
Insights into Domain Organization and Regulatory Mechanism of Cystathionine Beta-Synthase from Toxoplasma gondii

 , , and
, , and
Abstract
:1. Introduction
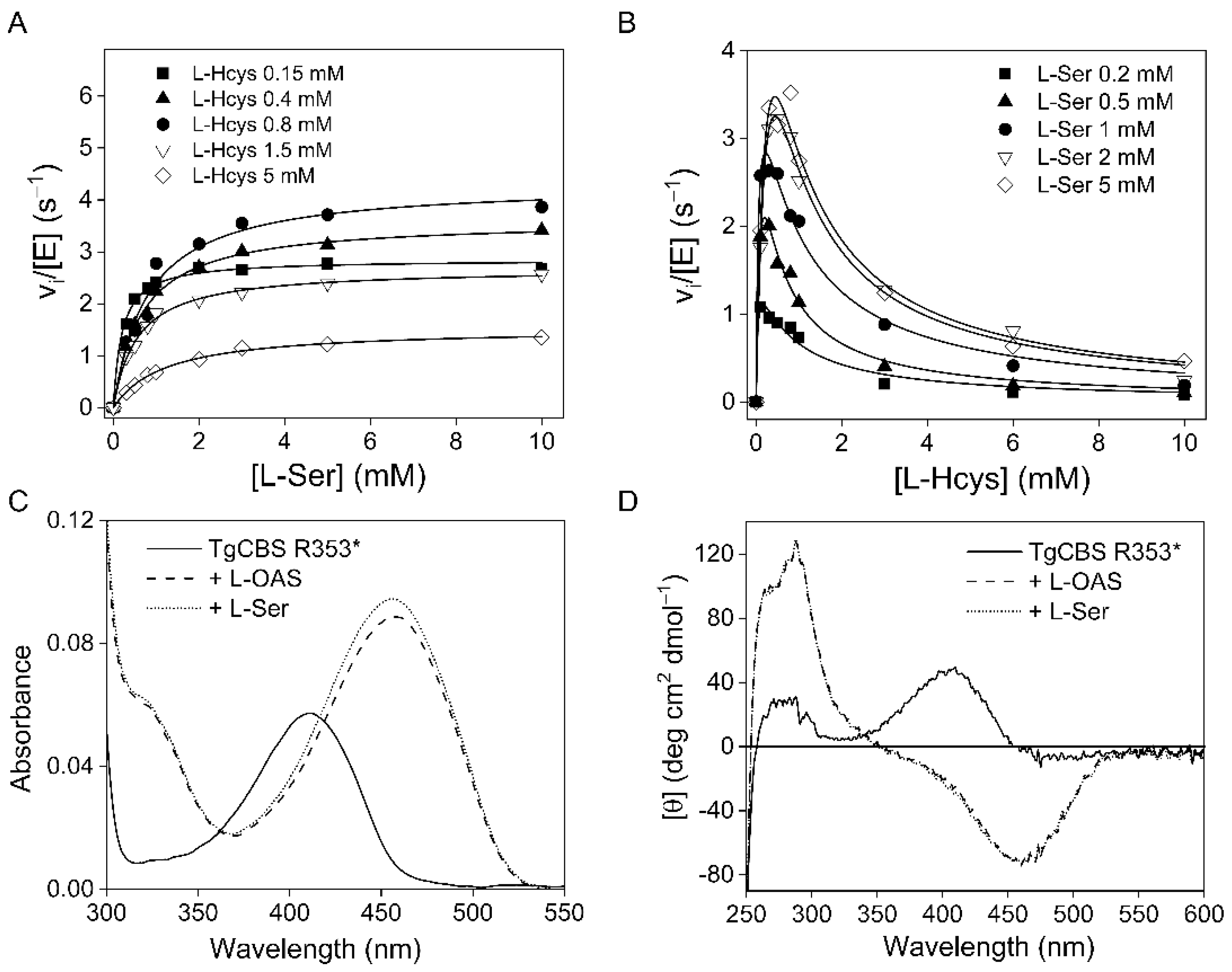
2. Results
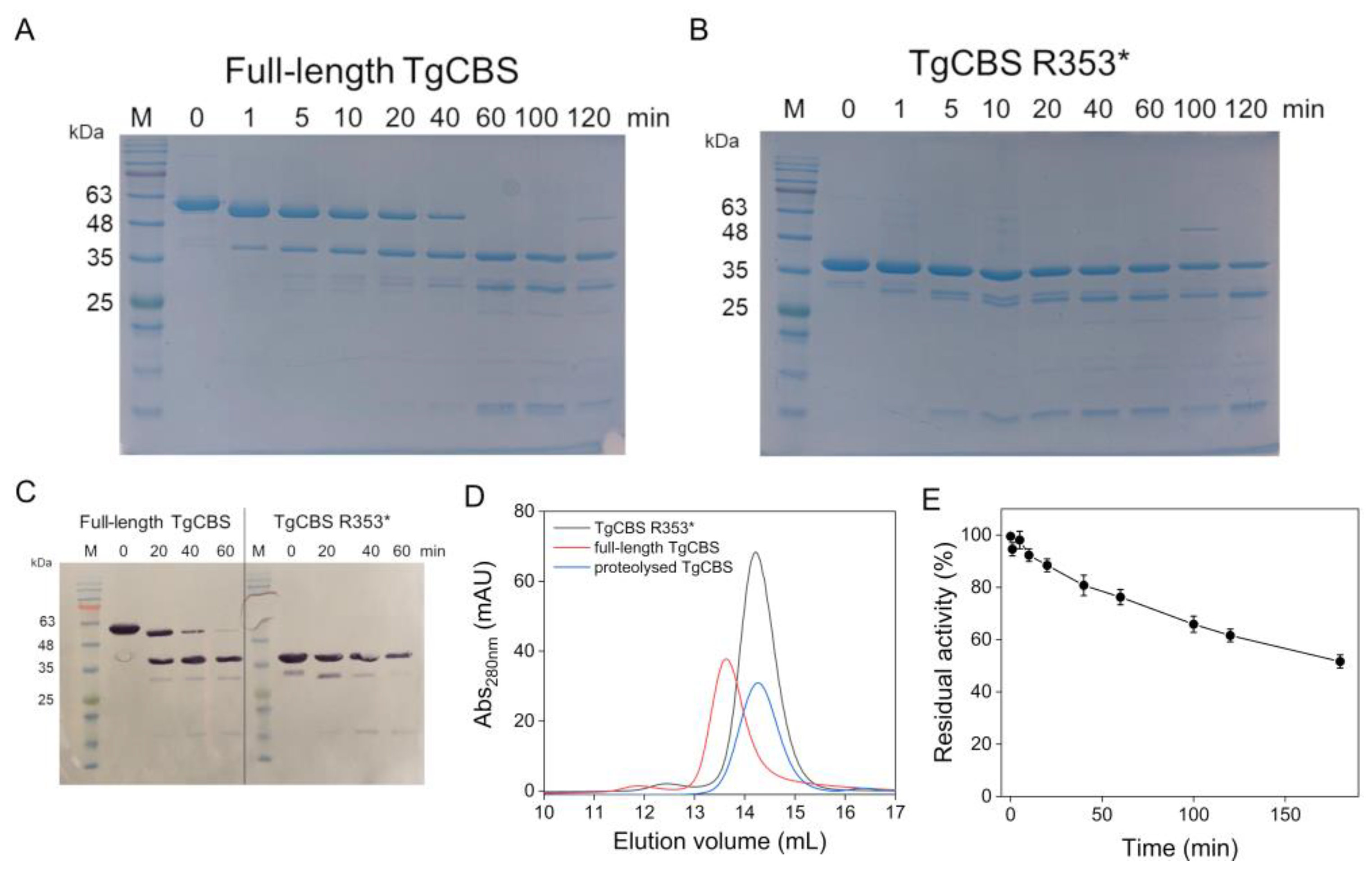
2.1. Production of Recombinant TgCBS Lacking the C-Terminal Domain
2.2. Trypsin Cleavage of Recombinant TgCBS
2.3. Thermal Denaturation
2.4. Structural Analysis of Cavities in TgCBS
3. Discussion
4. Materials and Methods
4.1. Protein Production
4.2. Steady-State Kinetics
4.3. Spectroscopic Measurements
4.4. Gel Filtration
4.5. Trypsin Cleavage of the Enzyme
4.6. Ferguson Plot
4.7. Differential Scanning Calorimetry
4.8. Structural Analysis of TgCBS and Its Homologs
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, R. Two’s Company, Three’s A Crowd: Can H2s Be the Third Endogenous Gaseous Transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, M.; Janosik, M.; Kery, V.; Kraus, J.P.; Burkhard, P. Structure of Human Cystathionine Beta-Synthase: A Unique Pyridoxal 5′-Phosphate-Dependent Heme Protein. EMBO J. 2001, 20, 3910–3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutmos, M.; Kabil, O.; Smith, J.L.; Banerjee, R. Structural Basis for Substrate Activation and Regulation by Cystathionine Beta-Synthase (Cbs) Domains in Cystathionine Β-Synthase. Proc. Natl. Acad. Sci. USA 2010, 107, 20958–20963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majtan, T.; Pey, A.L.; Fernández, R.; Fernández, J.A.; Martínez-Cruz, L.A.; Kraus, J.P. Domain Organization, Catalysis and Regulation of Eukaryotic Cystathionine Beta-Synthases. PLoS ONE 2014, 9, E105290. [Google Scholar] [CrossRef] [Green Version]
- González-Recio, I.; Fernández-Rodríguez, C.; Simón, J.; Goikoetxea-Usandizaga, N.; Martínez-Chantar, M.L.; Astegno, A.; Majtan, T.; Martinez-Cruz, L.A. Current Structural Knowledge on Cystathionine Β-Synthase, A Pivotal Enzyme in The Transsulfuration Pathway. In eLS; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2020; Volume 1, pp. 453–468. [Google Scholar] [CrossRef]
- Taoka, S.; Lepore, B.W.; Kabil, O.; Ojha, S.; Ringe, D.; Banerjee, R. Human Cystathionine Beta-Synthase Is a Heme Sensor Protein. Evidence That the Redox Sensor Is Heme and Not the Vicinal Cysteines in The Cxxc Motif Seen in The Crystal Structure of The Truncated Enzyme. Biochemistry 2002, 41, 10454–10461. [Google Scholar] [CrossRef]
- Janosík, M.; Oliveriusová, J.; Janosíková, B.; Sokolová, J.; Kraus, E.; Kraus, J.P.; Kozich, V. Impaired Heme Binding and Aggregation of Mutant Cystathionine Beta-Synthase Subunits in Homocystinuria. Am. J. Hum. Genet. 2001, 68, 1506–1513. [Google Scholar] [CrossRef] [Green Version]
- Majtan, T.; Singh, L.R.; Wang, L.; Kruger, W.D.; Kraus, J.P. Active Cystathionine Beta-Synthase Can Be Expressed in Heme-Free Systems in The Presence of Metal-Substituted Porphyrins or A Chemical Chaperone. J. Biol. Chem. 2008, 283, 34588–34595. [Google Scholar] [CrossRef] [Green Version]
- Weeks, C.L.; Singh, S.; Madzelan, P.; Banerjee, R.; Spiro, T.G. Heme Regulation of Human Cystathionine Β-Synthase Activity: Insights from Fluorescence and Raman Spectroscopy. J. Am. Chem. Soc. 2009, 131, 12809–12816. [Google Scholar] [CrossRef] [Green Version]
- Christen, P.; Mehta, P.K. From Cofactor to Enzymes. The Molecular Evolution of Pyridoxal-5′Phosphate-Dependent Enzymes. Chem. Rec. 2001, 1, 436–447. [Google Scholar] [CrossRef]
- Ereno-Orbea, J.; Majtan, T.; Oyenarte, I.; Kraus, J.P.; Martinez-Cruz, L.A. Structural Basis of Regulation and Oligomerization of Human Cystathionine Beta-Synthase, The Central Enzyme of Transsulfuration. Proc. Natl. Acad. Sci. USA 2013, 110, E3790–E3799. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A. The Structure of a Domain Common to Archaebacteria and The Homocystinuria Disease Protein. Trends Biochem. Sci. 1997, 22, 12–13. [Google Scholar] [CrossRef]
- Baykov, A.A.; Tuominen, H.K.; Lahti, R. The Cbs Domain: A Protein Module with An Emerging Prominent Role in Regulation. ACS Chem. Biol. 2011, 6, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Anashkin, V.A.; Baykov, A.A.; Lahti, R. Enzymes Regulated Via Cystathionine Β-Synthase Domains. Biochemistry 2017, 82, 1079–1087. [Google Scholar] [CrossRef]
- Mccorvie, T.J.; Kopec, J.; Hyung, S.J.; Fitzpatrick, F.; Feng, X.; Termine, D.; Strain-Damerell, C.; Vollmar, M.; Fleming, J.; Janz, J.M.; et al. Inter-Domain Communication of Human Cystathionine Β-Synthase: Structural Basis of S-Adenosyl-L-Methionine Activation. J. Biol. Chem. 2014, 289, 36018–36030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ereño-Orbea, J.; Majtan, T.; Oyenarte, I.; Kraus, J.P.; Martínez-Cruz, L.A. Structural Insight into The Molecular Mechanism of Allosteric Activation of Human Cystathionine Β-Synthase By S-Adenosylmethionine. Proc. Natl. Acad. Sci. USA 2014, 111, E3845–E3852. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.; Kreinbring, C.A.; Hill, M.; Liu, C.; Petsko, G.A.; Mccune, C.D.; Berkowitz, D.B.; Liu, D.; Ringe, D. Crystal Structures of Cystathionine Beta-Synthase from Saccharomyces Cerevisiae: One Enzymatic Step at A Time. Biochemistry 2018, 57, 3134–3145. [Google Scholar] [CrossRef]
- Devi, S.; Abdul Rehman, S.A.; Tarique, K.F.; Gourinath, S. Structural Characterization and Functional Analysis of Cystathionine Beta-Synthase: An Enzyme Involved in The Reverse Transsulfuration Pathway of Bacillus anthracis. FEBS J. 2017, 284, 3862–3880. [Google Scholar] [CrossRef] [Green Version]
- Devi, S.; Tarique, K.F.; Ali, M.F.; Abdul Rehman, S.A.; Gourinath, S. Identification and Characterization of Helicobacter Pylori O-Acetylserine-Dependent Cystathionine Β-Synthase, A Distinct Member of The Plp-Ii Family. Mol. Microbiol. 2019, 112, 718–739. [Google Scholar] [CrossRef]
- Matoba, Y.; Yoshida, T.; Izuhara-Kihara, H.; Noda, M.; Sugiyama, M. Crystallographic and Mutational Analyses of Cystathionine Beta-Synthase in The H2 S-Synthetic Gene Cluster in Lactobacillus plantarum. Protein Sci. 2017, 26, 763–783. [Google Scholar] [CrossRef] [Green Version]
- Conter, C.; Fruncillo, S.; Fernández-Rodríguez, C.; Martínez-Cruz, L.A.; Dominici, P.; Astegno, A. Cystathionine Β-Synthase is Involved in Cysteine Biosynthesis and H(2)S Generation in Toxoplasma gondii. Sci. Rep. 2020, 10, 14657. [Google Scholar] [CrossRef]
- Fernández-Rodríguez, C.; Oyenarte, I.; Conter, C.; González-Recio, I.; Núñez-Franco, R.; Gil-Pitarch, C.; Quintana, I.; Jiménez-Osés, G.; Dominici, P.; Martinez-Chantar, M.L.; et al. Structural Insight into The Unique Conformation of Cystathionine Β-Synthase from Toxoplasma gondii. Comput. Struct. Biotechnol. J. 2021, 19, 3542–3555. [Google Scholar] [CrossRef] [PubMed]
- Janošík, M.; Kery, V.; Gaustadnes, M.; Maclean, K.N.; Kraus, J.P. Regulation of Human Cystathionine Β-Synthase By S-Adenosyl-L-Methionine: Evidence for Two Catalytically Active Conformations Involving an Autoinhibitory Domain in the C-Terminal Region. Biochemistry 2001, 40, 10625–10633. [Google Scholar] [CrossRef] [PubMed]
- Kery, V.; Poneleit, L.; Kraus, J.P. Trypsin Cleavage Of Human Cystathionine Beta-Synthase Into An Evolutionarily Conserved Active Core: Structural And Functional Consequences. Arch. Biochem. Biophys. 1998, 355, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Kery, V.; Poneleit, L.; Meyer, J.D.; Manning, M.C.; Kraus, J.P. Binding of Pyridoxal 5′-Phosphate to The Heme Protein Human Cystathionine Beta-Synthase. Biochemistry 1999, 38, 2716–2724. [Google Scholar] [CrossRef] [PubMed]
- Jhee, K.-H.; Mcphie, P.; Miles, E.W. Domain Architecture of The Heme-Independent Yeast Cystathionine Β-Synthase Provides Insights into Mechanisms of Catalysis and Regulation. Biochemistry 2000, 39, 10548–10556. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.A. Starch-Gel Electrophoresis—Application to The Classification of Pituitary Proteins and Polypeptides. Metab. Clin. Exp. 1964, 13, 985–1002. [Google Scholar] [CrossRef]
- Astegno, A.; Giorgetti, A.; Allegrini, A.; Cellini, B.; Dominici, P. Characterization Of C-S Lyase from C. Diphtheriae: A Possible Target for New Antimicrobial Drugs. Biomed. Res. Int. 2013, 2013, 701536. [Google Scholar] [CrossRef] [Green Version]
- Astegno, A.; Allegrini, A.; Piccoli, S.; Giorgetti, A.; Dominici, P. Role of Active-Site Residues Tyr55 and Tyr114 in Catalysis and Substrate Specificity of Corynebacterium Diphtheriae C-S Lyase. Proteins 2015, 83, 78–90. [Google Scholar] [CrossRef]
- Oliveriusova, J.; Kery, V.; Maclean, K.N.; Kraus, J.P. Deletion Mutagenesis of Human Cystathionine Beta-Synthase. Impact On Activity, Oligomeric Status, And S-Adenosylmethionine regulation. J. Biol. Chem. 2002, 277, 48386–48394. [Google Scholar] [CrossRef] [Green Version]
- Pey, A.L.; Majtan, T.; Sanchez-Ruiz, J.M.; Kraus, J.P. Human Cystathionine Β-Synthase (Cbs) Contains Two Classes of Binding Sites For S-Adenosylmethionine (Sam): Complex Regulation of Cbs Activity and Stability by Sam. Biochem. J. 2013, 449, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.D.; Murphy, K.P. Protein Structure and The Energetics of Protein Stability. Chem. Rev. 1997, 97, 1251–1268. [Google Scholar] [CrossRef]
- Giménez-Mascarell, P.; Majtan, T.; Oyenarte, I.; Ereño-Orbea, J.; Majtan, J.; Klaudiny, J.; Kraus, J.P.; Martínez-Cruz, L.A. Crystal Structure of Cystathionine Β-Synthase from Honeybee Apis mellifera. J. Struct. Biol. 2018, 202, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Gut, H.; Dominici, P.; Pilati, S.; Astegno, A.; Petoukhov, M.V.; Svergun, D.I.; Grütter, M.G.; Capitani, G. A Common Structural Basis for Ph- And Calmodulin-Mediated Regulation in Plant Glutamate Decarboxylase. J. Mol. Biol. 2009, 392, 334–351. [Google Scholar] [CrossRef] [PubMed]
- Aitken, S.M.; Kirsch, J.F. Kinetics of The Yeast Cystathionine Beta-Synthase Forward and Reverse Reactions: Continuous Assays and The Equilibrium Constant for The Reaction. Biochemistry 2003, 42, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Astegno, A.; Capitani, G.; Dominici, P. Functional Roles of The Hexamer Organization of Plant Glutamate decarboxylase. Biochim. Biophys. Acta 2015, 1854, 1229–1237. [Google Scholar] [CrossRef]
- Allegrini, A.; Astegno, A.; La Verde, V.; Dominici, P. Characterization Of C-S Lyase from Lactobacillus Delbrueckii Subsp. Bulgaricus Atcc Baa-365 and Its Potential Role in Food Flavour Applications. J. Biochem. 2017, 161, 349–360. [Google Scholar] [CrossRef]
- Astegno, A.; Maresi, E.; Bertoldi, M.; La Verde, V.; Paiardini, A.; Dominici, P. Unique Substrate Specificity of Ornithine Aminotransferase from Toxoplasma gondii. Biochem. J. 2017, 474, 939–955. [Google Scholar] [CrossRef] [Green Version]
- La Verde, V.; Trande, M.; D’onofrio, M.; Dominici, P.; Astegno, A. Binding of Calcium and Target Peptide to Calmodulin-Like Protein Cml19, The Centrin 2 Of Arabidopsis thaliana. Int. J. Biol. Macromol. 2018, 108, 1289–1299. [Google Scholar] [CrossRef]
- Astegno, A.; Conter, C.; Bertoldi, M.; Dominici, P. Structural Insights into the Heme Pocket and Oligomeric State of Non-Symbiotic Hemoglobins from Arabidopsis thaliana. Biomolecules 2020, 10, 1615. [Google Scholar] [CrossRef]
- Schrödinger, L.D.; Pymol, W. Available online: http://www.Pymol.Org/Pymol (accessed on 7 July 2022).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. Ucsf Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. Castp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pey, A.L.; Martínez-Cruz, L.A.; Kraus, J.P.; Majtan, T. Oligomeric status of human cystathionine beta-synthase modulates AdoMet binding. FEBS Lett. 2016, 590, 4461–4471. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinetic Parameter | Full-Length b | R353* |
|---|---|---|
| L-Ser + L-Hcys→L-Cth + H2O | ||
| kcat (s−1) | 6.3 ± 0.4 | 5.7 ± 0.8 |
| KmL-Ser (mM) | 0.42 ± 0.04 | 0.6 ± 0.1 |
| KmL-Hcys(mM) | 0.23 ± 0.03 | 0.20 ± 0.04 |
| kcat/KmL-Ser (mM−1 s−1) | 15 ± 2 | 10 ± 2 |
| kcat/KmL-Hcys (mM−1 s−1) | 27 ± 4 | 29 ± 6 |
| KiL-Hcys (mM) | 1.0 ± 0.1 | 1.3 ± 0.4 |
| L-OAS + L-Hcys→L-Cth + acetate | ||
| kcat (s−1) | 5.5 ± 0.1 | 5.6 ± 0.6 |
| KmL-OAS (mM) | 1.3 ± 0.2 | 2.8 ± 0.5 |
| KmL-Hcys (mM) | 0.20 ± 0.05 | 0.27 ± 0.04 |
| kcat/KmL-OAS (mM−1 s−1) | 4.2 ± 0.7 | 2.0 ± 0.4 |
| kcat/KmL-Hcys (mM−1 s−1) | 28 ± 7 | 21 ± 4 |
| KiL-Hcys (mM) | 1.4 ± 0.2 | 1.4 ± 0.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conter, C.; Fruncillo, S.; Favretto, F.; Fernández-Rodríguez, C.; Dominici, P.; Martínez-Cruz, L.A.; Astegno, A. Insights into Domain Organization and Regulatory Mechanism of Cystathionine Beta-Synthase from Toxoplasma gondii. Int. J. Mol. Sci. 2022, 23, 8169. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158169
Conter C, Fruncillo S, Favretto F, Fernández-Rodríguez C, Dominici P, Martínez-Cruz LA, Astegno A. Insights into Domain Organization and Regulatory Mechanism of Cystathionine Beta-Synthase from Toxoplasma gondii. International Journal of Molecular Sciences. 2022; 23(15):8169. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158169
Chicago/Turabian StyleConter, Carolina, Silvia Fruncillo, Filippo Favretto, Carmen Fernández-Rodríguez, Paola Dominici, Luis Alfonso Martínez-Cruz, and Alessandra Astegno. 2022. "Insights into Domain Organization and Regulatory Mechanism of Cystathionine Beta-Synthase from Toxoplasma gondii" International Journal of Molecular Sciences 23, no. 15: 8169. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158169