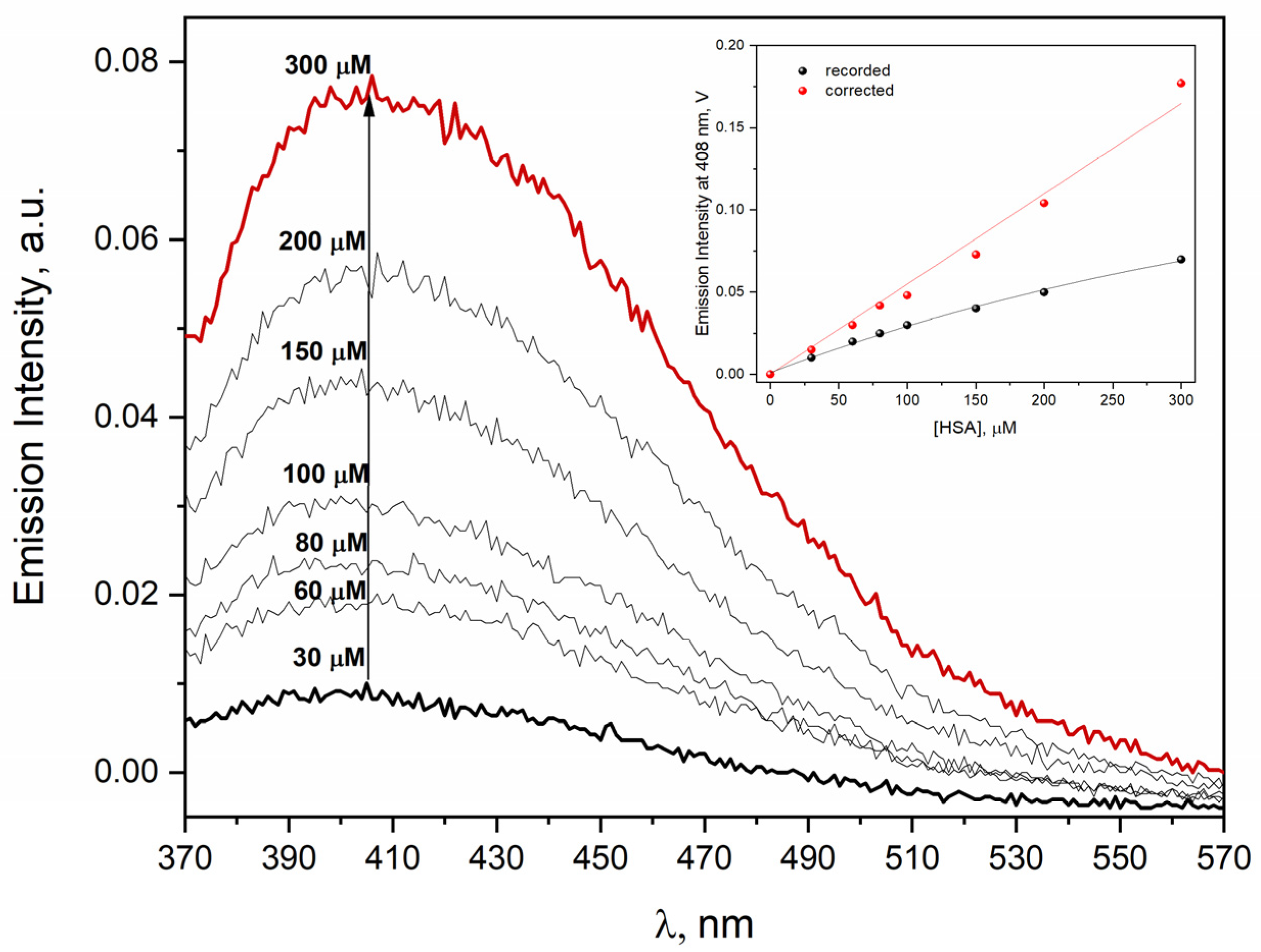
The emission spectra of neat HSA solutions in the concentration range of 30–300 µM has been registered to study the self-aggregation process of human serum albumin. The results of these experiments are shown in
Figure 1 and clearly revealed that the light emission of protein aggregates depended almost linearly on the concentration of HSA. The excitation wavelength was 320 nm for exciting HSA self-aggregates. Our measurements do not support the thesis [
1] that there is a concentration threshold (around 10 µM) above which self-aggregates of HSA are formed.
Helpful information on the process of albumin aggregation can be obtained by analyzing the fluorescence excitation spectra of the HSA solution. The fluorescence excitation spectrum of HSA (80 uM) in a buffer solution exhibits a maximum at 290 and 345 nm for the λ
em = 410 nm (
Figure 2). The band centered at 290 nm can be attributed to the tryptophan residue Trp214. Analysis of the emission excitation spectra of neat HSA solution (80 µM) for λ
em = 450 nm suggest that in relation to the emission spectrum for λ
em = 410 nm, there is an additional, third band with a maximum near 390 nm. The clearly observed band at 290 nm indicates the inactivation of the excited state of the tryptophan molecule as a result of energy transfer from *Trp214 to HSA aggregates (FRET—Förster Resonance Energy Transfer). The excitation emission spectrum of the HSA solution shows two additional bands with a maximum at 350 and 390 nm, indicating the presence of two different types of aggregates. Time resolution measurements also show two main groups of aggregates. The excitation of HSA solutions with the laser light with a wavelength of 337 nm allows registration of the emission of excited protein aggregates. This light is not absorbed by monomeric HSA. Decay at 400 nm is characterized by an average lifetime close to 3 ns, for 450 nm, the analogous lifetime is about 4.2 ns. By changing the concentration of HSA in a wide range (30, 60, 80, 100, 150, 200, 300 µM), we registered slight changes in the lifetime of the emission of HSA aggregates as a function of protein concentration. For example, for the above-mentioned solutions, we recorded the average emission decay time for 450 nm, amounting to 4.18, 4.19, 4.3, 4.3, 4.49, 4.5, and 4.6 ns, respectively. Therefore, it can be postulated that in albumin solutions, even at a low concentration of HSA, there are species with different emission states. The new bands (at 350 and 390 nm) in the excitation spectrum are probably a consequence of the formation of the hydrogen bonds network and/or π-π interactions (van der Waals force) between HSA molecules.
The change in HSA concentration in the studied solutions causes changes in the intensity of the emission and excitation spectra, which seemingly are not a linear function of the protein concentration. As shown in
Figure 1, the correction for the inner filter effect creates a linear relationship between the intensity of the emission band and the concentration of HSA over a wide range of HSA concentrations (30–300 μM).
The emission of light after the excitation of protein aggregates is caused by the coupling of non-aromatic chromophores, which ensures the subsequent overlapping of electron clouds along with the stiffening of the conformation of the interpenetrating network of amino acids of adjacent HSA molecules. In the scientific literature, there is no well-established term for this type of deactivation of excited states of aggregates. Typically, the aggregation of classic chromophore groups leads to the quenching of excited states (self-quenching). The reverse process of self-quenching, called aggregation-induced emission (AIE), occurs in some organic compounds [
23]. In the literature, one can also find the term “blue autofluorescence in protein aggregates” [
24] or intrinsic blue fluorescence from oligomeric interfaces of human serum albumin [
1]. For the purposes of this work, we will call the light emitted by protein aggregates excited by light with a wavelength above 300 nm emission, not fluorescence.
We found a similar relationship for the excitation spectra of HSA for the concentration range of 30–300 μM of protein (see
Figure S1 in
Supplementary Materials). The fluorescence excitation spectrum of HSA neat solutions (30–300 μM) exhibits a maximum at ≈290 and 345 nm for the λ
em = 410 nm. The band centered at 290 nm can be attributed to the tryptophan residue Trp214, while the second band (λ
max = 345 nm) is characteristic of HSA aggregates. As the concentration of HSA increases, the value of the emission intensity increases in the maximum bands at 290 and 345 nm. The excitation spectra of albumin solutions confirmed that the intensity of emission of protein aggregates depends linearly on the concentration of HSA (it was confirmed by the dependence of emission intensity recorded and corrected at 345 nm as a function of HSA concentration).
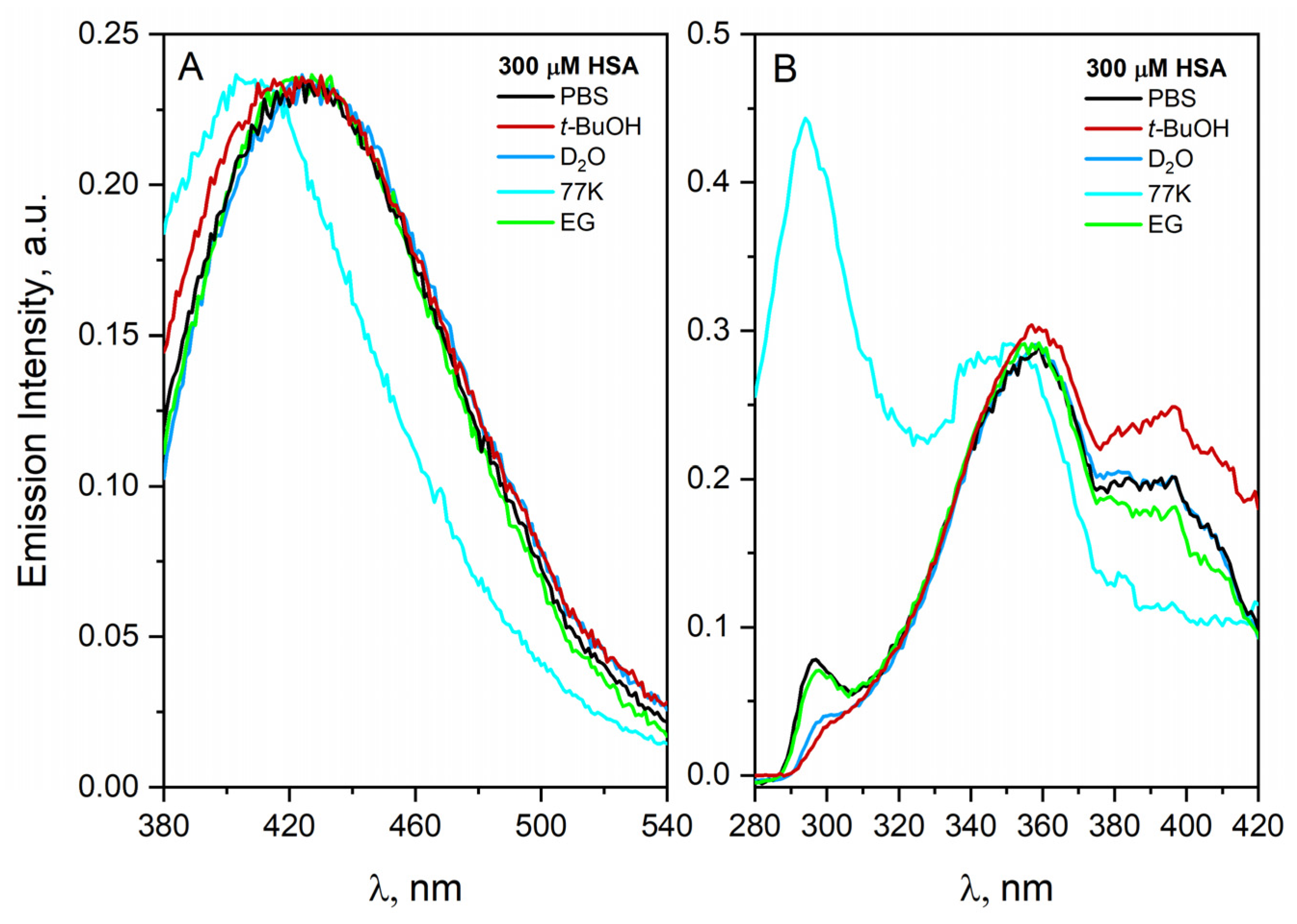
The influence of oxygen on the HSA aggregation process is negligible. It is important to note that dilution of the albumin solution leads to a disaggregation of the HSA. The HSA solution (300 µM) was characterized by an intense spectrum with an emission band in the visible region with a maximum at 440 nm (λexc = 360 nm). The emission spectrum of the HSA solution with a concentration of 300 µM recorded after tenfold dilution is very consistent with the spectrum recorded for a solution of HSA with a concentration of 30 µM prepared by dissolving HSA in PBS solution. In this “dilution” experiment, the existence of non-covalent HSA dimers in equilibrium with monomeric HSA was demonstrated.
We have performed fluorescence measurements of aqueous solution of 1 µM peptide MD26 (Pro–Glu–Pro–Thr–Ile–Asp–Glu–Ser), and we noted no emission after excitation of the solution with the 295 nm light. After increasing peptide concentration to 100 µM, light emission was observed with a maximum at near 425 nm (λ
exc = 337 nm). The structure of MD26 (no aromatic residues) clearly indicates that hydrogen and ionic bonds were sufficient to form aggregates of emission-active polypeptides. In order to clarify whether the formation of spontaneous aggregates (self-aggregates associated with an increase in concentration of protein) is unique to HSA only, we performed comparative measurements for other proteins. The results will be presented soon in a subsequent publication. Here, for illustration, we will compile the emissions data collected for HSA with those recorded for BSA and papain. For each analyzed protein, there were self-aggregates in non-irradiated protein solutions. The excitation of protein solutions with the 320 nm light results in the appearance of emissions at 410 nm (one band centered at 415 nm was observed for the papain solution). The excitation spectrum of the 430 nm emission band shows a sharp peak at 292 nm (Trp214) and around 350 nm (aggregates). It means that the tryptophan residue and the newly formed chromophore groups are close to each other. After filtering the protein solution, no changes in the emission and excitation spectra were observed, which proves the high stability of the self-aggregates (see
Figure 3 data presented only for HSA). The fluorescence lifetimes of HSA aggregates (C
HSA = 1.6 mM) varies between 4.3 and 4.75 ns, depending on detection wavelength. The emission lifetime values were calculated with the mono-exponential decay. Increasing the detection wavelength to 480 nm leads to an increase in the lifetime of the fluorescence of HSA aggregates. Above this value (480 nm), the lifetime of the HSA aggregates emission slightly decreases. The determined emission lifetime values confirmed that two populations of protein aggregates are presented in the concentrated HSA solution, which can be distinguished on the basis of time-resolved fluorescence measurements. The use of a more accurate measurement technique based on SPC single photon counting should help distinguish protein aggregates obtained by the oxidation of albumin molecules with azide or hydroxyl radicals. For accurate analysis of the emission of HSA aggregates, we always perform a series of measurements for different excitation and emission wavelengths and do not use only the DT fluorescence detection procedure (λ
exc = 320 nm; λ
det = 410 nm).
Using spectroscopic techniques, we have confirmed that HSA, BSA and papain undergoes reversible aggregation as a result of protein−protein interactions. Moreover, we have shown that two types of self-aggregates are formed. In our opinion, dimers are mainly responsible for blue emissions. We assume the possibility of formation of different dimer types (depending on the structural changes of the protein). Consequently, each solution will have a complex equilibrium between many variants of HSA dimers, which is not described by a unique equilibrium constant. Electrophoretic analysis confirmed that the presence of dimers, trimers and tetramers in the solution increases with increasing HSA concentration (described further). We called these higher-than-dimeric forms of HSA complexation spontaneous aggregates. It is also known from HPLC measurements that dimers and aggregates constitute about 16 and 7% of all HSA molecules in the non-irradiated albumin solution, respectively (concentration of HSA 15 µM) [
25,
26]. Unfortunately, fluorescence measurements cannot be used directly to estimate the percentage of dimers and other oligomers in the HSA solutions. It is due to the unknown value of the fluorescence quantum yield of HSA aggregates. It is not easy to determine the fluorescence quantum yield of protein aggregates due to their different structures (dimers, trimers, etc.) and scattering light in concentrated HSA solutions.
Our experiments confirmed also that pH has an influence on the aggregation process of HSA. The results show that the decrease in pH from 7.0 to 5.0 gradually unfolded the HSA structure to slightly increase the emission intensity of HSA aggregates in the aqueous solution.
An interesting observation from our experiments is quenching of the excited state of HSA aggregates by
N,N’-dimethyl-4,4′-bypyridinium (methylviologen; MV
2+). Fluorescence quenching of the excited state of aggregates by the MV
2+ has been studied by steady-state and time-resolved techniques. The quenching data were analyzed by using a conventional Stern–Volmer equation. On the basis of steady-state emission measurements and emission lifetime measurements of HSA aggregates with the addition of MV
2+ in various concentrations (0–90 mM), a Stern–Volmer plot was made. The SV dependence is shown in
Figure 4. The I
0 and I value for a given quencher concentration were read from the emission spectra recorded in the absence and presence of the methylviologen. The Stern–Volmer plot presented in
Figure 4 indicates a good linear relationship in the quencher concentration range up to 20 mM. Above this concentration value, the graphs deviate from linearity. It proved both static and dynamic quenching. The sublinear relationship of the Stern–Volmer curve was related to the formation of the MV
2+–HSA aggregate complex.
2.8. Radiolysis of Solutions Containing HSA
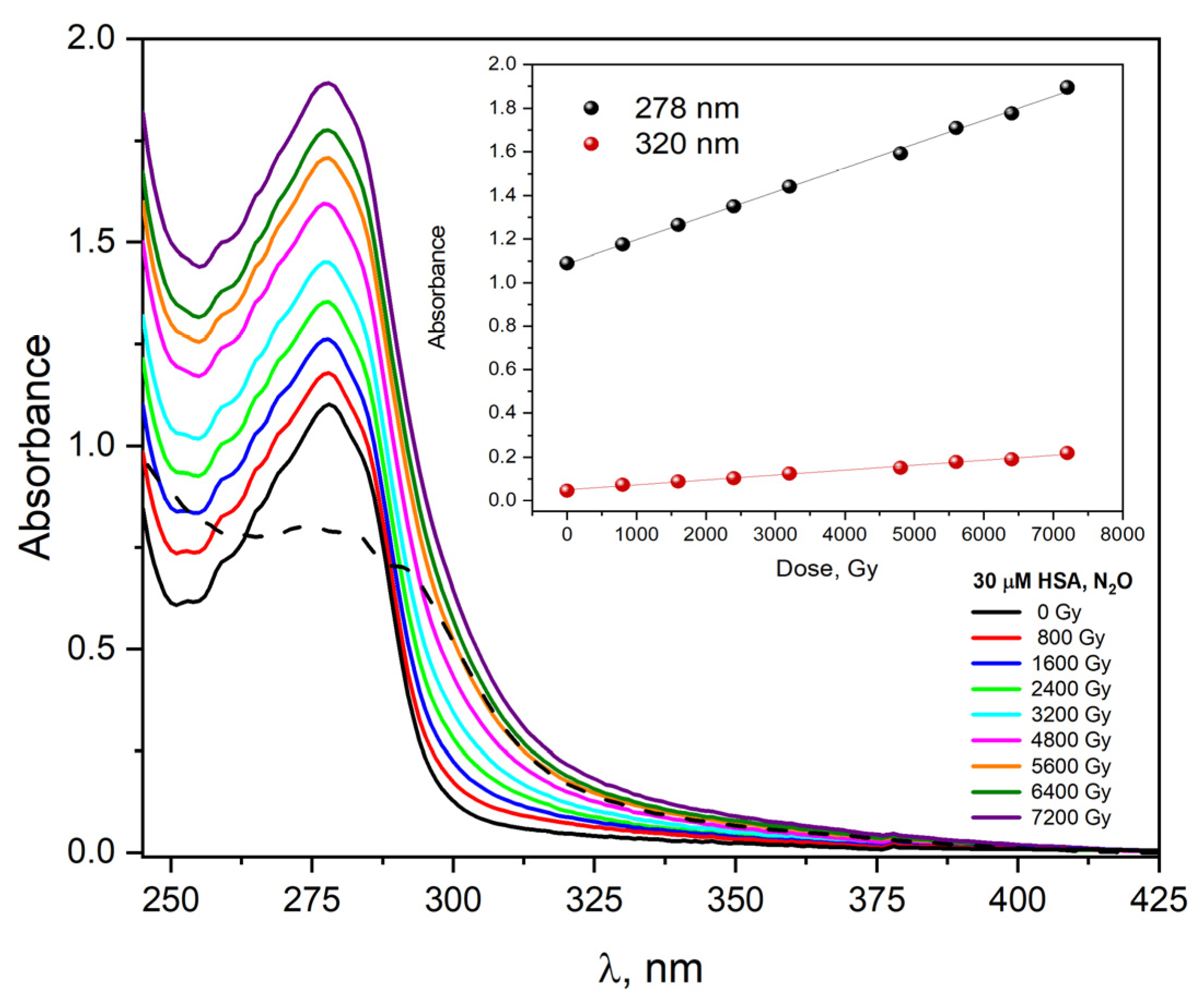
We also measured changes in absorption and emission spectra related to steady-state irradiation for HSA solutions. The absorption spectra of a saturated N
2O aqueous solution containing 30 μM HSA were recorded after irradiation with doses from 0 to 7200 Gy and shown in
Figure 13. The absorption spectrum of HSA before irradiation shows two bands, one with a maximum at 280 nm. The molar absorption coefficient of this band at 280 nm is equal to 35,500 M
−1 cm
−1 [
43] and the other monotonically rises to far UV. The molar absorption coefficient at 210 nm was estimated to be approximately 10
6 M
−1 cm
−1 [
43]. The absorption band peaking at 280 nm is mainly a consequence of the π→π * electronic transitions of two aromatic amino acids: Trp and Tyr residues. After irradiating this solution with multiple electron pulses, the band intensity (maximum at 280 nm) grows, and a new absorbance appears in the spectral range of 300–400 nm. The irradiation of HSA solution caused a marked increase in absorbance at 278 nm (A
800 Gy = 1.18→A
7200 Gy = 1.89). The new absorbance (between 300 and 400 nm) is attributed to the formation of HSA aggregates during oxidative stress. As seen from the insert of
Figure 13, the absorbance value increases in proportion to the absorbed dose by the albumin solution. The dependence of absorbance as a function of dose is linear, regardless of the analyzed wavelength (
Figure 13 shows dependence of A
278 nm and A
320 nm as a function of absorbed dose).
The newly formed absorbance (dash line in
Figure 13, dose absorbed 7200 Gy) is the result of light absorption by the generated HSA aggregates and is partly the result of light scattering by the solution.
It is reported in the literature that dityrosine formation is monitored by measuring the fluorescence at 325 nm excitation and 400–410 nm emission e.g., 400 nm [
44], 409 nm [
45], and 410 nm [
46]. The excitation of irradiated aqueous HSA solution with the 320 nm light leads to the emission of DT centered at 420 nm, as shown in
Figure 14. The emission intensity of HSA aggregates formed as a result of the reaction of albumin with
•OH radicals depends linearly on the absorbed dose (regardless of the excitation wavelength). HSA aggregates generated in the irradiated solution differ from self-aggregates (
Figure 14 shows the emission spectrum recorded for a concentrated HSA solution, 300 µM).
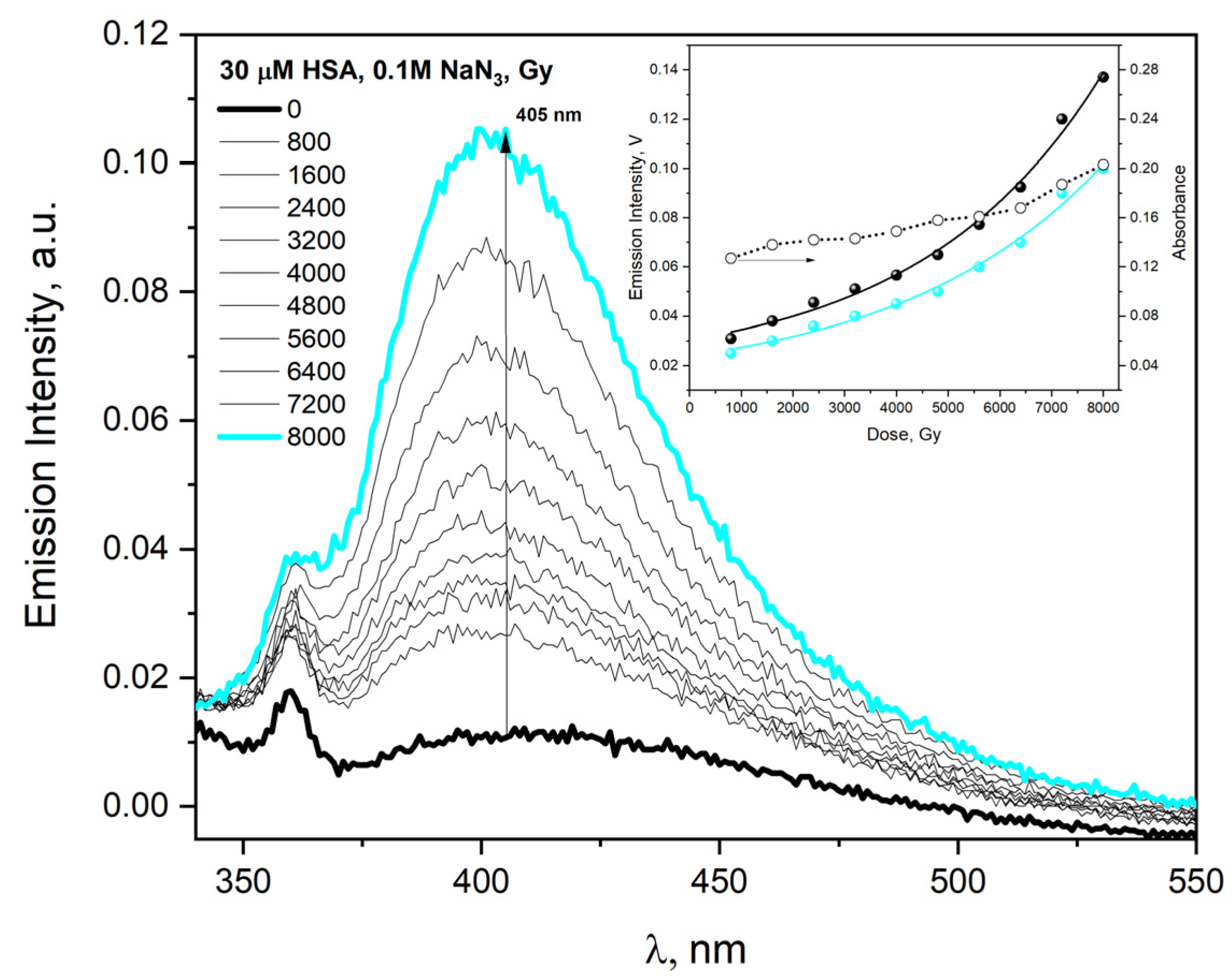
In the case of electron beam irradiation of HSA solution (30 µM) containing NaN
3 (0.1 M), the absorbance value and emission intensity increase monotonically to absorbed doses but non-linearly (
Figure 15, insert). The aggregates formed in the reaction of HSA with azide radicals were characterized by classic DT fluorescence (the emission spectrum recorded after excitation of buffer HSA solution with 320 nm light shows one band with maximum near 405 nm).
The conducted experiments confirmed that the mechanism of the azide and hydroxyl radical reactions with albumin differs significantly (
Figure 16). The hydroxyl radical often reacts simultaneously via several competing pathways, e.g., H atom abstraction, addition to a double bond or to an aromatic ring, or via electron transfer; therefore, a number of intermediates may initially be present. Contrary to hydroxyl radical,
radicals react mainly by one-electron transfer and are much more selective than
•OH. The reaction of
•OH with HSA is leading to a fluorescent product with a maximum at 420 nm.
Figure 16 shows the broad band due to the superposition of emission of DT and HSA aggregates (formed from different dimers: fluorescent and non-fluorescent). In the case of reaction of
radical with HSA, we observed the band characteristic of dityrosine. The diffusion of the azide radicals to the protein interior is postulated. The scavenging of
radicals by tyrosine residue within protein is a complex process. The HSA contains 17 tyrosine residues in a single polypeptide chain. The reactivity of azide radicals toward these Tyr residues differs significantly. Moreover, not every “meeting” of azide radicals with the tyrosine residue in the HSA structure leads to the formation of DT, and not every modification of tyrosine results in the formation of fluorescent products. According to the literature, the reaction of two TyrO
• radicals generated in different HSA molecules does not always lead to the formation of fluorescent tyrosine dimer [
47]. The overlapping of the emission bands of both DT and self-aggregates makes it very difficult to identify the ortho-tyrosine dimer in optical measurements.
The analysis of the HSA oxidation process with the use of a number of radiation-generated oxidants was performed. Combined pulse radiolysis and enzyme-activity studies allowed determining which amino acid residues within biomolecules are modified as a result of radical attack. Identification of the nature of damage caused by hydroxyl radicals responsible for the inactivation of proteins or enzymes was difficult due to the lack of selectivity of
•OH radicals in reactions with amino acids. For more detailed analysis of the modification of amino acid residue structures, specific inorganic anion radicals were used. Pulse radiolysis and enzyme-activity studies have shown that the radiation-induced inactivation of lysozyme in neutral aqueous solution is due principally to the reaction of hydroxyl radicals with tryptophan residues [
48]. In the case of trypsin, histidine damage leads to inactivation of the enzyme [
49]. The methodology described above (pulse radiolysis with simultaneous analysis of enzymatic activity) allowed determining which amino acid residues are modified in the presence of various oxidizing radicals [
50,
51].
The position of individual amino acids in the polypeptide chain within the protein is important in analyzing the oxidation and reduction process of albumin. Analysis of the crystal structure of HSA revealed that 40% of the aromatic amino acids are close to the surface in the native protein structure [
52]. Human serum albumin contains 31 phenylalanine residues, 1 tryptophan and 18 tyrosine residues in the peptide chain. Human serum albumin contains 31 phenylalanine residues, 1 tryptophan and 18 tyrosine residues in the peptide chain. In publication [
53], the quantum yield of singlet oxygen generated by excited states of aromatic amino acids included in the structure of HSA (Trp, Tyr, Phe) were determined by time-resolved phosphorescence measurements. Human serum albumin has a total of 21 surface exposed residues, 11 partially occluded (5 tyrosine and 6 phenylalanine residues), and 19 residues that are buried deeply within the HSA structure. If only surface residues could sensitize oxygen, the quantum yield of singlet oxygen would be smaller than that obtained with an equivalent number of free aromatic amino acids in solution. The expected quantum yield of singlet oxygen would be about 40% of the weighted average quantum yield. The determined value of quantum yield of singlet oxygen experimentally is in very good agreement with the assumption that readily available light-excited HSA aromatic residues are involved in the reactions with oxygen molecules. The close correlation between the efficiency of singlet oxygen and the availability of aromatic residues in the HSA structure for oxygen molecules is for us the most interesting aspect of the discussed publication. By using different oxidizing radicals in our pulsed radiolysis measurements, we can expect that certain aromatic groups of HSA will be more readily available for attack by the radical. In our publication on the reaction of reducing radicals (
, H
•,
) with HSA, we have shown that the Sudlow 1 site, and in particular Trp214 located here, is well protected against these radicals [
54]. Additionally, by generating radicals with different redox potential, we can to some extent decide which of the aromatic residues will be involved in the radical scavenging.
It was postulated that the amino acid residues that are modified by
•OH radicals were located on the protein surface regardless of the pH of the solution [
55]. Hydroxyl radicals react not only with aromatic amino acids but practically with all amino acid residues on the protein surface due to their high reactivity. It is known that
•OH radicals could generate radicals centered on the α-carbon atom and 27% of the total amount of radiation generated hydroxyl radicals in the BSA solution reacts with the protein, resulting in the formation of new carbonyl groups [
55].
Pulse radiolysis results provide helpful information on inter- and intramolecular electron transfer in protein solutions. Intramolecular long range electron transfer (LRET) in the hen egg-white lysozyme accompanying radical transformation Trp
•→TyrO
• was observed [
56]. In this study, the authors observed LRET between the following amino acid residues: Trp62/Tyr53, Trp63/Tyr53 and Trp123/Tyr23. On the other hand, HPLC and MALDI-TOF MS measurements that were made for a solution of lysozyme containing NaN
3 (0.1M) and γ irradiated indicate that Trp108 and/or 111 remain oxidized and that Tyr20 and 53 give DT [
57]. There is no evidence for the reaction of tyrosine at position 23 with
radicals, so LRET does not always take place in the protein structure. Other amino acids were probably involved in electron transfer (which are not always recorded in pulse radiolysis due to measurement difficulties, e.g., low molar absorbance coefficients).
Figure 17 shows the excitation spectra recorded after pulse radiolysis of a neat HSA solution (70 µM) and solutions containing additionally NaCl, LiBr or NaN
3. All solutions were saturated with N
2O. The salt concentration of the HSA solutions was 0.1 M. In the next stage, deaerated HSA solution (70 µM) with the addition of 5 mM (NH
4)
2S
2O
8 and 0.1 M
t-BuOH was irradiated. The water radiolysis results in the formation of three well-characterized reactive radical species used to initiate radical reactions:
,
•OH and H
+. A system containing only hydroxyl radicals can be obtained by saturating the aqueous solution with N
2O, while the hydrated electrons are converted into the
•OH radicals. The azide radicals were formed in irradiated N
2O-saturated aqueous solution containing NaN
3. The sulfate radical anions were formed in the reaction of
with (NH
4)
2S
2O
8. The dihalide radicals,
,
can be generated by pulse radiolysis carried out in LiBr or NaCl N
2O-saturated aqueous solution (hydroxyl radicals reacts with halide ions to form oxidizing species). The pulse radiolysis of aqueous solution allowed producing reactive oxidizing radicals, such as
•OH,
,
,
and
formed via the following reaction:
The HSA solutions containing NaCl, LiBr or NaN
3 irradiated with a dose of 420 Gy were analyzed spectroscopically by measuring the absorption and emission spectra. The emission spectra recorded for the excitation wavelength 320 nm before and after irradiation of the solutions are presented in
Figure 17. The emission spectra of irradiated HSA solutions differ markedly depending on the type of oxidant used. As a result of the reaction of radicals
,
,
with HSA, mainly tyrosine residues are modified and DT is formed, which is manifested by an intense emission band with a maximum of about 400 nm (after excitation of irradiated solution with the 320 nm light). A similar conclusion regarding the formation of DT was drawn in the case of irradiation of lysozyme oxidized with
radicals [
58,
59]. Consequently, of the reaction of anion radicals
with lysozyme, dimers of enzyme are formed, which are characterized by emission centered at about 400 nm (the dimer is formed as a result of the intermolecular recombination reaction of phenoxyl radicals). Albumin oxidation with radicals
also leads mainly to the formation of DT bridges. In this case, the emission spectrum is broader in comparison to the DT band. It is due to the superposition of fluorescence of dityrosine and the emission of aggregates formed by the network of hydrogen and/or ionic bonds, analogous to self-aggregates (
Figure 17).
The generation of
leads to a greater number of dityrosine bridges compared to DT obtained by other investigated oxidizing radicals. The emission intensity of HSA aggregates obtained in solutions in which the hydroxyl radical or chloride radical anion was generated is much lower compared to the emission of aggregates formed in the reaction of HSA with other oxidants. The more detailed analysis shows a red shift in the maximum of the band after the irradiation of N
2O-saturated HSA (
•OH) solution (403 nm→422 nm, ΔE = 0.1385 eV). The reaction of the
radical with HSA leads also mainly to species other than DT, as evidenced by a broad, less intense slightly red-shifted band (λ
max = 408 nm,
Figure 17). This means that in the case of reaction of
•OH or
radicals with HSA, aggregates were formed that were not stabilized by DT bridges. The high reactivity of the
•OH or radical
with amino acids contained within HSA is responsible for the formation of various covalent bonds (including C-C and S-S) between adjacent protein molecules [
60]. Hashimoto et al. explain the formation of DT in lysozyme solutions as a result of the reaction of this protein with
radical anion. It was also reported that in the reaction of lysozyme with hydroxyl radicals, the tryptophyl residues are the main sites to be attacked by the radicals [
58]. In a pulsed radiolysis experiment of lysozyme solution, the authors observed an absorption band around 400 nm typical for the phenoxy tyrosine band. It has been postulated that the intramolecular transfer of the radical site from the tryptophyl radical to the tyrosyl radical occurs in a lysozyme solution. This argument was confirmed spectrophotometrically; the lysozyme dimers produced by the
radicals show a similar fluorescence spectrum to dityrosine [
58]. On the basis of the reactivity of amino-acid residues located on the surface of lysozyme, the authors estimated that about 50% of the hydroxyl radicals attack aromatic amino acid residues, and 60% of these
•OH radicals (i.e., 30% of all OH radicals) react with tryptophan residues. The remaining amount of hydroxyl radicals could lead to the formation of aliphatic radicals.
Figure 18A shows the excitation spectra recorded for the emission bands at 410 or 450 nm for an HSA solution containing 0.1 M LiBr before and after irradiation with a dose of 420 Gy. In the case of recording the excitation spectrum for the detection of 410 nm before irradiation, we observe two bands with a maximum of 292 nm and about 345 nm. For a detection length of 450 nm, we have three peaks in the excitation spectrum with a maximum at 292, 356 and 390 nm.
After irradiation, the same number of bands of excitation spectra is observed, but the intensity of the bands centered at about 330 nm increases significantly. This is especially visible for the detection wavelength of 410 nm, which is typical for DT. The analogous excitation spectra were obtained for the solution containing NaN
3 (
Figure 18B). The reaction of the
radical with HSA results in similar spectral changes to those described for the
radical, but the DT band is less intense. These experiments clearly show that
and
penetrate to the HSA interior.
in particular reaches the tyrosine residues of the protein with high efficiency. The reaction of
radicals with HSA leads to the formation of DT in a small amount (
Figure 19A); however, in the reaction of albumin with
•OH radicals (
Figure 19B), the formation of tyrosine–tyrosine bridges was not observed. It is more precise to say that we do not rule out bridging DT but rather bridging by such dityrosine isomers that may not be fluorescent [
61]. An interesting observation is also the change in the amount of HSA intermolecular self-aggregates after the irradiation of N
2O saturated solutions containing HSA (70 µM) and 0.1 M NaN
3 or 0.1 M LiBr. In both cases, the number of light-emitting aggregates in the low-energy part of the band (an emission band extending over 375 nm, maximum band at ≈3.1 eV) decreased. This seems to be forced by the structure change of HSA dimers containing the DT covalent bond. The obtained results indicate that the DT formation efficiency does not correlate with the reactivity of the oxidizing radicals.
The oxidation process of HSA was monitored by pulse radiolysis measurements at 410 and 510 nm at the maximum of the absorbance band for TyrO
• and Trp
•, respectively. Pulse radiolysis measurements show a correlation between the amount of the generated TyrO
• radicals (measured by the absorbance at 410 nm) and the fluorescence intensity DT recorded in steady-state measurements. However, the differences in the concentrations of TyrO
• radicals were not large. The highest absorbance value of the TyrO
• was recorded for the
. The analysis of pulse radiolysis spectra for the TyrO
• at 410 nm is difficult due to broad absorption bands of the dihalide anion radicals in the spectral range at about 400 nm. The transient absorption spectra clearly revealed that for the reaction of
and
•OH with albumin, less TyrO
• and Trp
• are formed in compared to other radicals. By analyzing the rate constants of
•OH and
radicals with amino acids in a homogeneous solution, one can explain the reason for the significantly higher DT yield for the
reaction with HSA in relation to the
•OH reaction with HSA. The publication [
62] collects the values of the rate constants of
and
•OH radicals with tryptophan, tyrosine, phenylalanine, histidine, cysteine, methionine and valine. The work shows that in the reaction with amino acids, the
•OH radical is more reactive than
(which results from the difference in redox potential), but the difference is especially large in the case of non-aromatic amino acids (it is two or even three orders of magnitude). Of course, the
•OH radical can react with all the amino acids in HSA, but
reacts mostly with Tyr and Trp. Our own study confirms that hydroxyl radicals are not effective in the formation of DT in histone solutions, and this is in line with previous research by others [
63,
64]. It can therefore be assumed that as a rule, the high reactivity of
•OH radicals with protein amino acids results in a lower relative oxidation efficiency of aromatic protein residues and a lower probability of forming dityrosine bridges. In the case of N
2O-saturated and irradiated HSA solution, practically, we did not observe DT. It is probably related to the decay of TyrO
• radicals on the HSA surface. In order to induce DT formation, the TyrO
• radicals from different, modified protein molecules must react with each other. For non-selective, very reactive species, such as hydroxyl radicals, there is a high probability that the TyrO
• radical from one HSA molecule will react with a radical other than TyrO
• generated in another radiation-modified albumin molecule. The use of mild oxidizing species leads to the formation of only radicals centered on aromatic amino acid residues, mainly on or nearly the surface of proteins. In this case, the probability of recombination of TyrO
• radicals generated on the surfaces of two different HSA molecules increases, which leads to the formation of intermolecular DT. This does not exclude the participation of radicals generated within the HSA (e.g., at a tryptophan residue) in the formation of DT on the protein surface (or near the surface of the HSA where some Tyr residues are found).
It has long been known that within polypeptides, enzymes or proteins exposed to ionizing radiation, long-range electron transfer from tyrosine residues to semi-oxidized tryptophan moiety occurs. It can therefore be assumed that in the case of HSA, the residue of Trp214 will be involved indirectly in the formation of DT bridges. Such a hypothesis is confirmed by the observation of long-range electron transfer in HSA Tyr residue to the neutral tryptophan radical generated in pulse radiolysis [
65]. This mechanism was proposed by Hashimoto et al. [
59] to explain DT formation in lysozyme solutions as a result of the reaction of this protein with the radicals
. Of course, due to the presence of only one tryptophan group in the structure of HSA, this reaction channel may be of less importance in our case.
It is interesting to compare changes in the emission spectra of HSA solutions caused by the reaction of hydroxyl or azide radicals with the tested protein used in high concentration. For this purpose, four HSA solutions with a concentration of 300 µM were studied, all saturated with N
2O, and two additionally containing 0.1 M NaN
3. The irradiated solutions (two out of four) absorbed a dose of 27.8 kGy. The excitation of the solutions with light with a wavelength of 320 nm leads to a wide emission band of HSA concentration (self-aggregates) aggregates (λ
max = 415 nm). The intensity of this band in the presence of azide is slightly lower, which is the result of the quenching of the excited states of aggregates by NaN
3. This applies to all used excitation light wavelengths (range 295–410 nm). This effect has already been described by us. In the case of the reaction of
radicals with HSA, an intermolecular DT bridge is generated, as evidenced by an intense fluorescence band with a maximum intensity at about 403 nm (see
Figure 20).
The reaction of the
•OH radical with HSA leads mainly to species other than DT, as evidenced by a broad, less intense red-shifted band (λ
max = 414 nm,
Figure 20). These species are the result of a recombination of radicals generated in a non-specific manner as a result of the recombination of radicals generated on the surface of HSA molecules. This leads to the formation of numerous covalent bridges (including C-C, it is also possible to generate C-S bonds), and consequently HSA dimers and aggregates. The resulting HSA nanostructures lead to light emission similar to that typical for concentration aggregates. This is clearly illustrated by
Figure 21 summarizing the emission spectra recorded after excitation with light with a wavelength of 370 nm. The use of the correction procedure for the emission spectra (λ
exc = 320 nm) for the inner filter effect for HSA solutions (300 μM) exposed to the
or
•OH radicals increases the intensity of the emission band by about five and four times, respectively, compared with the spectra without correction (
Figure S3 in
Supplementary Materials. In the case of applying the correction procedure of the emission spectrum resulting from the reaction of the
•OH radical with HSA, the shift of the band maximum from 414 nm (before correction) to 407 nm (after correction) is visible.
For the HSA solution, in which the
•OH radical reacted with the protein, the emission band significantly increases and differs in intensity from the remaining “concentration” bands, which are practically unchanged. This indicates that DT bridging does not increase the number of concentration aggregates (the high-energy ones, related to an excitation band with a maximum of about 350 nm; maximum band at 3.6 eV). An additional confirmation of this is the lower scattering of the excitation light by the solution irradiated in the presence of NaN
3 as compared to the one in which the
•OH radical is generated, as shown in
Figure 21 for the range 285–392 nm (gray circle). Of course, this is also observed in the excitation spectrum recorded at 475 nm. DT bridging is accompanied by an increase in the number of low-energy HSA self-aggregates emitting light above 500 nm; the maximum band at 3.1 eV (see
Figure S4 for λ
det = 525 nm in
Supplementary Materials). There are much fewer of them than those initiated by
•OH radicals.
Excitation of the aqueous HSA solution (300 µM) with 390 nm light leads to the emission band centered at 455 nm. After irradiating this HSA solution saturated with N
2O, the emission band was recorded with the maximum at 445 nm. The intensity of this band is 2.9 times higher with respect to the intensity of the emission spectrum of neat solution (see
Figure S5 in in
Supplementary Materials). The reaction of
with HSA resulted in an emission band with a maximum of 470 nm (λ
exc = 390 nm). The intensity of this band is similar to the intensity of emission of neat solution. The use of the inner filter effect compensation procedure leads to an approx. 2-fold increase in the intensity of emission bands in irradiated solutions.
The HSA solution was analyzed by SDS-PAGE electrophoresis before and after irradiation with electron pulses (27.8 kGy) to detect the presence of monomer, dimer and aggregate molecules. First, 20 µL samples containing 30, 70, 300, and 600 µM HSA were tested before irradiation and N
2O-saturated solution containing 300 µM HSA or 0.1 M NaN
3 was tested after irradiation with a dose of 27.8 kGy. The electrophoresis results of non-irradiated solutions of HSA were consistent with emission measurements and showed a concentration-dependent aggregation. The aggregation of a commercial HSA was investigated by two different techniques, high-performance liquid chromatography and gel electrophoresis [
66]. The higher the human serum albumin concentration, the more the monomer proportion decreases. Within the range of HSA concentrations of 58–435 µM, the dimer content in relation to the remaining protein fractions is about 24%. Our results, although not as precise as in [
66], indicate a similar dimer contribution to other forms of HSA. The dimer is the most abundant object next to the monomer in HSA solutions in a very wide range of concentrations. It is important that in the conclusion from work [
66], the HSA shows reversible aggregation with increasing concentration, which has been confirmed by us in spectroscopic measurements as a result of dilution of concentrated protein solutions.
It was evident that HSA aggregates increase in size after irradiation, regardless of the type of oxidant. The SDS-PAGE date indicates that the irradiation of HSA solution leads to the generation of aggregates with high MW (these aggregates are not able to enter in the running gel).
After the irradiation of the solutions, the absorbance of the tested solutions increases. When HSA aggregates are formed, the increase in absorbance consists of two components of actual absorbance and “absorbance” due to scattering of the analyzing light. The correction is especially recommended for concentrated HSA solutions (above 300 µM). An example of the emission spectrum after correction for the inner filter effect is shown in
Figure S5. To illustrate the effect of emission correction on the intensity and position of the emission spectrum, uncorrected and corrected spectra of HSA after oxidation by
•OH and
radicals are compared (λ
exc = 390 nm). These experiments clearly revealed that after emission correction, the relative intensity of the HSA emission band in both (
•OH and
) cases increases. In the case of HSA aggregates, which emits deeper into the red after excitation of the albumin solution with 390 nm light, the use of emission correction shifts the spectrum by 15 nm (445 nm→460 nm, ΔE = 0.09 eV) and 25 nm (470 nm→495 nm, ΔE = 0.0566 eV) for
•OH and
radicals, respectively.
In this study, we focused on the qualitative characteristics of HSA dimers and aggregates; therefore, it is not always necessary to correct the emission intensity values. Moreover, the absorption/emission spectra of albumin solutions with concentrations above 300 µM were verified by measuring the spectra of these solutions in a cuvette with an optical path length of 0.2 mm. We have found that 2 × 10 mm fluorescent cuvettes (excitation along the 2 mm optical path) significantly reduce the internal filter effect.
The scavenging of hydroxyl and azide radicals by protein is a complex process. The HSA consists of a single polypeptide chain of 585 amino acids. The free radicals, namely
•OH and
, may react with amino acids residues within albumin. We conducted a series pulse radiolysis experiment with aqueous solutions containing amino acids, including L-tyrosine, L-tryptophan, phenylalanine, methionine, histidine and tyrosine-tryptophan.
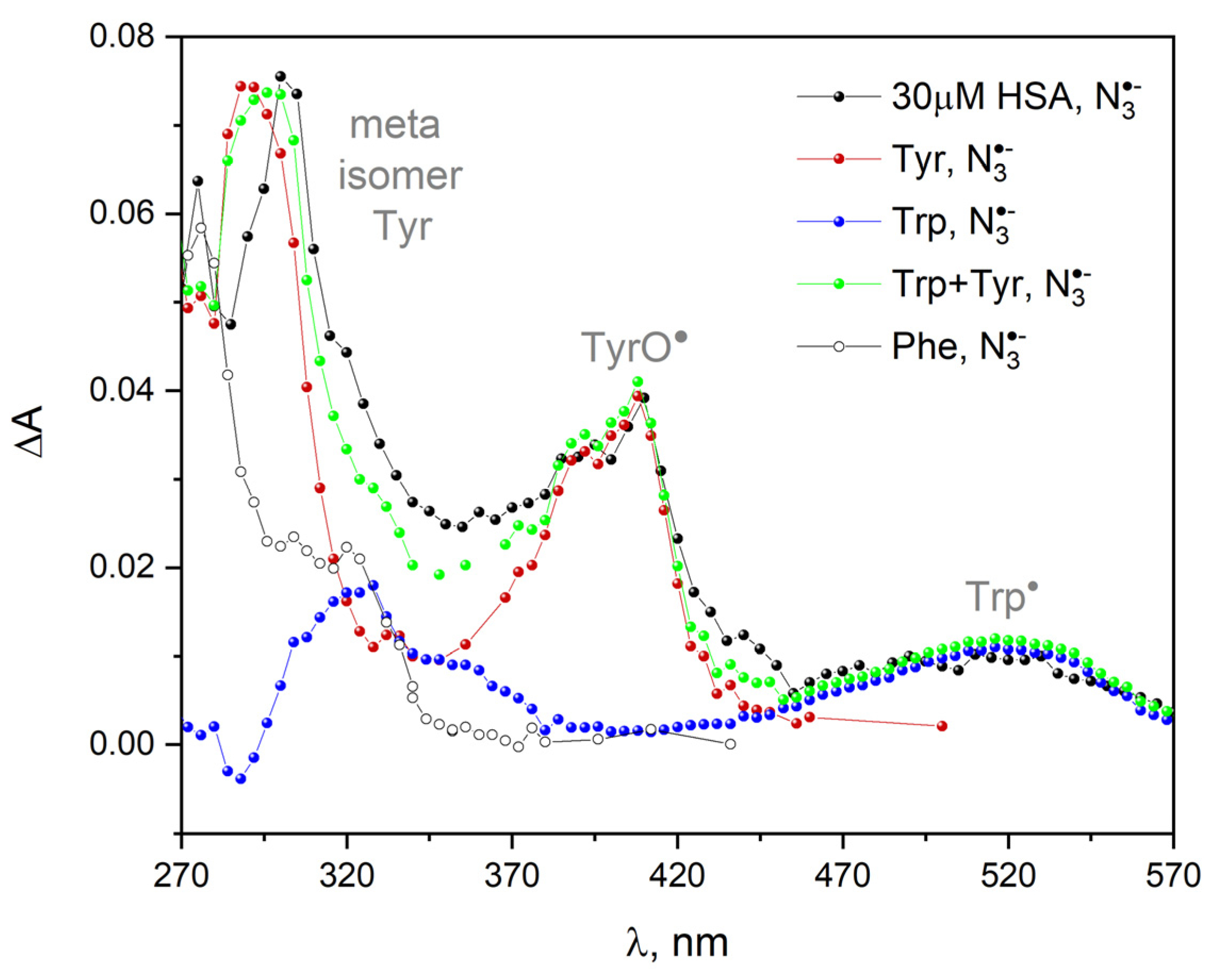
Figure 22 presents the transient absorption spectra of primary products of HSA one-electron oxidation by azide radical (N
2O-saturated 30 µM HSA solution containing 0.1 M NaN
3). These spectra are compared with the spectra obtained after the oxidation of various amino acids in a series of pulse radiolysis experiments, each with one selected amino acid at a concentration of 1 mM. The measurements were carried out under anaerobic conditions to avoid subsequent reactions of the generated radicals with oxygen. ΔA denotes differentia absorbance with respect to that of the non-irradiated sample (before the pulse). We can distinguish three spectral regions in the transient absorption spectra of HSA: with maximum at 300, 410 and 520 nm. We are able to assign the aforementioned absorption bands to the products obtained as a result of the oxidation of Tyr and Trp within albumin by azide radicals: the band of the Tyr meta isomer centered at 300 nm and phenoxyl radicals TyrO
• with a maximum near 410 nm. The band with a maximum at 520 nm is attributed to the oxidation of the tryptophan molecule,
:
The reaction of with histidine and methionine in neutral solution is much slower than with other amino acids. The reaction between azide radicals and His or Met molecules is not observed.
When BSA reacts with
radicals, similar transient absorption spectra were obtained as in the case of the reaction of azide radical with HSA. A significant difference was observed in the 450–570 nm region. The higher absorbance in the transition spectra for BSA after one-electron oxidation, observed above 500 nm (
Figure S6 in
Supplementary Materials), results from the presence of two tryptophan residues in the BSA structure and not one as for HSA. For BSA, the absorption band associated with Trp is about twice as intense as the tryptophan radical in HSA. It should be noted that the transient absorption in the 400 nm region (TyrO
•) for the
radical-induced oxidation of the HSA and BSA is the same.
The comparison of the absorption spectra recorded during the pulse radiolysis of aqueous solutions of several amino acids with analogous spectrum for HSA indicate that the mechanism of protein oxidation by hydroxyl and azide radicals differs significantly.
Figure 23 shows the transient absorption spectra recorded after the electron-pulsed irradiation of N
2O-saturated buffer solutions containing various amino acids or human serum albumin from a concentration of 30 µM. The transient absorption spectrum of HSA solution under oxidative conditions shows a maximum at 290 nm and in the spectral range 370–480 nm. The band with a maximum at 290 nm is due to products obtained as a result of the oxidation of Trp, Tyr, Phe, and His by hydroxyl radical. A broad absorption in the spectral range 370–480 nm can be attributed to the absorption of several oxidation products: Trp, Met, and His (superposition of absorption spectra of various amino acid oxidation products). The literature data indicate that methionine is oxidized to sulfoxide. However, this reaction is of minor importance in the process of peptide/protein oxidation in a N
2O-saturated solution. The lack of a band with the maximum at approx. 510 nm attributed to the Trp
• in the transient absorption spectrum of HSA may indicate that hydroxyl radicals do not react with Trp214 within protein. It is less likely that the OH radical will reach Trp 214 and will react but not oxidize the tryptophan moiety. Referring to our earlier work [
54] in which we showed that the hydrated electron does not reach the Sudlow 1 site, it can be concluded that this densely packed and hydrophobic domain is not directly exposed to radical attack. We noticed that which is assigned to the TyrO
• radical is formed in a longer time scale after the OH radical reaction with HSA (TyrO
• band is visible in the HSA spectrum after 170 µs after the electron pulse; see
Figure S7). It can be assumed that this is the result of the transformation of the OH-adduct hydroxycyclohexadienyl radical) in the ortho position to the OH group into the phenoxyl radical of tyrosine: TyrO
•. The conversion described above for the reaction of free tyrosine with the OH radical in the protein is slower. The amount of the TyrO
• radical generated in HSA is small, but it indicates that DT bridging in vivo as a result of OH radical attack on albumin is highly likely.
The literature describes that more than 94% of hydroxyl radicals form adducts on the aromatic ring of phenylalanine as a result of scavenging of
•OH [
67]. The reaction of phenylalanine with a hydroxyl radical leads to the formation of 4-, 3-, and 2-hydroxyl-phenylalanine (para-, meta-, and ortho-tyrosine, respectively) and other Phe derivatives. Solar has proposed the preference position of
•OH addition to the phenylalanine ring: ortho > para > meta.
Figure S8 in
Supplementary Materials shows transient absorption spectra recorded after electron pulse irradiation of N
2O-saturated buffer solution containing 1 mM phenylalanine. We can distinguish three spectral regions: broad absorption bands with maxima near 250, 270 and 320 nm. These bands were observed immediately after the irradiation of N
2O-saturated Phe solution. The reaction of
•OH radicals with phenylalanine mainly forms an OH adduct in the ortho position. The absorption centered near 270 nm is probably due to 3,4-dihydroxyphenylalanine (DOPA). In the transient absorption spectrum of an HSA solution saturated with N
2O exposed to an electron beam, it is difficult to recognize the bands derived from phenylalanine-OH adducts (
Figure S8). This is due to the fact that other components of the protein attacked by the hydroxyl radical absorb light in this spectral range.
In our opinion, the absorption spectrum of the protein after the reaction with the •OH radicals was more difficult to analyze due to the lack of selectivity of the hydroxyl radical. The •OH radicals can react with protein molecules in three different ways (preferentially on the surface of albumin molecules) in contrast to a specific one-electron oxidant, the azide radical. Taking into account the rate constants of the and •OH radicals’ reaction with individual amino acids, it is safe to postulate that the absorption spectrum of the oxidized protein does not have to correspond to the absorption spectra of the isolated oxidized amino acids.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






















